萜类化合物及其制备方法和应用
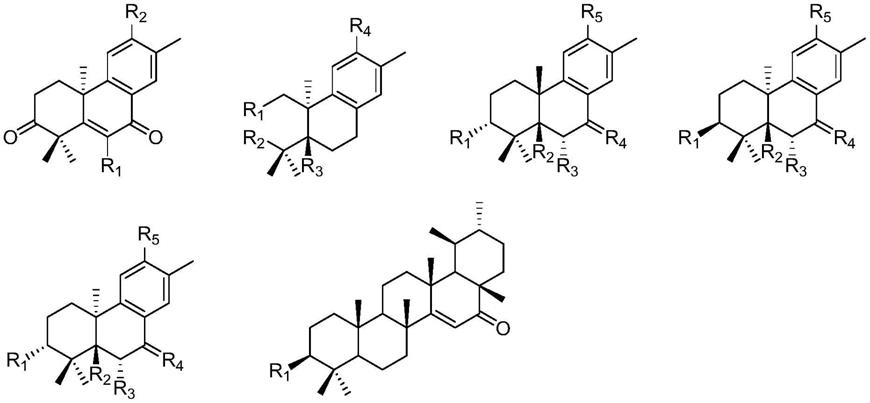
1.本发明涉及萜类化合物及其制备方法和应用,具体涉及麻疯树干燥茎枝中的abietane二萜类及ursane三萜类化合物及其制备方法和应用,属于医药技术领域。
背景技术:
2.麻疯树(jatropha curcas l.)是大戟科(euphorbiaceae)麻疯树属(jatropha)植物。麻疯树属植物全球有175多种,主产于美洲热带、亚热带地区,少数产子非洲。中国栽培或逸为野生的有5种。
3.麻疯树为小乔木或灌木,主产于我国福建、台湾、广东、海南、广西、贵州、四川、云南等地。《广西中草药》及《常用中草药彩色图谱》中记载其具有清热解毒、散瘀消肿、止血止痒、止吐止痛等功效;外用治疗跌打肿痛、关节挫伤、皮肤瘙痒、创伤止血等,内服有泻下、催吐的作用。现代药理学研究表明麻疯树具有抗炎作用,其主要化学成分是二萜、三萜、黄酮、木脂素等。
技术实现要素:
4.本发明的目的在于提供一系列萜类化合物及其药学上可接受的盐,具体为一系列abietane二萜类及ursane三萜类化合物及其制备方法和新的医药用途。
5.萜类化合物及其药学上可接受的盐,所述萜类化合物具有下述通式之一:
[0006][0007]
其中,r1为-oh或-cooh;r2为-h、-cooh或-och3;r3为-h、或-och3;r4为-oh、-h2或-o;r5为-oh或-och3。
[0008]
本发明优选如下abietane二萜类和ursane三萜类化合物及其药学上可接受的盐,
[0009][0010]
其中,r1为-oh或-cooh;r2为-h、-cooh或-och3;r3为-h、或-och3;r4为-oh、-h2或-o;r5为-oh或-och3。
[0011]
本发明具体公布了如下六个具体化合物:
[0012][0013]
本发明还提供了所述abietane二萜类化合物1,2,3,4,5和ursane三萜类化合物6的制备方法,该方法包括如下步骤:
[0014]
(1)麻疯树(jatropha curcas l.)茎枝用乙醇或甲醇提取、或超声提取,回收提取液得粗提物;
[0015]
(2)上述步骤(1)所得粗提物经水溶解,有机溶剂萃取,得到不同极性的萃取物;
[0016]
(3)上述步骤(2)所得的萃取物经硅胶柱色谱法分离,以石油醚和乙酸乙酯混合溶剂、或石油醚和丙酮混合溶剂、或氯仿和丙酮混合溶剂、或二氯甲烷和丙酮混合溶剂、或氯仿和甲醇混合溶剂、或二氯甲烷和甲醇混合溶剂梯度洗脱;
[0017]
(4)上述步骤(3)中所得流分经ods柱色谱分离,以甲醇-水或乙腈-水混合溶剂为流动相梯度洗脱;
[0018]
(5)上述步骤(4)中所得甲醇-水或乙腈-水洗脱物经hplc进一步分离,以甲醇和水混合溶剂或以乙腈和水混合溶剂为流动相梯度洗脱,得到化合物1、2、3、4、5和6。
[0019]
本发明所述abietane二萜类化合物1,2,3,4,5和ursane三萜类化合物6的制备方法,一个优选的技术方案为:
[0020]
abietane二萜类化合物1,2,3,4,5和ursane三萜类化合物6的制备方法,该方法包括如下步骤:
[0021]
(1)麻疯树(jatropha curcas l.)茎枝用70%~95%的乙醇或60%~90%的甲醇加热回流提取,回收提取液得粗提物;
[0022]
(2)步骤(1)所得粗提物经水溶解后,按照水相和有机相的体积比1:1~1:5,用石油醚或环己烷、二氯甲烷或氯仿、乙酸乙酯、正丁醇依次萃取3~5次,得到不同极性的萃取物;
[0023]
(3)上述步骤(2)所得的萃取物经硅胶柱色谱法分离,以石油醚和乙酸乙酯混合溶剂100:10~1:1,或石油醚和丙酮混合溶剂100:10~1:1,或二氯甲烷和丙酮混合溶剂100:1~1:1,或氯仿和丙酮混合溶剂100:1~1:1,或氯仿和甲醇混合溶剂100:1~1:1,或二氯甲烷和甲醇混合溶剂100:1~1:1梯度洗脱;
[0024]
(4)上述步骤(3)中所得流分经ods柱色谱分离,以甲醇-水1:9~9:1或乙腈-水2:8~8:2的混合溶剂为流动相梯度洗脱;
[0025]
(5)上述步骤(4)中所得甲醇和水、乙腈和水洗脱物经hplc进一步分离,以甲醇和水混合溶剂4:6~9:1或以乙腈和水3:7~8:2混合溶剂为流动相梯度洗脱,得到化合物1、2、3、4、5和6。
[0026]
本发明提供的所述abietane二萜类1,2,3,4,5和ursane三萜类化合物6的制备方法,步骤(1)所述提取方法为加热回流乙醇提取、加热回流甲醇提取或加热超声提取2~5次,所用溶剂为70%~95%的乙醇或60%~90%的甲醇,优选80%~95%的乙醇或70%~90%的甲醇。料液比为1:8~1:20g/ml,优选1:10~1:15。
[0027]
本发明提供的所述abietane二萜类1,2,3,4,5和ursane三萜类化合物6的制备方法,步骤(2)所述有机溶剂萃取法,采用水溶解粗提物,按照水相和有机相的体积比1:1~1:5,优选1:1~1:3,分别使用石油醚或环己烷、二氯甲烷或氯仿、乙酸乙酯、正丁醇依次萃取3~5次,优选4次,减压回收以上有机溶剂。
[0028]
本发明提供的所述abietane二萜类1,2,3,4,5和ursane三萜类化合物6的制备方法,步骤(3)所述的石油醚和乙酸乙酯,或石油醚和丙酮混合溶剂的体积比例为100:5~1:1,优选100:10~1:1;二氯甲烷和丙酮,或氯仿和丙酮,或二氯甲烷和甲醇,或氯仿和甲醇的混合溶剂的体积比例为100:1~3:1,优选100:3~5:1。
[0029]
本发明提供的所述abietane二萜类1,2,3,4,5和ursane三萜类化合物6的制备方法,步骤(4)所述的甲醇和水混合溶剂的体积比例为1:9~9:1,优选4:6~9:1,乙腈和水混合溶剂的体积比例为2:8~8:2,优选3:7~8:2。
[0030]
本发明提供的所述abietane二萜类1,2,3,4,5和ursane三萜类化合物6的制备方法,步骤(5)所述的甲醇和水混合溶剂的体积比例为4:6~9:1,优选5:5~9:1,乙腈和水混合溶剂的体积比例为3:7~8:2,优选4:6~8:2。
[0031]
本发明的另一目的是提供包含萜类化合物的组合物。一种药物组合物,所述组合物包含所述萜类化合物及其药学上可接受的盐和药学上可接受的载体,
[0032]
所述萜类化合物具有下述通式之一:
[0033][0034]
其中,r1为-oh或-cooh;r2为-h、-cooh或-och3;r3为-h、或-och3;r4为-oh、-h2或-o;r5为-oh或-och3。
[0035]
本发明的又一目的是提供所述萜类化合物及其药学上可接受的盐或上述药物组合物在制备预防或治疗神经性退行性疾病药物中的应用。
[0036]
本发明以lps诱导bv-2小胶质细胞过度活化模型,对制备得到的abietane二萜类1,2,3,4,5和ursane三萜类化合物6的抗神经炎症进行了评价。结果显示,新化合物1,6能够抑制lps诱导的过度活化的bv-2小胶质细胞no的释放,表现出显著的抗神经炎症活性。因此,本发明中制备的新abietane二萜类和ursane三萜类化合物可在开发治疗神经退行性疾病药物方面应用。
[0037]
本发明首次提供了以麻疯树干燥茎枝为原料,制备、鉴定5个abietane二萜类和1个ursane三萜类衍生物的方法,并且系统评价了其神经保护方面的活性,阐明了其在开发和治疗神经退行性疾病药物方面的应用。
具体实施方式
[0038]
下述非限制性实施例可以使本领域的普通技术人员更全面地理解本发明,但不以任何方式限制本发明。
[0039]
下述实施例中所述试验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径获得。
[0040]
实施例1
[0041]
(1)麻疯树干燥茎枝1000g用90%乙醇加热回流提取3次(用量:10l),减压回收提取液得粗提物;
[0042]
(2)上述步骤(1)所得90%乙醇粗提物用水溶解,经乙酸乙酯、正丁醇依次萃取,每个有机相萃取3次,每次水相和有机相的体积比1:1,得到不同极性部位的萃取物;
[0043]
(3)步骤(2)中乙酸乙酯萃取物,经硅胶柱色谱分离,依次以石油醚和乙酸乙酯混合溶剂:100:5,10:1,8:1,5:1,3:1,1:1洗脱;
[0044]
(4)上述步骤(3)中所得的石油醚:乙酸乙酯8:1~1:1流分经ods色谱,用甲醇-水3:7,5:5,7:3,9:1的混合溶剂梯度洗脱;
[0045]
(5)上述步骤(4)中所得甲醇-水(5:5~9:1)流分经hplc-uv色谱分离制备,210nm检测,流速为3ml/min,流动相为甲醇:水=48:52,得到abietane二萜类化合物3(tr=12min)(收率为0.0005
‰
)。
[0046]
(6)上述步骤(4)中所得甲醇-水(5:5~9:1)流分经hplc-uv色谱分离制备,210nm检测,流速为3ml/min,流动相为甲醇:水=68:32,得到到abietane二萜类化合物2(tr=19min)(收率为0.00024
‰
)、化合物4(tr=28min)(收率为0.00076
‰
)、化合物5(tr=30min)
(收率为0.00083
‰
)和化合物1(tr=56min)(收率为0.00019
‰
)。
[0047]
(7)上述步骤(4)中所得甲醇-水(5:5~9:1)流分经hplc-uv色谱分离制备,210nm检测,流速为3ml/min,流动相为甲醇:水=88:12,得到ursane三萜类化合物6(tr=35min)(收率为0.0011
‰
)。
[0048]
根据化合物1~6的理化性质和波谱数据鉴定了其结构。
[0049]
化合物1的结构鉴定数据如下:
[0050]
白色无定型粉末(甲醇),-40.4(c 0.3,ch3oh)。hr-esi-ms给出准分子离子峰m/z 313.1434[m-h]-(calcd.313.1440for c
19h21
o4),提示分子式为c
19h22
o4,不饱和度为9。1h nmr(600mhz,cdcl3)给出11个氢信号,低场区δ
h 7.98(1h,d,j=0.7hz,h-14),6.84(1h,s,h-11)为苯环上的氢信号;δ
h 3.93(12-och3)为甲氧基氢信号;δ
h 2.27(15-ch3),1.59(18-ch3),1.55(19-ch3)和1.32(20-ch3)为4个甲基氢信号;δ
h 2.84(1h,ddd,j=18.8,9.7,1.8hz,h-2α),2.77(1h,dt,j=18.8,9.7hz,h-7β),2.49(1h,ddd,j=13.8,9.0,1.8hz,h-1β),2.09(1h,dt,j=13.8,9.7hz,h-1α)为两组亚甲基氢信号。
13
c nmr(150mhz,cdcl3)给出19个碳信号,其中δ
c 214.5(c-3),179.7(c-7)为两个羰基碳信号;δ
c 162.4(c-12),151.2(c-9),129.0(c-14),127.3(c-13),120.7(c-8),106.2(c-11)为一组苯环碳信号;δ
c 142.5(c-6),137.3(c-5)为一组双键碳信号;δ
c 55.7(12-och3)为甲氧基碳信号,其余均为饱和脂肪碳信号。将化合物1的nmr数据与文献
[1]
报道的gosneilone的nmr数据对比,发现多了一组甲氧基信号,且文献中未提供明确的证据确定化合物的绝对构型。
[0051]
利用hsqc将所有的氢碳信号进行归属(表1,表2),并通过hmbc谱确认了甲氧基的连接位置。在hmbc中,δ
h 3.93(-och3)与δ
c 162.4(c-12)存在远程相关,证明甲氧基在c-12被取代。从而确定该化合物结构为12-methoxy-gossweilone,进一步通过实验和计算ecd数据对比确定了化合物1的绝对构型为10s。
[0052]
综上鉴定化合物1为(10r)-12-methoxy-gossweilone,经检索为一未见文献报道的新化合物。
[0053]
化合物2的结构鉴定数据如下:
[0054]
淡黄色无定形粉末(甲醇),hr-esi-ms给出准分子离子峰m/z 319.1540[m-h]-(calcd.319.1545for c
18h23
o5),提示分子式为c
18h24
o5。1h nmr(600mhz,cd3od)给出12个氢信号,低场区δ
h 6.69(2h,s,h-11,14)为苯环上的氢信号;δ
h 2.73(2h,m,h-7),2.72(1h,dd,j=14.1,5.8hz,h-1β),2.51(1h,d,j=14.1hz,h-1α),2.18(1h,m,h-6α),1.89(1h,dt,j=12.1,6.1hz,h-6β)为三组亚甲基氢信号;δ
h 2.10(15-ch3),1.50(20-ch3),1.24(19-ch3)和0.96(18-ch3)为4个甲基氢信号。
13
c nmr(150mhz,cd3od)给出18个碳信号,结合hsqc谱,确定δ
c 184.1(c-3),175.1(c-2)为两个羧基碳信号;δ
c 154.5(c-12),144.1(c-9),131.9(c-14),128.2(c-8),123.2(c-13),112.2(c-11)为一组苯环碳信号;δ
c 46.3(c-4),41.9(c-10)为两个季碳信号;δ
c 45.4(c-5)为次甲基碳信号;δ
c 50.3(c-1),27.9(c-7),22.1(c-6)为三个亚甲基碳信号;δ
c 28.6(19-ch3),24.8(20-ch3),22.7(18-ch3),15.7(15-ch3)为四个甲基碳信号(表1,表2)。
[0055]
在hmbc谱中,根据δ
h 1.24(19-ch3)/0.96(18-ch3)与c-3、c-4和c-5存在远程相关,确定偕二甲基位于c-4位。通过1h-1
h cosy谱进一步确定了化合物2中c5-c6-c7的结构片段的存在,结合hmbc谱中h-5与c-7/c-9、h-14与c-10/c-12、15-ch3与c-13/c-14、20-ch3与c-1/
9/c-12和20-ch3与c-5存在远程相关,确定了化合物4为abietane二萜。同时,根据且3.89(-och3)与δ
c 162.8(c-12)存在远程相关,证明甲氧基在c-12位。在noesy谱中,通过h-3与h-1β/h-5/19-ch3、20-ch3与h-1α/18-ch3远程相关,确定了化合物4的相对构型:h-3/h-5/19-ch3为β取向,18-ch3/20-ch3为α取向。进一步通过实验和计算ecd数据对比确定了化合物4的绝对构型为3r,5s,10r。
[0064]
综上鉴定化合物4为(3r,5s,10r)-jastrocurcasidione c,经检索为一未见文献报道的新化合物。
[0065]
化合物5的结构鉴定数据如下:
[0066]
淡黄色无定型粉末(甲醇),hr-esi-ms给出准分子离子峰m/z at at z 301.1798(calcd.301.1804for c
19h25
o3),提示分子式为c
19h26
o3。化合物5的氢谱和碳谱与4十分相似(表1、表2),唯一不同的是noesy谱图上,其中化合物5的h-3与20-ch3与h-1α/18-ch3、而h-5与h-1β/19-ch3远程相关,确定了化合物5的相对构型:h-5/19-ch3为β取向,h-3/18-ch3/20-ch3为α取向。且化合物5的核磁数据与文献报道的3-hydroxy-12-methoxy-13-methylpodopcarpa-8,11,13-trien-7-one的nmr数据对比一致,但旋光值相反。进一步通过实验和计算ecd数据对比确定了化合物5的绝对构型为3s,5s,10r,与文献报道的绝对构型相反。
[0067]
综上所述,鉴定化合物5为(3s,5s,10r)-3-hydroxy-12-methoxy-13-methylpodopcarpa-8,11,13-trien-7-one,经检索为一未见文献报道的新化合物。
[0068]
表1 化合物1~5的氢谱核磁数据归属
[0069][0070][0071]
a:in cdcl3;b:cd3od
[0072]
表2 化合物1~5的碳谱核磁数据归属
[0073][0074]
a:in cdcl3;b:cd3od
[0075]
化合物6的结构鉴定数据如下:
[0076]
淡黄色无定型粉末(甲醇),hr-esi-ms给出准分子离子峰m/z 881.7407[2m+h]
+
(calcd.881.7387for c
60h97
o4),提示分子式为c
30h48
o2。1h nmr(600mhz,cdcl3)中低场区δ
h 5.81(1h,s,h-15)存在一个烯氢信号;δ
h 1.27(3h,s,28-ch3),1.15(3h,s,26-ch3),1.11(3h,s,27-ch3),1.07(3h,d,j=6.8hz,29-ch3),0.98(3h,s,23-ch3),0.95(3h,d,j=6.2hz,30-ch3),0.94(3h,s,25-ch3),0.80(3h,s,24-ch3)为8个甲基氢信号。
13
c nmr(150mhz,cdcl3)给出30个碳信号,包括一组α,β不饱和酮信号δ
c 208.0(c-16),183.5(c-14)和119.5(c-15);以及8个甲基碳信号δ
c 34.7(28-ch3),28.1(23-ch3),26.0(26-ch3),25.1(29-ch3),22.9(27-ch3),22.1(30-ch3),15.6(24,25-ch3);其余均为饱和脂肪碳信号,氢碳信号归属见表3。
[0077]
将化合物6的nmr数据与文献报道的3β-acetyl ursa-14(15)-en-16-one的nmr数据对比,发现化合物6在c-3上没有乙酰基取代。结合2d nmr数据,鉴定化合物6被鉴定为ursane型五环三萜。据文献报道,当3位取代基为α时,最大偶合常数应在8hz左右,δ
h-3
在5.00-5.48之间;当3位取代基为β时,最大偶合常数在12hz左右,δ
h-3
在4.00-4.75之间。根据δ
h 3.21(1h,dd,j=11.5,4.5hz,h-3)判断3位羟基取代基为β构型。
[0078]
综上所述,鉴定化合物6为3β-ursa-14(15)-en-16-one,经检索为一未见文献报道的新化合物。
[0079]
表3 化合物6的氢谱和碳谱核磁数据归属
[0080][0081]
实施列2
[0082]
(1)麻疯树干燥茎枝500g用95%乙醇加热回流提取2次(用量:8l),减压回收提取液得粗提物;
[0083]
(2)上述步骤(1)中所得乙醇提取物经有机溶剂萃取,依次用石油醚、乙酸乙酯、正丁醇以水相和有机相的体积比1:2进行萃取,分别萃取3次,得到不同极性部位的萃取物;
[0084]
(3)上述步骤(2)中所得的乙酸乙酯萃取物,经硅胶柱色谱分离,依次以二氯甲烷和丙酮混合溶剂100:1,100:3,100:5,10:1,8:1,5:1,3:1洗脱;
[0085]
(4)上述步骤(3)中所得二氯甲烷:丙酮100:3~8:1流分经ods色谱,用甲醇-水3:7,5:5,7:3,9:1的混合溶剂梯度洗脱;
[0086]
(5)上述步骤(4)中所得甲醇-水(5:5~9:1)流分经hplc-uv色谱分离制备,210nm检测,流速为3ml/min,流动相为甲醇:水=45:55,得到abietane二萜类化合物3(tr=17min)(收率为0.00045
‰
)。
[0087]
(6)上述步骤(4)中所得甲醇-水(5:5~9:1)流分经hplc-uv色谱分离制备,210nm检测,流速为3ml/min,流动相为甲醇:水=70:30,得到到abietane二萜类化合物2(tr=18min)(收率为0.00022
‰
)、化合物4(tr=25min)(收率为0.00075
‰
)化合物5(tr=27min)(收率为0.00079
‰
)和化合物1(tr=52min)(收率为0.00018
‰
)。
[0088]
(7)上述步骤(4)中所得甲醇-水(5:5~9:1)流分经hplc-uv色谱分离制备,210nm检测,流速为3ml/min,流动相为甲醇:水=90:10,得到到ursane三萜类化合物6(tr=32min)(收率为0.0010
‰
)。
[0089]
abietane二萜类1,2,3,4,5和ursane三萜类化合物6的结构鉴定方法见实施例1。
[0090]
实施例3
[0091]
(1)麻疯树干燥茎枝1000g用80%乙醇加热回流提取3次(用量:15l),减压回收提取液得粗提物;
[0092]
(2)上述步骤(1)所得乙醇提取物经有机溶剂萃取,依次用环己烷、乙酸乙酯、正丁醇以水相和有机相的体积比1:3进行萃取,分别萃取5次,得到不同极性部位的萃取物;
[0093]
(3)上述步骤(2)所得的乙酸乙酯萃取物,经硅胶柱色谱分离,依次以氯仿和甲醇混合溶剂100:1,100:3,100:5,10:1,8:1,5:1,3:1洗脱;
[0094]
(4)上述步骤(3)中所得氯仿:甲醇100:3~8:1流分经ods色谱,用甲醇-水3:7,5:5,7:3,9:1的混合溶剂梯度洗脱;
[0095]
(5)上述步骤(4)中所得甲醇-水(5:5~9:1)流分经hplc-uv色谱分离制备,210nm检测,流速为3ml/min,流动相为乙腈:水=38:62,得到abietane二萜类化合物3(tr=11min)(收率为0.00046
‰
)。
[0096]
(6)上述步骤(4)中所得甲醇-水(5:5~9:1)流分经hplc-uv色谱分离制备,210nm检测,流速为3ml/min,流动相为乙腈:水=58:42,得到到abietane二萜类化合物2(tr=20min)(收率为0.00023
‰
)、化合物4(tr=28min)(收率为0.00076
‰
)、化合物5(tr=31min)(收率为0.00081
‰
)和化合物1(tr=47min)(收率为0.00017
‰
)。
[0097]
(7)上述步骤(4)中所得甲醇-水(5:5~9:1)流分经hplc-uv色谱分离制备,210nm检测,流速为3ml/min,流动相为乙腈:水=80:20,得到到ursane三萜类化合物6(tr=21min)(收率为0.0009
‰
)。
[0098]
abietane二萜类1,2,3,4,5和ursane三萜类化合物6的结构鉴定方法见实施例1。
[0099]
实施例4
[0100]
(1)麻疯树干燥茎枝1500g用80%甲醇加热回流提取4次(用量:20l),减压回收提取液得粗提物;
[0101]
(2)上述步骤(1)所得甲醇提取物经有机溶剂萃取,依次用石油醚、二氯甲烷、乙酸乙酯、正丁醇以水相和有机相的体积比1:3进行萃取,分别萃取4次,得到不同极性部位的萃取物;
[0102]
(3)上述步骤(2)所得的乙酸乙酯萃取物,经硅胶柱色谱分离,依次以氯仿和丙酮混合溶剂100:1,100:3,100:5,10:1,8:1,5:1,3:1洗脱;
[0103]
(4)上述步骤(3)中所得的氯仿:丙酮100:3~8:1流分经ods色谱,用乙腈-水2:8,4:6,6:4,8:2的混合溶剂梯度洗脱;
[0104]
(5)上述步骤(4)中所得乙腈-水(4:6~8:2)流分经hplc-uv色谱分离制备,210nm检测,流速为3ml/min,流动相为乙腈:水=18:82,得到螺环萘类化合物2(tr=26min)(收率为0.00014
‰
)和化合物3(tr=34min)(收率为0.00023
‰
)。
[0105]
(5)上述步骤(4)中所得乙腈-水(4:6~8:2)流分经hplc-uv色谱分离制备,210nm检测,流速为3ml/min,流动相为乙腈:水=40:60,得到abietane二萜类化合物3(tr=10min)(收率为0.00038
‰
)。
[0106]
(6)上述步骤(4)中所得乙腈-水(4:6~8:2)流分经hplc-uv色谱分离制备,210nm检测,流速为3ml/min,流动相为乙腈:水=60:40,得到到abietane二萜类化合物2(tr=18min)(收率为0.00017
‰
)、化合物4(tr=24min)(收率为0.00070
‰
)、化合物5(tr=29min)(收率为0.00071
‰
)和化合物1(tr=41min)(收率为0.00011
‰
)。
[0107]
(7)上述步骤(4)中所得乙腈-水(4:6~8:2)流分经hplc-uv色谱分离制备,210nm检测,流速为3ml/min,流动相为乙腈:水=82:18,得到到ursane三萜类化合物6(tr=17min)(收率为0.0008
‰
)。
[0108]
abietane二萜类1,2,3,4,5和ursane三萜类化合物6的结构鉴定方法见实施例1。
[0109]
实施例5
[0110]
(1)麻疯树干燥茎枝2000g用80%甲醇加热回流提取3次(用量:20l),减压回收提
取液得粗提物;
[0111]
(2)上述步骤(1)所得甲醇提取物经有机溶剂萃取,依次用环己烷、氯仿、正丁醇以水相和有机相的体积比1:1进行萃取,分别萃取5次,得到不同极性部位的萃取物;
[0112]
(3)上述步骤(2)所得的氯仿萃取物,经硅胶柱色谱分离,依次以石油醚和丙酮混合溶剂:100:5,10:1,8:1,5:1,3:1,1:1洗脱;
[0113]
(4)上述步骤(3)中所得石油醚:丙酮8:1~1:1流分经ods色谱,用甲醇-水3:7,5:5,7:3,8:2,9:1的混合溶剂梯度洗脱;
[0114]
(5)上述步骤(4)中所得甲醇-水(5:5~9:1)流分经hplc-uv色谱分离制备,210nm检测,流速为3ml/min,流动相为甲醇:水=52:48,得到abietane二萜类化合物3(tr=10min)(收率为0.00046
‰
)。
[0115]
(6)上述步骤(4)中所得甲醇-水(5:5~9:1)流分经hplc-uv色谱分离制备,210nm检测,流速为3ml/min,流动相为甲醇:水=72:28,得到到abietane二萜类化合物2(tr=15min)(收率为0.00021
‰
)、化合物4(tr=22min)(收率为0.00071
‰
)化合物5(tr=25min)(收率为0.00077
‰
)和化合物1(tr=47min)(收率为0.00017
‰
)。
[0116]
(7)上述步骤(4)中所得甲醇-水(5:5~9:1)流分经hplc-uv色谱分离制备,210nm检测,流速为3ml/min,流动相为甲醇:水=92:8,得到到ursane三萜类化合物6(tr=24min)(收率为0.0009
‰
)。
[0117]
abietane二萜类1,2,3,4,5和ursane三萜类化合物6的结构鉴定方法见实施例1。
[0118]
实施例6
[0119]
(1)麻疯树干燥茎枝2500g用90%甲醇加热回流提取3次(用量:25l),减压回收提取液得粗提物;
[0120]
(2)上述步骤(1)所得甲醇提取物经有机溶剂萃取,依次用石油醚、乙酸乙酯、正丁醇以水相和有机相的体积比1:2进行萃取,分别萃取4次,得到不同极性部位的萃取物;
[0121]
(3)上述步骤(2)所得的乙酸乙酯萃取物,经硅胶柱色谱分离,依次以氯仿和甲醇混合溶剂100:1,100:3,100:5,10:1,8:1,5:1,3:1洗脱;
[0122]
(4)上述步骤(3)中所得的氯仿:甲醇100:3~8:1流分经ods色谱,用乙腈-水2:8,4:6,6:4,8:2,9:1的混合溶剂梯度洗脱;
[0123]
(5)上述步骤(4)中所得乙腈-水(4:6~8:2)流分经hplc-rid色谱分离制备,流速为3ml/min,流动相为乙腈:水=25:75,得到螺环萘类化合物2(tr=31min)(收率为0.00013
‰
)和化合物3(tr=42min)(收率为0.00021
‰
)
[0124]
(5)上述步骤(4)中所得乙腈-水(4:6~8:2)流分经hplc-uv色谱分离制备,210nm检测,流速为3ml/min,流动相为乙腈:水=42:58,得到abietane二萜类化合物3(tr=9min)(收率为0.00042
‰
)。
[0125]
(6)上述步骤(4)中所得乙腈-水(4:6~8:2)流分经hplc-uv色谱分离制备,210nm检测,流速为3ml/min,流动相为乙腈:水=62:38,得到到abietane二萜类化合物2(tr=17min)(收率为0.00019
‰
)、化合物4(tr=22min)(收率为0.00072
‰
)、化合物5(tr=26min)(收率为0.00079
‰
)和化合物1(tr=40min)(收率为0.00016
‰
)。
[0126]
(7)上述步骤(4)中所得乙腈-水(4:6~8:2)流分经hplc-uv色谱分离制备,210nm检测,流速为3ml/min,流动相为乙腈:水=79:21,得到到ursane三萜类化合物6(tr=
31min)(收率为0.0009
‰
)。
[0127]
abietane二萜类1,2,3,4,5和ursane三萜类化合物6的结构鉴定方法见实例1。
[0128]
实施例7
[0129]
实施例1-6中制备得到的abietane二萜类1,2,3,4,5和ursane三萜类化合物6的抗神经炎症活性测试。
[0130]
(1)实验原理:小胶质细胞过度活化介导的慢性炎症反应是神经退行性疾病的发生、发展过程中的重要环节,因此,抑制过度活化的小胶质细胞可能成为发现药物的一个新的靶点。lps可激活小胶质细胞释放no、tnf-α,il-6,il-1β等。本实验通过建立体外lps诱导的异常活化的bv-2小胶质细胞筛选模型,以no释放量为指标,评价新abietane二萜类1,2,3,4,5和ursane三萜类化合物6的抗神经炎症活性。
[0131]
(2)实验方法:
[0132]
①
小鼠小胶质细胞系bv-2的培养
[0133]
细胞培养和模型建立中使用的所有玻璃器皿及金属器械(培养瓶,移液管,溶液瓶等),均经过121℃高压灭菌30min,以彻底去除污染的lps。以dmem培养基作为基础配制成内含10%胎牛血清的细胞培养液。小胶质细胞以约2.0
×
105cells/ml的浓度在5%co2,37℃培养瓶中传代培养,至第三天贴壁细胞约占培养瓶底面积70-80%,以胰酶消化贴壁细胞,传代至另一培养瓶。以-80℃超低温冰箱冻存复苏后的bv-2作为第一代,选择第3-8代bv-2细胞进行实验。
[0134]
②
药物配制方法
[0135]
待测化合物均用dmso溶解。配成母液(100mm),储存于-20℃。临用时用dmem培养液将其进行稀释,依次稀释为100μm、30μm、10μm、1μm。dmso终浓度《1
‰
。
[0136]
③
griess法检测化合物对lps激活小胶质细胞的抑制作用
[0137]
取对数生长期的bv-2小胶质细胞,用含10%胎牛血清的新鲜dmem培养液将细胞密度调至2.0
×
105cells/ml,接种于96孔板内,100μl/well,于37℃,5%co2的培养箱内培养。细胞贴壁培养24h后换成无血清的新鲜培养液,同时进行加药处理。化合物测试浓度为100μm、30μm、10μm、1μm与lps共同作用。同时设空白对照。各给药组中lps终浓度为100ng/ml。细胞加药后继续培养24h后,收集上清液,griess比色法检测上清液中no
2-含量。
[0138]
④
mtt法检测化合物对小胶质细胞细胞成活率的影响
[0139]
取对数生长期培养的bv-2小胶质细胞,用含10%胎牛血清的新鲜dmem培养基将细胞密度调至2.0
×
105cells/ml,接种于96孔板内,100μl/well,于37℃,5%co2的培养箱内培养。细胞贴壁培养24h后换成新鲜培养液,同时进行加药处理。化合物剂量100μm、30μm、10μm、1μm与lps共同作用。同时设空白对照。各给药组中lps终浓度为100ng/ml。细胞加药后继续培养24h,然后向细胞液中加入mtt溶液,10μl/well,将细胞与0.25mg/ml mtt于37℃下共同孵育3h,吸除培养液,然后加入150μl的dmso溶液,测定其光密度od值。数据处理,利用酶标仪软件进行数据处理,计算每一种样品3个孔od值的平均值,利用平均值按如下公式计算细胞成活率(cell viability,cv%)。
[0140]
细胞成活率%=[样品组od值的平均值/空白对照组od值的平均值]
×
100%
[0141]
⑤
统计方法
[0142]
全部资料采用spss(19.0)统计软件包进行检验分析。结果用平均值
±
标准误表
示,评价整体性差异,组间均数采用one-way anova分析法进行方差齐性分析,并结合dunnett’s test分析方法进行组间比较。多样本方差齐性检验采用levene检验,当p》0.05,方差是齐的,采用dunnett’s双侧t检验多组间均数的差异,当p《0.05,方差不齐,采用dunnett t3检验多组间均数的差异。
[0143]
⑥
ic
50
的计算方法
[0144]
将各剂量和抑制率等参数用非线性回归拟合计算ic
50
。
[0145]
(3)实验结果:
[0146]
实验结果见表4。
[0147]
表4 化合物1-6对lps激活bv-2小胶质细胞释放no的影响实验结果
[0148][0149]
注:*p《0.05,**p《0.01,***p《0.001与lps诱导组相比;
###
p《0.001与对照组相比。
[0150]
结果可知,实施条例1-6中制备得到的新abietane二萜类1(30μm,100μm)、2(30μm,100μm)、3(30μm,100μm)、4(10μm,30μm,100μm)、5(30μm,100μm)和ursane三萜类化合物6(1μm,10μm,30μm,100μm)能够显著抑制lps诱导的过度活化的bv-2小胶质细胞no的释放。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1