一种可降解的共聚物及其制备方法与流程
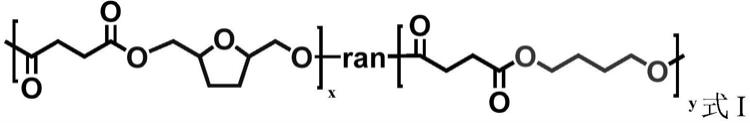
1.本技术涉及一种可降解的共聚物及其制备方法,属于共聚物合成领域。
背景技术:
2.几十年来,传统高分子材料在众多领域得到了广泛的应用。其中,包装领域的应用占有相当大的市场份额。然而,每年有数百万吨的塑料包装被填埋造成了相当严重的环境污染问题。在此背景下,生物可降解高分子材料作为传统高分子材料的替代品,在近些年来得到了长足的发展。聚丁二酸丁二醇酯(pbs)是一种生物可降解的脂肪族聚合物,具有较高的熔化温度、良好的韧性和加工能力。但pbs玻璃化转变温度较低,室温下分子链容易运动形成更加规整的排列,从而导致pbs结晶性能随时间延长而提高,进而导致材料韧性变差。因此,其作为包装材料单独使用时,随着货架期的延长,材料性能会发生变化。另外pbs的降解速率会受到其结晶程度的影响。
3.通过引入第三单体,制备pbs基共聚酯材料以获得理想的性能,如优异的的机械性能、良好的生物降解性和加工性能等,扩大其应用领域。pbs基共聚酯种类繁多,其中引入含有醚键的第三单体能够有效提高聚酯的亲水性,降低聚酯的结晶性,从而达到提高pbs聚酯的韧性,延长货架期,提高降解速率的目的。但是含有醚键的线性结构的第三单体的引入往往对聚酯的强度和热性能都有负面影响。
技术实现要素:
4.根据本技术的一个方面,提供了一种可降解的共聚物,该共聚物采用同时含醚键和环状结构的二元醇thfdm单体引入到pbs中,具有良好的韧性,长的货架期,降解速率高的优势。
5.本技术中提供了一种可降解的共聚物,所述共聚物选自具有式i所示结构式的共聚物中的至少一种;
[0006][0007]
其中,x,y分别为对应结构单元的摩尔分数;ran代表无规共聚;
[0008]
x+y=1,x=0.1~0.7。
[0009]
可选地,x独立的选自0.11、0.22、0.29、0.38、0.46、0.70中的任意值或任意两者之间的范围值。
[0010]
可选地,式i中:
[0011]
x+y=1,x=0.1~0.3。
[0012]
可选地,式i中x=0.3~0.7。
[0013]
可选地,式i中x=0.1~0.29。
[0014]
可选地,式i中x=0.3~0.5。
[0015]
可选地,所述共聚物的特性粘度为1.03dl/g~1.71dl/g。
[0016]
可选地,所述共聚物的特性粘度为1.53dl/g~1.71dl/g。
[0017]
可选地,所述共聚物的断裂强度为20~24mpa。
[0018]
可选地,所述共聚物的断裂伸长率为690~1120%。
[0019]
根据本技术的又一个方面,提供了一种可降解共聚物的制备方法,所述方法包括:
[0020]
将含有四氢呋喃二甲醇,1,4-丁二醇,丁二酸的原料进行熔融缩聚,得到所述可降解共聚物;
[0021]
其中,所述熔融缩聚包括酯化和缩聚。
[0022]
所述原料中四氢呋喃二甲醇,1,4-丁二醇和丁二酸的醇酸比为1.0~2.5;
[0023]
其中,醇以四氢呋喃二甲醇和1,4-丁二醇的摩尔之和计算;
[0024]
酸以丁二酸的摩尔数计算。
[0025]
所述原料中还包括催化剂;
[0026]
其中,所述催化剂选自钛系催化剂、锡类催化剂中的一种;
[0027]
所述钛系催化剂选自钛酸四丁酯、钛酸二乙二脂、钛酸叔丁酯中的一种;
[0028]
所述锡类催化剂选自辛酸亚锡、二醋酸二丁基锡、二丁基锡二月桂酸酯中的一种。
[0029]
所述酯化的条件包括阶段升温酯化;
[0030]
所述阶段升温酯化包括:160~220℃反应i1~3h,然后升温至220~230℃反应ii 0.5~2h;
[0031]
其中反应ii的温度高于反应i的温度。
[0032]
可选地,所述反应i的温度上限选自170℃、180℃、190℃、200℃、210℃或220℃;下限选自160℃、170℃、180℃、190℃、200℃或210℃。
[0033]
可选地,所述反应ii的温度上限选自221℃、225℃或230℃;下限选自220℃、221℃或225℃。
[0034]
所述缩聚的条件包括阶段缩聚;
[0035]
所述阶段缩聚包括:1000~3000pa的条件下220~230℃反应iii 0.5~1.5h,然后50~100pa的条件下继续反应iv 3~5h。
[0036]
所述熔融缩聚之前所述原料进行打浆处理;
[0037]
可选地,所述反应iii的反应压力上限选自3000pa;下限选自1000pa。
[0038]
可选地,所述反应iv的反应压力上限选自100pa;下限选自50pa。
[0039]
所述打浆处理的条件包括:100~150℃打浆0.5~1h。
[0040]
作为其中一种具体的实施方式,所述可降解的共聚物的制备方法包括:
[0041]
(1)将含有四氢呋喃二甲醇,1,4-丁二醇,丁二酸及钛酸四丁酯的原料混合(加入到反应器中),打浆;
[0042]
(2)将原料升温至反应i的温度,恒温反应后升温至反应ii的温度,恒温反应,完成酯化;
[0043]
(3)酯化结束后,进行缩聚:将反应体系的真空度降至反应iii的反应压力,并升温至反应iii的温度,进行反应;然后将反应体系的压力降低至反应iv的反应压力,反应,得到
所述共聚物。
[0044]
可选地,步骤(3)中反应iv结束后,进行后处理。
[0045]
可选地,所述后处理包括:反应结束后,向体系中充入惰性气体(如氮气)至体系压力升高(如0.2mpa,釜底温度150℃,铸带头温度120℃),出料,冷却后成型(如造粒);干燥后备用。
[0046]
本技术的又一方面,提供了上述可降解的共聚物和/或上述制备方法制备得到的可降解的共聚物在食品包装膜袋材料、医用无纺布中的应用。
[0047]
本技术的另一方面,上述可降解的共聚物和/或上述制备方法制备得到的可降解的共聚物,在酶催化条件下,70天的质量损失率为6~60%。
[0048]
本技术中各物质缩写如下:
[0049]
聚丁二酸丁二醇酯缩写为pbs;
[0050]
聚(丁二酸四氢呋喃二甲酯)缩写为pts;
[0051]
聚(丁二酸丁二醇酯-共-丁二酸四氢呋喃二甲酯)缩写为pbts;
[0052]
四氢呋喃二甲醇缩写为thfdm;
[0053]
钛酸四丁酯缩写为tbt;
[0054]
1,4-丁二醇缩写为bdo;
[0055]
1,4-丁二酸缩写为sa。
[0056]
本技术能产生的有益效果包括:
[0057]
1)本技术中所述可降解的共聚物,共聚物特性粘度较高,结晶度降低,玻璃化转变温度提高,能够满足实际应用的需求;且共聚酯中thfdm实际含量比投料量偏低。
[0058]
2)本技术中所述可降解的共聚物的制备方法,具有提高pbs聚酯的韧性,延长货架期,提高降解速率的作用。
[0059]
3)本技术所提供的可降解的共聚物,制备方法简单,获得的可降解的共聚酯具有比pbs聚酯更优的韧性,延长货架期,降解速率高,同时具有优异强度和热性能的作用。
附图说明
[0060]
图1为本技术一种实施方式中合成路线图。
[0061]
图2(a)为实施例5特征峰归属(b)为实施例1~6和对比例1、2的1h nmr谱图。
[0062]
图3(a)为实施例1~6和对比例1、2的降温dsc曲线(b)为实施例1~6和对比例1、2的二次升温dsc曲线。
[0063]
图4为实施例1~6和对比例1、2退火后的x射线衍射图。
[0064]
图5为对比例1(a)、实施例1(b)和实施例2(c)在不同退火温度下dsc熔融曲线。
[0065]
图6为对比例1(a)、实施例1(b)和实施例2(c)的不同样品热分级dsc曲线。
[0066]
图7为对比例1、实施例1、实施例2和实施例3的力学性能表征:(a)应力-应变曲线;(b)聚合物力学性能对比。
[0067]
图8为对比例1、实施例1、实施例2和实施例3在酶催化降解不同时间下的sem图。
[0068]
图9为对比例1、实施例1、实施例2和实施例3在酶催化降解下的质量随时间变化趋势。
具体实施方式
[0069]
下面结合实施例详述本技术,但本技术并不局限于这些实施例。
[0070]
如无特别说明,本技术的实施例中的原料和催化剂均通过商业途径购买,其中四氢呋喃二甲醇(thfdm)购于浙江糖能科技有限公司,1,4-丁二醇(bdo)购于康辉新材料科技有限公司,1,4-丁二酸(sa)购于康辉新材料科技有限公司,钛酸四丁酯(tbt)购于百灵威化学。
[0071]
本技术的实施例中分析方法如下:
[0072]
利用乌氏粘度计进行聚合物特性粘度分析。
[0073]
利用核磁共振波谱仪进行共聚酯的实际组成及微观结构分析。
[0074]
利用差示扫描量热仪进行聚合物热性能分析。
[0075]
利用广角x射线衍射进行聚合物waxd表征。
[0076]
利用拉伸试验机进行拉伸性能测试分析。
[0077]
利用热失重分析仪进行聚合物失重性能分析。
[0078]
利用冲击试验机进行冲击性能测试分析。
[0079]
利用钨灯丝扫描电镜进行降解性能分析。
[0080]
本技术的实施例中共聚酯中两种结构单元(丁二酸丁二醇酯和丁二酸四氢呋喃二甲酯单元)的含量可以通过以下公式进行计算:
[0081]
其中i
2.60
和i
3.90~4.26
分别表示化学位移在2.60ppm和3.90~4.26ppm处的峰面积,和分别代表共聚酯中丁二酸四氢呋喃二甲酯单元和丁二酸丁二醇酯单元的摩尔含量。
[0082][0083][0084][0085][0086]
实施例1
[0087]
向5l反应釜中加入185.0g的四氢呋喃二甲醇,908.4g的1,4-丁二醇,826.7g的丁二酸及5.71g的钛酸四丁酯,在120℃下打浆0.5h;
[0088]
随后将釜内温度逐渐升至160℃,反应2h后逐渐升温至220℃,继续反应1h;
[0089]
关闭酯化系统,打开缩聚系统,将体系真空度由常压逐渐降至2000pa,并逐渐升温至230℃,反应40min;
[0090]
将体系真空度逐渐降至50pa以下,230℃条件下反应4h;
[0091]
反应结束后,向体系中充入n2至釜内压力升高至0.2mpa,打开出料口,经水冷后通过切粒机造粒,将聚合物粒料放入真空烘箱30℃干燥24h备用,样品记为pbts-11。
[0092]
实施例2
[0093]
向5l反应釜中加入370g的四氢呋喃二甲醇,807.5g的1,4-丁二醇,826.7g的丁二酸及5.71g的钛酸四丁酯,其他步骤同实施例1,此处不再赘述,样品记为pbts-22。
[0094]
实施例3
[0095]
向5l反应釜中加入555.1g的四氢呋喃二甲醇,706.5g的1,4-丁二醇,826.7g的丁二酸及5.71g的钛酸四丁酯,其他步骤同实施例1,此处不再赘述,样品记为pbts-29。
[0096]
实施例4
[0097]
向5l反应釜中加入740.1g的四氢呋喃二甲醇,605.6g的1,4-丁二醇,826.7g的丁二酸及5.71g的钛酸四丁酯,其他步骤同实施例1,此处不再赘述,样品记为pbts-38。
[0098]
实施例5
[0099]
向5l反应釜中加入925.1g的四氢呋喃二甲醇,504.7g的1,4-丁二醇,826.7g的丁二酸及5.71g的钛酸四丁酯,其他步骤同实施例1,此处不再赘述,样品记为pbts-46。
[0100]
实施例6
[0101]
向5l反应釜中加入1387.7g的四氢呋喃二甲醇,252.3g的1,4-丁二醇,826.7g的丁二酸及5.71g的钛酸四丁酯,其他步骤同实施例1,此处不再赘述,样品记为pbts-70。
[0102]
对比例1
[0103]
向5l反应釜中加入1009.3g的1,4-丁二醇,826.7g的丁二酸及5.71g的钛酸四丁酯,在120℃下打浆0.5h;
[0104]
随后将釜内温度逐渐升至160℃,反应2h后逐渐升温至220℃,继续反应1h;
[0105]
关闭酯化系统,打开缩聚系统,将体系真空度由常压逐渐降至2000pa,并逐渐升温至230℃,反应40min;
[0106]
将体系真空度逐渐降至50pa以下,230℃条件下反应4h;
[0107]
反应结束后,向体系中充入n2至釜内压力升高至0.2mpa,打开出料口,经水冷后通过切粒机造粒,将聚合物粒料放入真空烘箱30℃干燥24h备用,样品记为pbs。
[0108]
对比例2
[0109]
向5l反应釜中加入1850.2g的四氢呋喃二甲醇,826.7g的丁二酸及5.71g的钛酸四丁酯,其他步骤同对比例2,此处不再赘述,样品记为pts。
[0110]
分析例1共聚酯的结构表征
[0111]
采用1h nmr对共聚酯及两种均聚酯的化学结构和组成进行了表征,共聚酯的实际组成及微观结构通过bruker avance 400mhz核磁共振波谱仪进行测试。将聚合物样品7mg溶于0.5ml氘代氯仿(chcl3,δ=7.26ppm)中,测试温度为25℃。
[0112]
对于聚(丁二酸四氢呋喃二甲醇酯)(pts)即对比例2,化学位移在2.60ppm的峰代表丁二酸中两个亚甲基的质子(a)特征峰;而化学位移在3.90~4.05ppm和1.60~2.05ppm处的峰分别为四氢呋喃环上的ch(c)和ch2(d+d’),此外,在3.95-4.11ppm处的峰属于四氢呋喃二甲醇单元中与酯基相连的的另一个ch2(b)质子峰。对于聚(丁二酸丁二醇酯)(pbs),化学位移在2.60ppm的峰代表丁二酸中两个亚甲基的质子(a)特征峰;而化学位移在3.95~4.11ppm处的峰代表丁二醇单元中与酯基相连的ch2(e)的特征峰,此外,在1.53~1.73ppm处的峰属于丁二醇中与酯基间接相连的ch2(f)质子特征峰。在共聚酯即实施例1-6中,两种醇与酯基相连的亚甲基的特征峰和thfdm中的次甲基ch(c)的特征峰发生重叠(3.90~4.26ppm),四氢呋喃中的亚甲基特征峰与丁二醇中的ch2(f)特征峰发生重叠(1.60~1.80ppm);由此可以看出共聚酯中的丁二酸丁二醇酯单元(bs单元)和丁二酸四氢呋喃二甲醇酯单元)(ts单元)的含量可以通过以下公式进行计算:
[0113][0114][0115][0116][0117]
其中i
2.60
和i
3.90~4.26
分别表示化学位移在2.60ppm和3.90~4.26ppm处的峰面积,和分别代表共聚酯中丁二酸四氢呋喃二甲酯单元和丁二酸丁二醇酯单元的摩尔含量。
[0118]
分析例2粘度表征
[0119]
对上述实施例和对比例中制备得到的聚合物进行特性粘度([h])测试。
[0120]
特性粘度测试采用的是乌氏粘度计,具体包括:将待测聚合物溶解于氯仿溶剂中,分别配制浓度为10g/l,7.5g/l,5g/l和2.5g/l的聚合物溶液,将聚合物溶液分别加入乌氏粘度计中,在25℃恒温水浴中稳定5min,测定不同浓度聚合物溶液流出时间及纯溶剂流出时间,通过外推法得到聚合物的特性粘度。
[0121]
测试结果参见表1。
[0122]
表1 pbs、pts和pbts共聚酯的合成投料比及结构表征
[0123][0124]
a投料二元醇中thfdm的摩尔分数;
[0125]
b采用1hnmr计算得到的共聚酯中ts单元的摩尔分数;
[0126]
c聚合物特性粘度测试条件为25℃,溶剂为氯仿。
[0127]
从表1中可以看出随着thfdm投料量的提高,实施例1-6的粘度呈下降趋势,这是由于thfdm存在比bdo更高的位阻效应。
[0128]
分析例3热性能分析
[0129]
对对比例1-2和实施例1-6的热性能和结晶进行了研究。
[0130]
其中,热性能测试采用差示扫描量热仪进行测试(非等温dsc),具体包括聚合物热性能采用美国ta公司的q25差示扫描量热仪进行测试。在降温过程中测定了结晶温度(tc)和结晶熵(δhc),在二次升温过程中测定了熔融温度(tm)、冷结晶温度(tcc)和相应的结晶熵(δhm和δhcc)。将聚合物样品5~10mg样品置于铝坩埚中,聚合物在氮气环境下进行测
试(氮气流速为50ml/min)。具体测试程序为一次升温:25℃升温至150℃(10℃/min);150℃等温3min消除热历史;降温:由150℃降温至-60℃(-10℃/min);-60℃等温3min以达到热力学平衡;二次升温:-60℃升温至150℃(10℃/min)。
[0131]
由图3可知,对比例1即pbs结晶能力较强,随着thfdm的引入,实施例1-6的结晶峰强度降低,冷结晶峰出现,说明共聚酯结晶能力下降;随着thfdm含量进一步提高,结晶峰和熔融峰消失,当共聚酯中thfdm含量高于29%时,共聚酯表现为无定形态,这主要是由于thfdm中的环状结构阻碍了分子链的规整排列,从而导致结晶能力下降,达到提高实施例的韧性,延长货架期,提高降解速率的目的。
[0132]
同时,对比例1-2和实施例1-6的热性能参数(tg、tm、tc、tcc及其对应的焓值δhm、δhc、δhcc)如表2所示,聚合物失重性能采用美国ta公司的q500热失重分析仪进行测试。二次升温过程中测定了聚合物失重5%,10%时的温度(td,5%,td,10%);聚合物最快热分解速率温度(td,max);及残碳量(rw)。
[0133]
将聚合物样品5~10mg样品置于样品盘中,聚合物在氮气环境下进行测试(氮气流速为50ml/min)。具体测试程序为室温升温至650℃。
[0134]
pts的tg为0.1℃。pbs的tg、tm、tc分别为-35.7℃、70.3℃和114.1℃(δhc=54.7j/g)、267.6℃(δhm=68.5j/g)。
[0135]
由图3和表2所知,随着thfdm含量进一步提高,实施例1-6结晶焓和熔融焓下降,证明了共聚酯结晶能力下降。当共聚酯中ts单元含量超过30%时,共聚酯表现为无定形态。在玻璃化转变温度方面,对比例1相比于对比例2的玻璃化转变温度更低,随着thfdm的引入,共聚酯tg有明显的提高(由-35.7℃提升至-0.1℃),从而提高共聚酯的韧性。基于此,说明虽然thfdm含有醚键,但是其环状结构还是使得其均聚酯分子链表现出更高的刚性。
[0136]
表2 pbs、pts和pbts聚酯的热性能参数
[0137][0138]
分析例4结晶性能研究
[0139]
共聚酯的结晶性能对聚合物的力学性能,降解性能等都有着重要的影响。采用waxd和dsc探究了聚酯的结晶行为。图4所示为waxd图谱,对制备的聚合物进行waxd表征,采用配有ni过滤,cu靶kα射线(λ=0.15418nm)的rigaku d/max-ultima型x射线衍射仪。测试条件:室温,5
°
~45
°
,5
°
/min。
[0140]
聚合物膜的制备:将样品(1-2g)置于1cm
×
1cm不锈钢膜具中,放到两个聚酰亚胺
iii区域。同样的,不同共聚酯的domain区域及最佳初始自成核退火温度(domain ii区域内的最低温度)总结于表3,随着共聚酯中thfdm含量的提高,其domain ii区域的温度范围变窄,这主要是由于共聚酯结晶能力变差,从而达到提高pbs聚酯的韧性,延长货架期,提高降解速率的目的。
[0147]
表3 domain区域温度范围及各样品理想初始自成核退火温度
[0148][0149]
在确定每种聚合物样品的最佳初始自成核退火温度(ts,ideal)后,对样品进行了ssa表征,其热分级结果如图6所示。由图可知,三个样品均能够实现较好的热分级效果,在经过ssa处理后pbs均聚酯的熔融峰列分为五个强度不同的熔融峰,实施例1即pbts-11样品熔融峰列分为六个强度不同的熔融峰,实施例2即pbts-22样品熔融峰列分为七个强度不同的熔融峰,随着thfdm含量的提高,热分级产生的熔融峰越多,这是由于thfdm的引入破坏了结晶,从而导致不完善的晶体含量提高。熔融温度越高表示片晶厚度越大,熔融峰的强度则代表对应片晶厚度晶体在聚酯材料中的含量。对于对比例1pbs均聚酯材料,其熔融温度最高的峰出现在115.8℃,并且其也是强度最高的熔融峰,说明对于对比例1均聚酯材料,其晶体中相对完善的晶体含量占主导;对于实施例1聚酯材料,其熔融温度最高的峰出现在106.4℃,虽然也是强度最高的熔融峰,但是温度略低于它的熔融峰(103.9℃)强度较高,说明对于pbts均聚酯材料,虽然其晶体中相对完善的晶体含量占主导,但是完善程度较低的晶体含量相比于pbs明显提高;对于实施例2聚酯材料,其熔融温度最高的峰出现在90.8℃,但是其并非强度最高的熔融峰,温度略低于它的熔融峰(87.8℃)强度较高,说明对于pbts均聚酯材料,晶体中相对完善的晶体含量并非占主导,完善程度较低的晶体含量相比于对比例1或实施例1明显提高。综合ssa热分级结果可以得到结论,实施例1-6的共聚酯随着thfdm含量的提高,其结晶能力变差,具有不完善结构的晶体含量提高,与前文dsc结果一致。
[0150]
分析例5力学性能测试
[0151]
图7为通过万能拉伸试验机对对比例1及实施例1、实施例2和实施例3的力学性能进行的表征结果(从上到下(应变在300%或20%处的)依次为对比例1、实施例1、实施例2和实施例3),拉伸样条制备:采用haake minijet注塑机制备了厚度为2.0mm、宽度为5.0mm的哑铃形样条,物料腔温度150℃,注射压力0.2mpa。
[0152]
拉伸性能测试:采用instron 5565试验机进行拉伸试验,拉伸速率5mm/min的,对每个样品进行5个试样的测量,取平均值。
[0153]
冲击样条制备:采用haake minijet注塑机制备了厚度为4.0mm、宽度为10.0mm的冲击样条,物料腔温度285℃,注射压力0.4mpa。
[0154]
冲击性能测试:采用冲击试验机(ceast 9050)进行冲击试验,对每个样品进行5个试样的测量,取平均值。
[0155]
应力-应变曲线如图7(a)所示,所有样品均表现出典型的热塑性材料拉伸行为。在室温下,对比例1表现出应变硬化和第二次屈服现象。此外,pbs基共聚酯材料在拉伸过程中也表现出不同程度的拉伸震荡(stress oscillation,so),这种现象常见于非晶和半晶聚合物的拉伸过程,其特征是垂直于形变方向周期性地形成明显的透明和不透明带。在最近的研究中发现,pbs的拉伸震荡主要是由于在拉伸过程中,细颈区产生微纤维和裂纹进一步形成微空洞诱导了不透明区域的出现,并降低了拉伸应力。
[0156]
不同样品的屈服强度及断裂伸长率如图7(b)所示。随着thfdm含量的提高屈服强度降低,断裂伸长率提高。这主要是由于随着thfdm含量的提高,pbs结晶完善程度降低,结晶度下降。
[0157]
表4总结了对比例1和实施例1-3的共聚酯的屈服强度(σy)、拉伸强度(σb)和断裂伸长率(εb)。对比例1的σy值为33.2mpa,εb值为494.5%,对共聚酯而言,随着共聚酯中ts含量的提高,屈服强度从26.5mpa降低至15.5mpa,而断裂伸长率则呈相反的趋势,由696.8%提升至1112.5%,这表明通过调节共聚酯的组成实现了共聚酯韧性的提升。
[0158]
表4 pbs和部分pbts共聚物的力学性能比较
[0159][0160]
分析例6降解效果测试
[0161]
薄膜样品制备:将样品(5g)置于10cm
×
10cm不锈钢膜具中,放到两个聚酰亚胺薄膜之间,将样品放置于平板硫化机中,上下板温度设定为150℃,在0.2mpa的压力下保温2min。最后,在这样的压力下(30分钟)将薄膜冷却到25℃。将薄膜裁成1cm
×
1cm的正方形,25℃真空干燥24h备用。
[0162]
将每个样片称重后置于5ml浓度为0.1m的磷酸盐(pbs)缓冲液(ph=7.2)中,缓冲液中脂肪酶浓度为0.025mg/ml,置于30℃恒温摇床中约60天,每七天更换降解液。间隔固定时间取出样品,去离子水冲洗后置于真空烘箱干燥至恒重,测量样品剩余质量,并进行后续测试表征。
[0163]
对比例1及实施例1-3共聚酯的薄膜的表面形貌变化,降解时间到第7天时,实施例3膜表面形貌发生相对明显的变化,逐渐有孔洞形成,而其他样品表面比较平滑,无明显孔洞形成;当降解时间为14天时,实施例2样品薄膜逐渐变得粗糙,并且出现了孔洞,而实施例3孔洞逐渐扩大,实施例1与对比例1薄膜表面未出现明显变化;当降解时间为14天时,对比例1表面逐渐粗糙,但未出现明显孔洞。实施例1出现了少量孔洞。实施例2的孔洞逐渐变大。实施例3薄膜表面出现大量具有不规则轮廓的孔洞。
[0164]
图9为对比例1及实施例1-3共聚酯的质量损失随降解时间的变化,降解聚酯薄膜表面形貌采用配有jem-101显微镜的quanta 450扫描电镜观察聚合物降解薄膜样品表面形貌,所有样品均于测试前进行喷金处理。
[0165]
在初始降解阶段,不同样品降解速率差别不大,thfdm含量较高的聚酯材料质量损
失率略高,这主要是由于酶分子量较高,其只能附着在膜表面,初始阶段样品主要以聚酯水解为主,酶催化作用并不明显,而thfdm中含有的醚键提高了聚酯亲水性,在初始阶段降解速率相对较高;实施例3的降解速率在28天后出现明显提高,因此实施例3表现出明显高于其他样品的降解速率,这主要是由于在酶的作用下,孔洞的逐渐形成和发展导致了与酶接触的聚酯薄膜的表面积增大,并且更多的聚酯分子链发生断裂并与酶接触,并发生新陈代谢作用,加快了材料的降解速率。降解时间达到35天时,实施例3质量损失接近20%,而此时其他样品的质量损失低于5%,并且随着聚酯样品中thfdm含量的提高,在相同降解时间下聚酯质量损失提高,这主要可以归因于thfdm的引入降低了聚酯的结晶度,提高了亲水性。
[0166]
以上所述,仅是本技术的几个实施例,并非对本技术做任何形式的限制,虽然本技术以较佳实施例揭示如上,然而并非用以限制本技术,任何熟悉本专业的技术人员,在不脱离本技术技术方案的范围内,利用上述揭示的技术内容做出些许的变动或修饰均等同于等效实施案例,均属于技术方案范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1