1.本发明涉及光刻胶技术领域,具体涉及一种酸敏感光刻胶树脂单体的制备方法。
背景技术:2.193nm光刻胶基本为化学放大胶,化学放大胶是一种基于化学放大原理的光刻胶,其主要成分是聚合物树脂、光致产酸剂以及相应的添加剂(碱性添加剂、溶解抑制剂等)和溶剂。其中,聚合物树脂是由不同侧链结构的树脂单体之间共聚形成的,侧链结构是赋予聚合物树脂所需功能的关键组分,通常为聚合物树脂提供极性集团和酸敏基团。极性基团可以平衡树脂的亲疏水性,改善树脂与基底间的粘附性,并为主体树脂提供可显影性。酸敏基团可以在光致产酸剂作用下从侧链脱离,使树脂由不溶转变为碱可溶,实现曝光区域与未曝光区域的溶解度反差。
3.聚(甲基)丙烯酸酯体系是一种广泛应用于193nm光刻胶的树脂聚合物,但是现有(甲基)丙烯酸酯类树脂单体的制备方法纯度不高。
技术实现要素:4.本发明的目的就是为了克服上述现有技术存在的缺陷而提供一种酸敏感光刻胶树脂单体的制备方法。
5.为了实现本发明之目的,本技术提供以下技术方案。
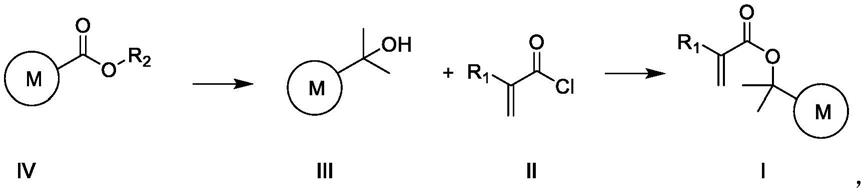
6.在第一方面中,本技术提供一种酸敏感光刻胶树脂单体的制备方法,所述制备方法包括如下反应路线:
[0007][0008]
其中,m为碳数5~7的环烷基,r1为氢原子或烷基,r2为烷基;
[0009]
所述制备方法包括如下步骤:
[0010]
c)向第一溶剂中通入卤甲烷,得到含卤甲烷的第一溶剂溶液;
[0011]
优选的,所述第一溶剂为四氢呋喃;
[0012]
d)在惰性气体条件下,将金属锂和式ⅳ化合物加入到第一溶剂中混合,滴加步骤c)制备的含卤甲烷的第一溶剂溶液,搅拌,反应,纯化,得到式ⅲ化合物;
[0013]
f)将式ⅲ化合物和缚酸剂加入到第二溶剂中混合,并滴加式ⅱ化合物,经酯化反应,纯化,得到式ⅰ化合物;
[0014]
优选的,所述第二溶剂为二氯甲烷。
[0015]
在第一方面的一种实施方式中,步骤c)中还包括如下特征:
[0016]
c1)所述通入卤甲烷所用的温度为-10~-20℃,如-10℃或-15℃或-20℃等;
[0017]
c2)所述卤甲烷选自氯甲烷或溴甲烷。
[0018]
在第一方面的一种实施方式中,步骤d)中,还包括如下技术特征中的至少一项:
[0019]
d1)所述式ⅳ化合物、卤甲烷与金属锂的摩尔比为1:(2~5):(2~5),如1:3:3或1:2:2或1:5:5等;
[0020]
d2)所述滴加含卤甲烷的第一溶剂溶液的温度为-20~10℃,如-15℃或-10℃或0℃等;
[0021]
d3)所述滴加含卤甲烷的第一溶剂溶液的时间为0.5~1.5h,如1h或1.5h等;
[0022]
d4)所述惰性气体为氮气;
[0023]
d5)所述反应的温度为20~30℃,如22℃或25℃或28℃等,反应的时间为1~4h,如2h或3h等。
[0024]
在第一方面的一种实施方式中,步骤f)中还包括如下特征:
[0025]
f1)所述式ⅲ化合物、式ⅱ化合物与缚酸剂的摩尔比为1:(1~2):(1~2),如1:1.2:1.2或1:1:1或1:2:2等;
[0026]
f2)所述缚酸剂选自三乙胺、n,n-二异丙基乙胺或吡啶中的一种;
[0027]
f3)所述滴加式ⅱ化合物的温度为-60~-65℃,如-60℃等;
[0028]
f4)所述滴加式ⅱ化合物的时间为0.5~1.5h,如0.5h或1h等;
[0029]
f5)所述酯化反应的温度为-60~-65℃,如-65℃等,反应的时间为0.5~1h,如0.5h等。
[0030]
在第一方面的一种实施方式中,所述步骤d)中的纯化e)包括如下步骤:
[0031]
e1)将步骤d)搅拌反应所得反应液倒入冰水中淬灭,然后加入酸调节ph至2~3,得到酸性溶液;
[0032]
e2)用正己烷提取所述酸性溶液,得到的有机相用水洗至中性,然后干燥并旋除溶剂。
[0033]
在第一方面的一种实施方式中,所述纯化e)还包括如下技术特征中的至少一项:
[0034]
e11)在步骤e1)中,所述酸为浓度为36wt%的盐酸;
[0035]
e21)在步骤e2)中,所述正己烷提取的次数为至少3次;
[0036]
e22)在步骤e2)中,所述水洗的次数为至少2次;
[0037]
e23)在步骤e2)中,所述干燥所用的干燥剂选自无水硫酸钠或无水硫酸镁;
[0038]
e24)在步骤e2)中,所述旋除溶剂所用的水浴温度为30~35℃,如32℃或34℃等。
[0039]
在第一方面的一种实施方式中,所述步骤f)中纯化g)包括如下步骤:
[0040]
g1)在步骤f)酯化反应所得反应液中加水搅拌0.5~1h,如0.5h等,静置分层得到第一水相和第一有机相;
[0041]
g2)水洗所述第一有机相,得到第二有机相;
[0042]
g3)所述第二有机相经干燥、过滤、滤液旋干后,得到油状物;
[0043]
g4)在所述油状物中加入正己烷,经硅胶过滤、淋洗硅胶、滤液旋干后得到产物粗品;
[0044]
g5)在粗品中加入阻聚剂,然后进行减压蒸馏,得到所述式ⅰ化合物。
[0045]
在第一方面的一种实施方式中,所述纯化g)还包括如下特征中的至少一项:
[0046]
g21)在步骤g2)中,所述水洗的次数为至少2次;
[0047]
g31)在步骤g3)中,所述干燥所用的干燥剂为无水硫酸钠或无水硫酸镁;
[0048]
g32)在步骤g3)中,所述滤液旋干所用的水浴温度为30~35℃,如32℃、35℃等;
[0049]
g41)在步骤g4)中,所述滤液旋干所用的水浴温度25~30℃,如30℃、35℃等;
[0050]
g42)在步骤g4)中,所述淋洗硅胶的溶剂为正己烷;
[0051]
g51)在步骤g5)中,所述减压蒸馏所用的油浴温度为85~95℃,如90℃或95℃等,馏分温度为45~50℃,如45℃或48℃等,压力为20~130pa,如30pa或70pa或100pa等;
[0052]
g52)在步骤g5)中,所述阻聚剂为吩噻嗪。
[0053]
在第一方面的一种实施方式中,所述制备方法还包括式ⅳ化合物的制备,反应路线如下:
[0054][0055]
其中,m为碳数5~7的环烷基,r2为烷基;
[0056]
所述式ⅳ化合物的制备包括如下步骤:
[0057]
a)将式
ⅴ
化合物和浓硫酸溶于醇类溶剂中混合,加热回流反应,纯化,得到式ⅳ化合物;
[0058]
优选的,所述醇类溶剂为甲醇。
[0059]
在第一方面的一种实施方式中,步骤a)中还包括如下技术特征中的至少一项:
[0060]
a1)所述浓硫酸的加入量为式
ⅴ
化合物的5~10wt%,如6wt%、8wt%等;
[0061]
a2)所述加热回流反应的油浴温度为85~95℃,如90℃等;
[0062]
a3)所述加热回流反应的时间为1.5~2.5h,如2h等;
[0063]
a4)所述浓硫酸的浓度为98wt%。
[0064]
在第一方面的一种实施方式中,所述步骤a)中的纯化b)包括如下步骤:
[0065]
b)将步骤a)回流反应所得反应液除去醇类溶剂,蒸馏,收集温度25~30℃的馏分,得到式ⅳ化合物。
[0066]
与现有技术相比,本发明的有益效果在于:该制备方法得到的树脂单体为一种酸敏感光刻胶树脂单体,得到的产物收率高,纯度高,杂质少。
附图说明
[0067]
图1为实施例1所得产品的核磁谱图。
[0068]
图2为实施例1所得产品的gc谱图。
具体实施方式
[0069]
除非另有说明、从上下文暗示或属于现有技术的惯例,否则本技术中所有的份数和百分比都基于重量,且所用的测试和表征方法都是与本技术的提交日期同步的。在适用的情况下,本技术中涉及的任何专利、专利申请或公开的内容全部结合于此作为参考,且其等价的同族专利也引入作为参考,特别这些文献所披露的关于本领域中的合成技术、产物
和加工设计、聚合物、共聚单体、引发剂或催化剂等的定义。如果现有技术中披露的具体术语的定义与本技术中提供的任何定义不一致,则以本技术中提供的术语定义为准。
[0070]
本技术中的数字范围是近似值,因此除非另有说明,否则其可包括范围以外的数值。数值范围包括以1个单位增加的从下限值到上限值的所有数值,条件是在任意较低值与任意较高值之间存在至少2个单位的间隔。例如,如果记载组分、物理或其它性质(如分子量等)是100至1000,意味着明确列举了所有的单个数值,例如100,101,102等,以及所有的子范围,例如100到166,155到170,198到200等。对于包含小于1的数值或者包含大于1的分数(例如1.1,1.5等)的范围,则适当地将1个单位看作0.0001,0.001,0.01或者0.1。对于包含小于10(例如1到5)的个位数的范围,通常将1个单位看作0.1。这些仅仅是想要表达的内容的具体示例,并且所列举的最低值与最高值之间的数值的所有可能的组合都被认为清楚记载在本技术中。还应指出,本文中的术语“第一”、“第二”等不限定先后顺序,只是为了区分不同结构的物质。
[0071]
关于化学化合物使用时,除非明确地说明,否则单数包括所有的异构形式,反之亦然(例如,“己烷”单独地或共同地包括己烷的全部异构体)。另外,除非明确地说明,否则用“一个”,“一种”或“该”形容的名词也包括其复数形式。
[0072]
术语“包含”,“包括”,“具有”以及它们的派生词不排除任何其它的组分、步骤或过程的存在,且与这些其它的组分、步骤或过程是否在本技术中披露无关。为消除任何疑问,除非明确说明,否则本技术中所有使用术语“包含”,“包括”,或“具有”的组合物可以包含任何附加的添加剂、辅料或化合物。相反,除了对操作性能所必要的那些,术语“基本上由
……
组成”将任何其他组分、步骤或过程排除在任何该术语下文叙述的范围之外。术语“由
……
组成”不包括未具体描述或列出的任何组分、步骤或过程。除非明确说明,否则术语“或”指列出的单独成员或其任何组合。
[0073]
实施例
[0074]
下面将对本发明的实施例作详细说明,本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施例。
[0075]
实施例1
[0076]
1、
[0077][0078]
把甲醇(720g)和环己基甲酸(150g,1.17mol)加入到四口反应瓶,搅拌下加入质量浓度为98%的浓硫酸9g(环己基甲酸的6wt%),加热回流反应2h(油浴温度90℃),gc跟踪至原料基本消失,旋蒸甲醇至无液滴后(水浴温度40℃),油泵简单蒸馏(馏分温度25~30℃,油浴50℃)得到产品133g,摩尔收率80%,纯度98.8%。
[0079]
2、
[0080][0081]
把四氢呋喃(450g)加入四口瓶,用封口膜密封塞子,称重。用干冰乙醇冷至-20℃,
开始通溴甲烷(钢瓶要记录重量),注意观察鼓泡器,通气结束后,再称量四口瓶的重量,确认溴甲烷只有少量逸出,共通入了200.3g(2.11mol)溴甲烷,将制备好的溴甲烷的四氢呋喃溶液放置待用。
[0082]
在另一四口瓶中氮气保护下加入金属锂(14.64g,2.11mol)、四氢呋喃(450g)和环己基甲酸甲酯(100g,0.703mol),降温至0℃左右,滴加溴甲烷的四氢呋喃溶液,很容易引发并有放热,控温在0~10℃,约1h滴完,滴完后在室温(20~30℃)下搅拌2h。反应结束,把反应液缓慢倒入约冰水(3.5l)中,用质量浓度为36%的盐酸(350ml)中和至呈酸性(ph为2~3),搅拌1h,用正己烷提取3次,每次使用正己烷(1500ml),合并有机相用水洗至中性(水洗2次,每次1500ml水),经无水硫酸钠干燥后,旋除溶剂(水浴温度30~35℃),得到黄色油状物118g。粗品直接用于下一步反应。
[0083]
3、
[0084][0085]
氮气保护下,加入二氯甲烷(650g)、2-环己基-2-丙醇(100g,0.703mol)和三乙胺(85g,0.840mol),搅拌降温至-60~-65℃,滴加甲基丙烯酰氯(88g,0.842mol),滴加时控温-60~-65℃,1h滴完,滴完后在此温度下保温反应0.5h。
[0086]
加入水(100ml)搅拌0.5h,静置分层后,下层有机相水洗两次,每次200ml水。有机层用无水硫酸钠(100g)干燥,过滤,滤液旋干(30~35℃)得到160g油状物(有大量悬浮物)。向油状物中加入正己烷(800ml),砂芯漏斗上铺硅胶(50g)过滤,正己烷(320ml)淋洗硅胶,滤液旋干后得到140g油状物(水浴温度25~30℃,油状物无悬浮物),残留物加入吩噻嗪(1.4g)后用油泵减压蒸馏(馏分温度45~50℃,油浴温度90℃)得到产品120g,摩尔收率81.2%,纯度98.7%,核磁谱图见图1,gc谱图见图2。
[0087]
实施例2
[0088]
1、
[0089][0090]
把甲醇(720g)和环己基甲酸(150g,1.17mol)加入到四口反应瓶,搅拌下加入质量浓度为98%的浓硫酸15g(环己基甲酸的10wt%),加热回流反应1.5h(油浴温度95℃),gc跟踪至原料基本消失,旋蒸甲醇至无液滴后(水浴温度40℃),油泵简单蒸馏(馏分温度25~30℃,油浴50℃)得到产品136g,摩尔收率83%,纯度98.6%。
[0091]
2、
[0092][0093]
把四氢呋喃(450g)加入四口瓶,用封口膜密封塞子,称重。用干冰乙醇冷至-10℃,开始通氯甲烷(钢瓶要记录重量),注意观察鼓泡器,通气结束后,再称量四口瓶的重量,确认氯甲烷只有少量逸出,共通入了106.5g(2.11mol)氯甲烷,将制备好的氯甲烷的四氢呋喃溶液放置待用。
[0094]
在另一四口瓶中氮气保护下加入金属锂(14.64g,2.11mol)、四氢呋喃(450g)和环己基甲酸甲酯(100g,0.703mol),降温至-20℃左右,滴加氯甲烷的四氢呋喃溶液,很容易引发并有放热,控温在-20~-10℃,约0.5h滴完,滴完后在室温(20~30℃)下搅拌1h,取样。反应结束,把反应液缓慢倒入约冰水(3.5l)中,用质量浓度为36%的盐酸(350ml)中和至呈酸性(ph为2~3),搅拌1h,用正己烷提取3次,每次使用正己烷(1500ml),合并有机相用水洗至中性(水洗2次,每次1500ml水),经无水硫酸镁干燥后,旋除溶剂(水浴温度30~35℃),得到黄色油状物109g,粗品直接用于下一步反应。
[0095]
3、
[0096][0097]
氮气保护下,加入二氯甲烷(650g)、2-环己基-2-丙醇(100g,0.703mol)和三乙胺(85g,0.840mol),搅拌降温至-60~-65℃,滴加甲基丙烯酰氯(88g,0.842mol),滴加时控温-60~-65℃,0.5h滴完,滴完后在此温度下保温反应1h,gc跟踪,加入水(100ml)搅拌1h。静置分层后,下层有机相水洗两次,每次200ml水。有机层用100g无水硫酸镁干燥,过滤,滤液旋干(30~35℃)得到160g油状物(有大量悬浮物)。向油状物中加入800ml正己烷,砂芯漏斗上铺50g硅胶过滤,320ml正己烷淋洗硅胶,滤液旋干后得到140g油状物(水浴温度25~30℃,油状物无悬浮物),残留物加入吩噻嗪(1.4g)后用油泵减压蒸馏(馏分温度45~50℃,油浴温度90℃)得到产品115g,摩尔收率77.8%,纯度99.2%。
[0098]
实施例3
[0099]
1、
[0100][0101]
把甲醇(720g)和环己基甲酸(150g,1.17mol)加入到四口反应瓶,搅拌下加入质量浓度为98%的浓硫酸7.5g(环己基甲酸的5wt%),加热回流反应2.5h(油浴温度85℃),gc跟踪至原料基本消失,旋蒸甲醇至无液滴后(水浴温度40℃),油泵简单蒸馏(馏分25~30℃,油浴50℃)得到产品130g,摩尔收率78.1%,纯度98.9%。
[0102]
2、
[0103][0104]
把四氢呋喃(450g)加入四口瓶,用封口膜密封塞子,称重。用干冰乙醇冷至-20℃,开始通溴甲烷(钢瓶要记录重量),注意观察鼓泡器,通气结束后,再称量四口瓶的重量,确认溴甲烷只有少量逸出,共通入了200.3g(2.11mol)溴甲烷,将制备好的溴甲烷的四氢呋喃溶液放置待用。
[0105]
在另一四口瓶中氮气保护下加入金属锂(14.64g,2.11mol)、四氢呋喃(450g)和环己基甲酸甲酯(100g,0.703mol),降温至-10℃左右,滴加溴甲烷的四氢呋喃溶液,很容易引发并有放热,控温在-10~0℃,约1h滴完,滴完后在室温(20~30℃)搅拌4h,取样。反应结
束,把反应液缓慢倒入约冰水(3.5l)中,用质量浓度为36%的盐酸(350ml)中和至呈酸性(ph为2~3),搅拌1h,用正己烷提取3次,每次使用正己烷(1500ml),合并有机相用水洗至中性(水洗2次,每次1500ml水),经无水硫酸钠干燥后,旋除溶剂(水浴温度30~35℃),得到黄色油状物121g,粗品直接用于下一步反应。
[0106]
3、
[0107][0108]
氮气保护下,加入二氯甲烷(650g)、2-环己基-2-丙醇(100g,0.703mol)和吡啶(66.6g,0.842mol),搅拌降温至-60~-65℃,滴加甲基丙烯酰氯(88g,0.842mol),滴加时控温-60~-65℃,1.5h滴完,滴完后在此温度下保温反应1h。
[0109]
加入100ml水搅拌0.5h,静置分层后,下层有机相水洗两次,每次200ml水。有机层用无水硫酸钠(100g)干燥,过滤,滤液旋干(30~35℃)得到160g油状物(有大量悬浮物)。向油状物中加入800ml正己烷,砂芯漏斗上铺50g硅胶过滤,正己烷淋(320ml)洗硅胶,滤液旋干后得到140g油状物(水浴温度25~30℃,油状物无悬浮物),残留物加入吩噻嗪(1.4g)后用油泵减压蒸馏(馏分温度45~50℃,油浴温度90℃)得到产品117g,摩尔收率79.2%,纯度99.4%。
[0110]
实施例4
[0111]
1、
[0112][0113]
把甲醇(720g)和环己基甲酸(150g,1.17mol)加入到四口反应瓶,搅拌下加入质量浓度为98%的浓硫酸15g(环己基甲酸的10wt%),加热回流反应2h(油浴温度90℃),gc跟踪至原料基本消失,旋蒸甲醇至无液滴后(水浴温度40℃),油泵简单蒸馏(馏分温度25~30℃,油浴50℃)得到产品136g,摩尔收率81.7%,纯度99.1%。
[0114]
2、
[0115][0116]
把四氢呋喃(450g)加入四口瓶,用封口膜密封塞子,称重。用干冰乙醇冷至-10℃,开始通溴甲烷(钢瓶要记录重量),注意观察鼓泡器,通气结束后,再称量四口瓶的重量,确认溴甲烷只有少量逸出,共通入了200.3g(2.11mol)溴甲烷,将制备好的溴甲烷的四氢呋喃溶液放置待用。
[0117]
在另一四口瓶中氮气保护下加入金属锂(14.64g,2.11mol)、四氢呋喃(450g)和环己基甲酸甲酯(100g,0.703mol),降温至-20℃左右,滴加溴甲烷的四氢呋喃溶液,很容易引发并有放热,控温在-20~-10℃,约1h滴完,滴完后在室温(20~30℃)搅拌2h,取样。反应结束,把反应液缓慢倒入约3.5l冰水中,用质量浓度为36%的盐酸350ml中和至呈酸性(ph为2~3),搅拌1h,用正己烷提取3次,每次使用正己烷(1500ml),合并有机相用水洗至中性(水
洗2次,每次1500ml水),经无水硫酸镁干燥后,旋除溶剂(水浴温度30~35℃),得到黄色油状物115g,粗品直接用于下一步反应。
[0118]
3、
[0119][0120]
氮气保护下,加入二氯甲烷(650g)、2-环己基-2-丙醇(100g,0.703mol)和n,n-二异丙基乙胺(109g,0.843mol),搅拌降温至-60~-65度,滴加甲基丙烯酰氯(88g,0.842mol),滴加时控温-60~-65度,1h滴完,滴完后在此温度下保温反应1h,gc跟踪,加入水(100ml)搅拌0.5h。静置分层后,下层有机相水洗两次,每次200ml水。有机层用无水硫酸镁(100g)干燥,过滤,滤液旋干(30~35℃)得到160g油状物(有大量悬浮物)。向油状物中加入正己烷(800ml),砂芯漏斗上铺硅胶(50g)过滤,320ml正己烷淋洗硅胶,滤液旋干后得到140g油状物(水浴温度25~30℃,油状物无悬浮物),残留物加入吩噻嗪(1.4g)后用油泵减压蒸馏(馏分温度45~50℃,油浴温度90℃)得到产品116g,摩尔收率78.5%,纯度99.1%。
[0121]
上述对实施例的描述是为了便于本技术领域的普通技术人员能理解和应用本技术。熟悉本领域技术的人员显然可以容易地对这些实施例做出各种修改,并把在此说明的一般原理应用到其它实施例中而不必付出创造性的劳动。因此,本技术不限于这里的实施例,本领域技术人员根据本技术披露的内容,在不脱离本技术范围和精神的情况下做出的改进和修改都在本技术的范围之内。