一种PCPN配体的制备、乙烯齐聚催化剂及其应用的制作方法
一种pcpn配体的制备、乙烯齐聚催化剂及其应用
技术领域
1.本发明属于乙烯齐聚反应技术领域,具体涉及一种pcpn配体的制备、乙烯齐聚催化剂及其应用。
背景技术:
2.线型α-烯烃是重要的化工原料,可用作聚烯烃共聚单体、pvc增塑剂、表面活性剂、润滑油添加剂等众多领域。其中共聚单体的用量占α-烯烃消耗量的一半以上,以1-辛烯和1-己烯的聚烯烃产品具有良好的力学性能和优异的加工性能,在工业生产中具有大量的需求。
3.α-烯烃的工艺主要有选择性齐聚工艺和非选择性齐聚工艺。乙烯非选择性齐聚工艺中1-辛烯的1-己烯的选择性都在10%到20%之间,该工艺对于制备1-辛烯和1-己烯的转化率非常低,且有数十种各类副产物,难以利用。
4.而选择性齐聚工艺1-辛烯和1-己烯的选择性占总产物的90%以上,选择性好,转化率高,适合工业化生产。选择性齐聚常见的催化剂为铬系催化剂体系,分别是pccp体系和pnp体系。其中pccp体系,必须要在络合金属铬之后,才具有聚合活性,因此在存在制备过程复杂的问题,且在络合以后,pccp-cr金属配合物不溶于常规聚合溶剂,如饱和烷烃、isoper-e、甲苯等,也存在进料系统复杂,定量困难,催化剂均匀性难以保证等问题。
5.因此,自2004年sasol公司首次提出pnp骨架后,该骨架一直是乙烯选择性齐聚的研究热点。该体系具有1-辛烯和1-己烯的总选择性在90%以上,使用前不需要提前络合等优点。
6.虽然经过十多年的研究,但1-辛烯和1-己烯的选择性并没有很大的提升,尤其是聚合物问题一直没能妥善解决。选择性齐聚工业化面临的主要问题之一就是副产聚合物挂壁挂釜引起的堵塞问题。反应釜一旦堵塞,势必影响连续反应,进行停车清理,影响产产品质量和装置的经济性,更甚至会引起管线憋压而引发更大的装置风险。
7.专利cn104961618a公布了一种添加酚类物质作为聚乙烯抑制剂的方法,来降低聚合物的生成,但由于酚类物质的添加影响了催化剂的活性,也对后期产品的纯化带来的其他问题。专利cn102850168a介绍了一种釜内进行聚四氟乙烯涂层的方法,减少聚乙烯的挂壁现象,而减少聚合物堵塞的问题。但该方法并没有从本质上减少聚合物生成,在管线拐角或其他位置依旧存在聚合物挂壁堵塞等问题。
8.因此,从目前公开的各类技术文献中,并没有提出一种可以从催化剂设计方面上减少聚合物生成且不影响催化剂活性,而解决聚合物挂壁,反应釜堵塞的问题。
技术实现要素:
9.本发明的目的在于提供一种pcpn配体的制备、乙烯齐聚催化剂及其在乙烯齐聚反应中的应用。本发明的催化剂提高了乙烯齐聚反应过程中1-己烯和1-辛烯的选择性,聚合物生成量更低,催化活性好,不易堵塞设备。
10.本发明提供一种pcpn配体,其结构如式i所示:
[0011][0012]
其中,r2独立地选自芳基及其衍生物,优选的,r2选自苯基、苄基、联苯基、萘基、蒽基、2-甲基苯基、4-甲基苯基、2,4-二甲基苯基,2,6-二甲基苯基,2-乙基苯基、4-乙基苯基、2,4-二乙基苯基、2,6-二乙基苯基、2-异丙基苯基、4-异丙基苯基、2,4-二异丙基苯基、2,6-二异丙基苯基、2-丁基苯基、4-丁基苯基、2,4-二丁基苯基、2,6-二丁基苯基、4-甲氧基苯基、邻甲氧基苯基、4-乙氧基苯基、邻乙氧基苯基、2-氟基苯基、3-氟基苯基、4-氟基苯基、2-(三甲基硅烷基)苯基、3-(三甲基硅烷基)苯基、4-(三甲基硅烷基)苯基、2-(三正丁基硅烷基)苯基、3-(三正丁基硅烷基)苯基、4-(三正丁基硅烷基)苯基。
[0013]
r1独立地选自烷基、芳基及其衍生物;优选甲基、乙基、丙基、异丙基、正丁基、叔丁基、乙烯基、丙烯基、环戊基、环己基、苯基、萘基、蒽基或联苯基,更优选自甲基、乙基、异丙基、正丁基、环己基。
[0014]
本发明还提供所述的配体的制备方法,包含如下步骤:
[0015]
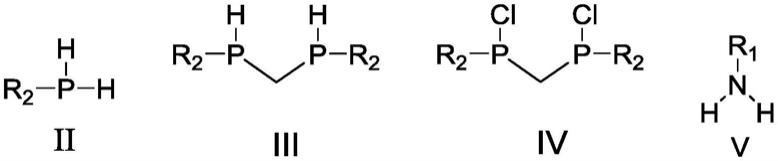
(1)取适量式ii所示化合物苯基膦、二氯甲烷,溶于溶剂a,冰水浴搅拌下,加入碱性溶液,滴加1-5h,滴加完毕后,冰水浴下反应3-10h,加入水淬灭反应,提纯处理反应液得到产物一,即为氢膦桥配体;其结构如式iii所示:
[0016]
(2)将化合物iii和六氯乙烷,溶于溶剂b,加热回流24-48h,经提纯处理反应液得到产物二,即为氯膦桥配体,其结构如式iv所示:
[0017]
(3)无水无氧条件下,将式v所示取代胺化合物溶解在溶剂a中得到反应液一;
[0018]
(4)无水无氧条件下,将式iv所示氯膦桥配体溶解在溶剂a中得到反应液二;
[0019]
(5)在-78℃下,搅拌下向反应液一中滴加反应液二,加入催化剂后缓慢升温至室温,搅拌下继续反应36-72h,提纯处理反应液得到产物,即为pcpn配体。
[0020]
步骤1)-5)中所述的式ii-式v化合物结构如下
[0021][0022]
其中,r1、r2定义与式i相同。
[0023]
优选的,所述步骤(1)中,取代苯基膦与二氯甲烷的摩尔比为1:0.3-0.7。
[0024]
优选的,所述碱性溶液为氢氧化钠或氢氧化钾水溶液,溶液的质量浓度为20%-80%。
[0025]
所述溶剂a为甲苯、甲基环己烷、乙腈、环己烷、正己烷、二氯甲烷、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺中的一种或多种。
[0026]
优选的,所述步骤(2)中,所述溶剂b选自甲苯、甲基环己烷、乙腈、环己烷、正己烷、乙醚、二氧六环中的一种或多种;
[0027]
优选的,氢膦桥配体与六氯乙烷的摩尔比为1:1-1:5。
[0028]
优选的,所述步骤(5)中,反应液一中的取代胺与反应液二中的氯膦桥配体的摩尔比为1:0.7-2。
[0029]
优选的,所述步骤(5)中,催化剂选自三乙胺,n,n-二甲基苯胺,二异丙基乙胺,4-二甲氨基吡啶。催化剂的加入量与式iv所示的氯膦桥配体的摩尔比为1:5-1:20。
[0030]
本发明所述提纯处理包括将反应液进行柱层析提纯得到目标产物以及将目标产物进行重结晶,所述柱层析提纯使用的层析柱高径比为5-10,停留时间10-60min,重结晶所用溶剂为乙醇和乙酸乙酯的混合溶剂。
[0031]
本发明的另一方面还提供一种乙烯齐聚催化剂,包括过渡金属化合物和本发明所述的pcpn配体和烷基铝助催化剂。
[0032]
本发明所述的过渡金属化合物选自铬、钼、钴、钛、钒、锆、镍和钯的化合物中的一种或两种以上,优选铬、锆和镍的化合物,过渡金属化合物包括过渡金属的有机盐、无机盐、配位络合物或者有机金属络合物,如可以为乙酰丙酮铬、氯化铬、三(四氢呋喃)三氯化铬、2-乙基己酸铬(iii)、辛酸铬(iii)、六羰基铬、(苯)三羰基铬中的一种或两种以上。
[0033]
本发明所述的烷基铝助催化剂选自三甲基铝、三乙基铝、三异丁基铝、二乙基乙氧基铝、一氯二乙基铝、二氯乙基铝、倍半乙基氯化铝、三辛基铝、甲基铝氧烷(mao)、改性甲基铝氧烷(mmao)或乙基铝氧烷中的一种或两种以上。
[0034]
在本发明所述的催化剂中,过渡金属化合物与pcpn配体的摩尔比为1:1-3,优选为1:1-2;烷基铝助催化剂与过渡金属化合物的摩尔比50-2000:1,优选90-800:1。
[0035]
本发明还提供了上述催化剂的应用,其用于乙烯齐聚反应。
[0036]
在本发明的一些优选实施方式中,所述乙烯齐聚反应的方法为:反应前需将反应釜加热至110-160℃,抽真空1-4h,采用氮气置换,待温度冷却至室温,乙烯置换,先加入溶剂c和烷基铝助催化剂,然后加入过渡金属化合物、pcpn配体,待温度达到反应温度后,依次通入0-0.8mpa氢气、2mpa-7mpa乙烯开始反应,反应温度35-90℃,优选40-70℃,反应时间10min-240min,优选20min-100min。
[0037]
所述的乙烯齐聚反应溶剂c选自正丁烷、异丁烷、正戊烷、环戊烷、甲基环戊烷、亚甲基环戊烷、正己烷、环己烷、甲基环己烷、正庚烷、正辛烷、正壬烷、苯、甲苯、二甲苯中的一种或两种以上。
[0038]
在一个更具体的实施方案中,本发明所述的催化剂组合物的聚合方法如下所示:在300ml高压反应釜中进行聚合,使用精制后的烷烃作为溶剂c。反应前将反应釜加热至130℃,抽真空1-3h,氮气置换三次。待温度冷却至室温,乙烯置换两次,先加入脱水脱氧的溶剂c和定量的烷基铝助催化剂,然后加入过渡金属化合物、pcpn配体,待温度恒定在反应温度,依次通入0.2-0.7mpa氢气、2mpa-7mpa乙烯开始反应。反应温度35-90℃,优选40-70℃,反应时间10min-240min,优选20min-100min。反应结束后,关闭乙烯进气阀门,用冰水浴或液氮迅速降温,缓慢泄压,卸釜得到乙烯齐聚产物。
[0039]
催化剂的加入量以过渡金属化合物加入量在乙烯齐聚反应体系的摩尔浓度为10-25μmol/l(溶剂),优选15-20μmol/l(溶剂)。
[0040]
目前对于pnp骨架活性的影响主要集中在两方面,一类是膦上的电子效应,另一类是n上取代基的位阻效应。pnp配体的键角一般是通过上述n上取代基的位阻,来间接调控键角的开合大小,n上取代基位阻大时,将两侧的p往两侧挤,使得pnp键角变小,改变催化活性。
[0041]
本发明通过将碳原子与两个磷原子桥连,可制备得到一种pnp得键角小,催化剂活性高的pcpn,而且通过实验发现,该pcpn配体制备得到的金属催化剂可明显降低反应中聚合物的生成量,延长催化剂使用时间,减少设备堵塞,对应工业化生产具有十分重要的意义。
[0042]
而且,与现有技术相比,本发明的乙烯齐聚催化剂体系对乙烯齐聚的活性可达1500000g/(gcr
·
h)以上,最为显著的是1-己烯和1-辛烯的总选择性可达91.5%,pe选择性低于0.05wt%,与经典异丙基pnp相比较有50%活性的提高,聚合物有非常明显的下降,1-己烯和1-辛烯的总选择性有明显的提升。
[0043]
附图说明图1为采用对比例1的方法进行乙烯齐聚反应,运行24小时后反应器中聚合物堵塞情况图。
[0044]
图2为采用实施例1的方法进行乙烯齐聚反应,运行24小时后反应器中聚合物堵塞情况图。
具体实施方式
[0045]
以下具体实施例只对本发明进行说明,但这些实例仅是本发明的部分内容,并不限制本发明在其他领域中的应用。
[0046]
实施例中所用原料均为本领域常规原料,所用的纯度规格为分析纯或化学纯。
[0047]
原料来源信息:
[0048]
异丙基pnp配体:上海药明康德有限公司
[0049]
烷氧基膦pcnp配体:上海药明康德有限公司
[0050]
苯基膦:98%,百灵威科技有限公司
[0051]
二氯甲烷:98%,上海易恩化学技术有限公司
[0052]
n,n-二甲基甲酰胺:99%,上海迈瑞尔化学科技有限公司
[0053]
n,n-二甲基乙酰胺:99%,上海迈瑞尔化学科技有限公司
[0054]
氢氧化钾:≥99.5%(gc),上海阿拉丁生化科技有限公司
[0055]
六氯乙烷:99%,北京伊诺凯科技有限公司
[0056]
乙醚:99%,上海阿拉丁生化科技股份有限公司
[0057]
二氧六环:99%,上海阿拉丁生化科技股份有限公司
[0058]
环己基甲基胺:95%,上海阿拉丁生化科技股份有限公司
[0059]
1-金刚烷甲胺:97%,上海阿拉丁生化科技股份有限公司
[0060]
对甲基苯胺:98%,百灵威科技有限公司
[0061]
苯基膦:97%,阿法埃莎(中国)化学品有限公司
[0062]
(2-甲基)苯基膦:97%,陕西缔都医药化工有限公司
[0063]
(2,4,6-三叔丁基)苯基膦:97%,陕西缔都医药化工有限公司
[0064]
(4-甲氧基)苯基膦:97%,陕西缔都医药化工有限公司
[0065]
(4-氟)苯基膦:97%,陕西缔都医药化工有限公司
[0066]
(2,5-二甲基)苯基膦:97%,陕西缔都医药化工有限公司
[0067]
乙酸乙酯:99.9%%,百灵威科技有限公司
[0068]
乙醇:分析纯,国药集团化学试剂有限公司
[0069]
齐聚反应的催化剂活性通过对反应液中的各组分进行定性定量分析,所用的gc分析仪器的条件如下:
[0070]
仪器型号:岛津gc2010
[0071]
色谱柱:db-5(30m 0.25mm 0.25μm)
[0072]
柱温程序:先在35℃下保持10min,然后以10℃/min的速度升到250℃,在此温度下保持10min。
[0073]
检测器温度:300℃
[0074]
载气:1bar
[0075]
空气:0.3bar
[0076]
燃气(h2):0.3bar
[0077]
样品质量分析利用内标法进行。应有:
[0078][0079]
式中m1为某种产品的质量,m为内标物质量,a1为该产品在气象色谱中检测到的峰面积,a为内标物峰面积。k是与被测物质和检测条件相关的一个校正系数。
[0080]
实施例1
[0081]
pcpn配体的制备:相关溶剂使用前除水氧。
[0082]
氯膦桥配体的制备:取适量150mmol苯基膦,75mmol二氯甲烷,溶于250mldmf,冰水浴搅拌下,加入520mol氢氧化钾水溶液(60wt%),滴加1.5h,滴加完毕后,冰水浴下反应6h。加入适量水淬灭反应,正己烷萃取反应液,经减压蒸馏得氢膦桥配体。将100mmol氢膦桥配体,200mmol六氯乙烷溶于240ml乙醚,回流24-48h,过滤除去不溶物,抽干滤液即为氯膦桥配体a,其化合物结构如下所示:
[0083][0084]
pcpn配体的制备:无水无氧条件下,将50mmol正丁胺,500mmol三乙胺溶解在180ml二氯甲烷中得到反应液一。在0℃下,将50mmol氯膦桥配体a滴加到反应液一中;滴加完毕后,使反应液缓慢升至室温,搅拌反应36h。待反应结束后,滤出反应清液,真空下除去溶剂,经柱层析处理后,得pcpn纯品1a,其结构如下式所示:
[0085][0086]
上述配体(1a)的核磁数据如下:1h nmr(400mhz,cdcl3):,7.36~7.48(m,10h),2.65(t,2h),1.55~1.31(m,8h),0.91(t,3h)
[0087]
乙烯齐聚:
[0088]
反应前将300ml反应釜加热至150℃,抽真空3h,氮气置换三次。待温度冷却至室温,乙烯置换两次,先加入100ml脱水脱氧的溶剂甲基环己烷和1ml的(al/cr=500)mmao-3a(7wt%al,正庚烷),然后加入4.2μmol pcpn配体1a和3.5μmol乙酰丙酮铬,待温度恒定在45℃,依次通入0.5mpa氢气、5mpa乙烯开始反应。反应温度45℃,反应时间60min。反应结束后,关闭乙烯进气阀门,用冰水浴或迅速降温至5℃以下,缓慢泄压,卸釜得到乙烯齐聚产物。
[0089]
采用gc分析产物,活性1623kg/gcr.h,(1-己烯+1-辛烯)选择性88.6wt%,聚合物选择性0.09wt%。
[0090]
实施例2
[0091]
pcpn配体的制备:相关溶剂使用前除水氧。
[0092]
氯膦桥配体的制备:取适量150mmol临氟苯基膦,50mmol二氯甲烷,溶于250mldmf,冰水浴搅拌下,加入520mol氢氧化钾水溶液(60wt%),滴加1.5h,滴加完毕后,冰水浴下反应6h。加入适量水淬灭反应,正己烷萃取反应液,经减压蒸馏得氢膦桥配体。将100mmol氢膦桥配体,100mmol六氯乙烷溶于240ml乙醚,回流24-48h,过滤除去不溶物,抽干滤液即为氯膦桥配体b,其化合物结构如下所示:
[0093][0094]
pcpn配体的制备:无水无氧条件下,将35mmol异丙胺,500mmol二异丙基乙胺溶解在180ml二氯甲烷中得到反应液二。在0℃下,将50mmol氯膦桥配体b滴加到反应液二中;滴加完毕后,使反应液缓慢升至室温,搅拌反应36h。待反应结束后,滤出反应清液,真空下除去溶剂,经柱层析处理后,得pcpn纯品1a,其结构如下式所示:
[0095][0096]
上述配体(1b)的核磁数据如下:1h nmr(400mhz,cdcl3):,7.75~7.22(m,8h),2.97(m,1h),1.4(t,2h),1.07(d,6h)
[0097]
乙烯齐聚:
[0098]
反应前将500ml反应釜加热至160℃,抽真空1.5h,氮气置换三次。待温度冷却至室温,乙烯置换两次,先加入200ml脱水脱氧的溶剂甲基环己烷和1.4ml(al/cr=600)mmao-3a(7wt%al,正庚烷),然后加入4.8μmol pcpn配体1b和3μmol乙酰丙酮铬,待温度恒定在55℃,依次通入0.4mpa氢气、4.5mpa乙烯开始反应。反应温度55℃,反应时间40min。反应结束后,关闭乙烯进气阀门,用冰水浴或迅速降温至5℃以下,缓慢泄压,卸釜得到乙烯齐聚产物。
[0099]
采用gc分析产物,活性2278kg/gcr.h,(1-己烯+1-辛烯)选择性90.2wt%,聚合物选择性0.07wt%。
[0100]
实施例3
[0101]
pcpn配体的制备:相关溶剂使用前除水氧。
[0102]
氯膦桥配体的制备:取适量150mmol对甲氧基苯基膦,105mmol二氯甲烷,溶于250mldmf,冰水浴搅拌下,加入520mol氢氧化钾水溶液(60wt%),滴加1.5h,滴加完毕后,冰水浴下反应6h。加入适量水淬灭反应,正己烷萃取反应液,经减压蒸馏得氢膦桥配体。将100mmol氢膦桥配体,300mmol六氯乙烷溶于240ml乙醚,回流24-48h,过滤除去不溶物,抽干滤液即为氯膦桥配体c,其化合物结构如下所示:
[0103][0104]
pcpn配体的制备:无水无氧条件下,将50mmol甲基环己胺,250mmol三乙胺溶解在180ml二氯甲烷中得到反应液二。在0℃下,将50mmol氯膦桥配体c滴加到反应液二中;滴加完毕后,使反应液缓慢升至室温,搅拌反应36h。待反应结束后,滤出反应清液,真空下除去溶剂,经柱层析处理后,得pcpn纯品1c,其结构如下式所示:
[0105][0106]
上述配体(1c)的核磁数据如下:1h nmr(400mhz,cdcl3):7.27-6.99(m,4h),3.83(s,6h),2.61(d,2h),1.4(t,2h),1.67-1.27(m,15h),乙烯齐聚:
[0107]
反应前将500ml反应釜加热至120℃,抽真空2h,氮气置换三次。待温度冷却至室温,乙烯置换两次,先加入200ml脱水脱氧的溶剂甲苯和0.93ml的(al/cr=400)mao(10wt%,甲苯),然后加入4.2μmol pcpn配体1c和3.5μmol乙酰丙酮铬,待温度恒定在60℃,依次通入0.3mpa氢气、4.5mpa乙烯开始反应。反应温度60℃,反应时间45min。反应结束后,关闭乙烯进气阀门,用冰水浴或迅速降温至5℃以下,缓慢泄压,卸釜得到乙烯齐聚产物。
[0108]
采用gc分析产物,活性1551kg/gcr.h,(1-己烯+1-辛烯)选择性91.5wt%,聚合物选择性0.05wt%。
[0109]
实施例4
[0110]
pcpn配体的制备:相关溶剂使用前除水氧。
[0111]
氯膦桥配体的制备:取适量150mmol苯基膦,75mmol二氯甲烷,溶于250ml n,n-二甲基乙酰胺,冰水浴搅拌下,加入520mol氢氧化钾水溶液(60wt%),滴加1.5h,滴加完毕后,冰水浴下反应6h。加入适量水淬灭反应,正己烷萃取反应液,经减压蒸馏得氢膦桥配体。将100mmol氢膦桥配体,100mmol六氯乙烷溶于240ml二氧六环,回流24-48h,过滤除去不溶物,抽干滤液即为氯膦桥配体a,其化合物结构如下所示:
[0112][0113]
pcpn配体的制备:无水无氧条件下,将100mmol对甲基苯胺,500mmol 4-二甲氨基
吡啶溶解在180ml二氯甲烷中得到反应液二。在0℃下,将50mmol氯膦桥配体a滴加到反应液二中;滴加完毕后,使反应液缓慢升至室温,搅拌反应36h。待反应结束后,滤出反应清液,真空下除去溶剂,经柱层析处理后,得pcpn纯品1d,其结构如下式所示:
[0114][0115]
上述配体(1d)的核磁数据如下:1h nmr(400mhz,cdcl3):7.45-7.38(m,10h),6.98(d,2h),6.51(d,2h),2.34(s,3h),1.4(t,2h)
[0116]
乙烯齐聚:
[0117]
反应前将500ml反应釜加热至160℃,抽真空2.5h,氮气置换三次。待温度冷却至室温,乙烯置换两次,先加入200ml脱水脱氧的溶剂甲苯和1.2ml的(al/cr=600)mmao(7wt%al,正庚烷),然后加入4.2μmol pcpn配体l4和4μmol乙酰丙酮铬,待温度恒定在45℃,依次通入0.5mpa氢气、4.5mpa乙烯开始反应。反应温度45℃,反应时间25min。反应结束后,关闭乙烯进气阀门,用冰水浴或迅速降温至5℃以下,缓慢泄压,卸釜得到乙烯齐聚产物。
[0118]
采用gc分析产物,活性1977kg/gcr.h,(1-己烯+1-辛烯)选择性89.8wt%,聚合物选择性0.08wt%。
[0119]
实施例5
[0120]
pcpn配体的制备:相关溶剂使用前除水氧。
[0121]
采用与实施例1相同的方法制备pcpn配体。
[0122]
乙烯齐聚:
[0123]
反应前将500ml反应釜加热至120℃,抽真空4h,氮气置换三次。待温度冷却至室温,乙烯置换两次,先加入200ml脱水脱氧的溶剂环己烷和0.18ml的(al/cr=90)mmao(7wt%al,正庚烷),然后加入3.5μmol本实施例制备的pcpn配体和3.5μmol乙酰丙酮铬,待温度恒定在55℃,依次通入0.5mpa氢气、4.5mpa乙烯开始反应。反应温度55℃,反应时间50min。反应结束后,关闭乙烯进气阀门,用冰水浴或迅速降温至5℃以下,缓慢泄压,卸釜得到乙烯齐聚产物。
[0124]
采用gc分析产物,活性1152kg/gcr.h,(1-己烯+1-辛烯)选择性88.8wt%,聚合物选择性0.07wt%。
[0125]
实施例6
[0126]
pcpn配体的制备:相关溶剂使用前除水氧。
[0127]
氯膦桥配体的制备:取适量150mmol临甲基苯基膦,90mmol二氯甲烷,溶于250ml乙腈,冰水浴搅拌下,加入520mol氢氧化钾水溶液(60wt%),滴加1.5h,滴加完毕后,冰水浴下反应6h。加入适量水淬灭反应,正己烷萃取反应液,经减压蒸馏得氢膦桥配体。将100mmol氢膦桥配体,100mmol六氯乙烷溶于240ml正己烷,回流24-48h,过滤除去不溶物,抽干滤液即为氯膦桥配体d,其化合物结构如下所示:
[0128][0129]
pcpn配体的制备:无水无氧条件下,将65mmol异丙胺,500mmol三乙胺溶解在180ml二氯甲烷中得到反应液二。在0℃下,将50mmol氯膦桥配体d滴加到反应液二中;滴加完毕后,使反应液缓慢升至室温,搅拌反应36h。待反应结束后,滤出反应清液,真空下除去溶剂,经柱层析处理后,得pcpn纯品1f,其结构如下式所示:
[0130][0131]
上述配体(1f)的核磁数据如下:1h nmr(400mhz,cdcl3):1h nmr(400mhz,cdcl3):,7.33~7.26(m,8h),2.97(m,1h),2.34(s,6h)1.41(m,2h),1.07(d,6h)
[0132]
乙烯齐聚:
[0133]
反应前将500ml反应釜加热至120℃,抽真空5h,氮气置换三次。待温度冷却至室温,乙烯置换两次,先加入200ml脱水脱氧的溶剂甲基环己烷和1.8ml的(al/cr=900)mmao(7wt%al,正庚烷),然后加入4.2μmol膦桥配体1f和3.5μmol乙酰丙酮铬,待温度恒定在60℃,依次通入0.5mpa氢气、4.5mpa乙烯开始反应。反应温度60℃,反应时间20min。反应结束后,关闭乙烯进气阀门,用冰水浴或迅速降温至5℃以下,缓慢泄压,卸釜得到乙烯齐聚产物。
[0134]
采用gc分析产物,活性1833kg/gcr.h,(1-己烯+1-辛烯)选择性89.7wt%,聚合物选择性0.11wt%。
[0135]
实施例7
[0136]
pcpn配体的制备:相关溶剂使用前除水氧。
[0137]
氯膦桥配体的制备:取适量150mmol临氟苯基膦,50mmol二氯甲烷,溶于250mldmf,冰水浴搅拌下,加入520mol氢氧化钾水溶液(60wt%),滴加1.5h,滴加完毕后,冰水浴下反应6h。加入适量水淬灭反应,正己烷萃取反应液,经减压蒸馏得氢膦桥配体。将100mmol氢膦桥配体,100mmol六氯乙烷溶于240ml甲基环己烷,回流24-48h,过滤除去不溶物,抽干滤液即为氯膦桥配体b,其化合物结构如下所示:
[0138][0139]
pcpn配体的制备:无水无氧条件下,将50mmol正丁胺,500mmol二异丙基乙胺溶解在180ml二氯甲烷中得到反应液二。在0℃下,将50mmol氯膦桥配体b滴加到反应液二中;滴加完毕后,使反应液缓慢升至室温,搅拌反应36h。待反应结束后,滤出反应清液,真空下除去溶剂,经柱层析处理后,得pcpn纯品1g,其结构如下式所示:
[0140][0141]
上述配体(1g)的核磁数据如下:1h nmr(400mhz,cdcl3)::7.76-7.22(m,8h),2.65(t,2h),1.55~1.31(m,6h),0.90(t,3h)
[0142]
乙烯齐聚:
[0143]
反应前将500ml反应釜加热至125℃,抽真空3.5h,氮气置换三次。待温度冷却至室温,乙烯置换两次,先加入200ml脱水脱氧的溶剂甲基环己烷和1.2ml的(al/cr=600)mmao(7wt%al,正庚烷),然后加入7μmol膦桥配体1g和3.5μmol乙酰丙酮铬,待温度恒定在55℃,依次通入0.5mpa氢气、4.5mpa乙烯开始反应。反应温度55℃,反应时间60min。反应结束后,关闭乙烯进气阀门,用冰水浴或迅速降温至5℃以下,缓慢泄压,卸釜得到乙烯齐聚产物。
[0144]
采用gc分析产物,活性2170kg/gcr.h,(1-己烯+1-辛烯)选择性88.9wt%,聚合物选择性0.13wt%。
[0145]
实施例8
[0146]
pcpn配体的制备:相关溶剂使用前除水氧。
[0147]
氯膦桥配体的制备:取适量150mmol对甲氧基苯基膦,105mmol二氯甲烷,溶于250ml甲基环己烷,冰水浴搅拌下,加入520mol氢氧化钾水溶液(60wt%),滴加1.5h,滴加完毕后,冰水浴下反应6h。加入适量水淬灭反应,正己烷萃取反应液,经减压蒸馏得氢膦桥配体。将100mmol氢膦桥配体,500mmol六氯乙烷溶于240ml乙醚,回流24-48h,过滤除去不溶物,抽干滤液即为氯膦桥配体c,其化合物结构如下所示:
[0148][0149]
pcpn配体的制备:无水无氧条件下,将75mmol正丁胺,750mmol n,n-二甲基苯胺溶解在180ml二氯甲烷中得到反应液二。在0℃下,将50mmol氯膦桥配体c滴加到反应液二中;滴加完毕后,使反应液缓慢升至室温,搅拌反应36h。待反应结束后,滤出反应清液,真空下除去溶剂,经柱层析处理后,得pcpn纯品1h,其结构如下式所示:
[0150][0151]
上述配体(1h)的核磁数据如下:1h nmr(400mhz,cdcl3):7.27(d,2h),6.99(d,2h),2.65(t,2h),1.55~1.31(m,6h),0.90(t,3h)
[0152]
乙烯齐聚:
[0153]
反应前将500ml反应釜加热至155℃,抽真空2h,氮气置换三次。待温度冷却至室温,乙烯置换两次,先加入200ml脱水脱氧的溶剂甲基环己烷和1.2ml的(al/cr=500)mmao(7wt%al,正庚烷),然后加入5.3μmol膦桥配体1h和3.5μmol乙酰丙酮铬,待温度恒定在50℃,依次通入0.5mpa氢气、4.5mpa乙烯开始反应。反应温度50℃,反应时间60min。反应结束后,关闭乙烯进气阀门,用冰水浴或迅速降温至5℃以下,缓慢泄压,卸釜得到乙烯齐聚产物。
[0154]
采用gc分析产物,活性1933kg/gcr.h,(1-己烯+1-辛烯)选择性89.1wt%,聚合物选择性0.09wt%。
[0155]
实施例9
[0156]
pcpn配体的制备:相关溶剂使用前除水氧。
[0157]
氯膦桥配体的制备:取适量150mmol临氟苯基膦,75mmol二氯甲烷,溶于250mldmf,冰水浴搅拌下,加入520mol氢氧化钾水溶液(60wt%),滴加1.5h,滴加完毕后,冰水浴下反应6h。加入适量水淬灭反应,正己烷萃取反应液,经减压蒸馏得氢膦桥配体。将100mmol氢膦桥配体,100mmol六氯乙烷溶于240ml乙腈,回流24-48h,过滤除去不溶物,抽干滤液即为氯膦桥配体b,其化合物结构如下所示:
[0158][0159]
pcpn配体的制备:无水无氧条件下,将50mmol异丙胺,500mmol三乙胺溶解在180ml二氯甲烷中得到反应液二。在0℃下,将50mmol氯膦桥配体b滴加到反应液二中;滴加完毕后,使反应液缓慢升至室温,搅拌反应36h。待反应结束后,滤出反应清液,真空下除去溶剂,经柱层析处理后,得pcpn纯品1i,其结构如下式所示:
[0160][0161]
上述配体(1i)的核磁数据如下:1h nmr(400mhz,cdcl3):7.75-7.22(m,8h),2.97(m,1h),1.41(t,2h),1.07(d,6h)
[0162]
乙烯齐聚:
[0163]
反应前将500ml反应釜加热至160℃,抽真空2h,氮气置换三次。待温度冷却至室温,乙烯置换两次,先加入200ml脱水脱氧的溶剂甲基环己烷和1.4ml的(al/cr=600)mmao(7wt%al,正庚烷),然后加入4.2μmol膦桥配体1i和3.5μmol乙酰丙酮铬,待温度恒定在53℃,依次通入0.5mpa氢气、4.5mpa乙烯开始反应。反应温度53℃,反应时间60min。反应结束后,关闭乙烯进气阀门,用冰水浴或迅速降温至5℃以下,缓慢泄压,卸釜得到乙烯齐聚产物。
[0164]
采用gc分析产物,活性1362kg/gcr.h,(1-己烯+1-辛烯)选择性88.5wt%,聚合物选择性0.06wt%。
[0165]
对比例1
[0166]
异丙基pnp配体的制备:购自上海药明康德有限公司,纯度:lc》98%
[0167][0168]
乙烯齐聚:
[0169]
反应前将500ml反应釜加热至160℃,抽真空2h,氮气置换三次。待温度冷却至室温,乙烯置换两次,先加入200ml脱水脱氧的溶剂甲基环己烷和1.4ml的(al/cr=600)mmao(7wt%al,正庚烷),然后加入4.2μmol膦桥配体1j和3.5μmol乙酰丙酮铬,待温度恒定在53℃,依次通入0.5mpa氢气、4.5mpa乙烯开始反应。反应温度53℃,反应时间60min。反应结束后,关闭乙烯进气阀门,用冰水浴或迅速降温至5℃以下,缓慢泄压,卸釜得到乙烯齐聚产物。
[0170]
采用gc分析产物,活性732kg/gcr.h,(1-己烯+1-辛烯)选择性71.2wt%,聚合物选择性2.16wt%。
[0171]
对比例2
[0172]
烷氧基膦pcnp配体的制备:购自上海药明康德有限公司,纯度:lc》98%
[0173][0174]
乙烯齐聚:
[0175]
反应前将500ml反应釜加热至160℃,抽真空2h,氮气置换三次。待温度冷却至室温,乙烯置换两次,先加入200ml脱水脱氧的溶剂甲基环己烷和1.4ml的(al/cr=600)mmao(7wt%al,正庚烷),然后加入4.2μmol膦桥配体1k和3.5μmol乙酰丙酮铬,待温度恒定在53℃,依次通入0.5mpa氢气、4.5mpa乙烯开始反应。反应温度53℃,反应时间60min。反应结束后,关闭乙烯进气阀门,用冰水浴或迅速降温至5℃以下,缓慢泄压,卸釜并未得到产物。
[0176]
采用gc分析产物,活性0.3kg/gcr.h,(1-己烯+1-辛烯)选择性0.02wt%,聚合物选择性0.1wt%。无乙烯齐聚活性。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1