一种天然蟾毒基内酯类化合物的制备方法
1.本发明属于药物合成技术领域,涉及制备名贵中药材蟾酥中的活性成分包括但不限于14,15β-环氧基-3β-羟基-5β,14β-蟾毒基-20,22-二烯内酯(又名脂蟾毒配基,resibufogenin)等蟾毒基内酯类化合物的方法。
背景技术:
2.蟾酥是蟾蜍科动物中华大蟾蜍bufo bufo gargarizans cantor或黑眶蟾蜍bufo melanostictus schneider的干燥分泌物,是我国传统名贵中药材,属国家二级保护野生药材物种。许多中医典籍记载蟾酥味甘、辛,性温,有毒;传统医学认为其具有解毒止痛、开窍醒神的功效,主治痈疽疔疮、瘰疬、咽喉肿痛、牙痛、痧胀腹痛、神昏吐泄。现代药理研究表明,蟾酥具有增强心肌收缩力、抗心肌缺血、升压、降压、抗肿瘤、抑菌抗炎、麻醉止痛,致幻等多种药理作用。《中国药典》2020版收录的多个成方制剂如牙痛一粒丸,血栓心脉宁片,梅花点舌丸和麝香保心丸等,均含有蟾酥成分。
3.在蟾酥已经探明的化学成分中,蟾蜍内酯类化合物具有强心、抗肿瘤、抗炎、局部麻醉等生理作用,与蟾酥的生物活性表现基本一致,迄今为止已有140多个结构多样的蟾蜍内酯类化合物从蟾蜍中分离鉴定出来。《中国药典》2020版以蟾毒灵(bufalin),华蟾酥毒基(cinobufagin)和脂蟾毒配基(resibufogenin)这三个蟾蜍内酯类化合物的总含量作为蟾酥的质量标准。因此,对蟾蜍内酯类化合物的相关研究是蟾酥研究的重点。然而,由于蟾酥制作工艺复杂,不同来源地的蟾酥有效成分含量也不同,使其生产过程中伴有大量的残次品,生物利用较低,而日益稀缺的野生动物资源也在很大程度上制约着蟾酥的药理研究和临床应用。蟾酥总体组成成分复杂,既有强心苷,又有致幻剂;一些组分的生理作用相互拮抗,另一些组分虽有较好的生理活性,但同时毒性较大。要解决上述的问题,对蟾酥中具有较强生理活性的化学组分进行人工合成就显得尤为重要。
4.迄今为止,脂蟾毒配基(resibufogenin)的合成仅有少量报道(nat.prod.rep.,2017,34,361
–
410;european journal of medicinal chemistry,2020,189,112038.),其合成策略主要分为两类:一是从蟾蜍内酯类化合物及其结构相近的天然产物出发进行天然产物之间的转化,如pettit等人从蟾毒灵(bufalin)出发实现对脂蟾毒配基(resibufogenin)的全合成(j.org.chem.1971,36,3736
–
3739);二是从简单的甾体化合物出发,经过多步化学转化得到蟾蜍内酯类化合物,例如inoue等人对蟾毒灵bufalin,bufogenin b以及蟾毒它灵(bufotalin)等的全合成(org.lett.2020,22,8652
–
8657)。无论是哪种合成策略都无法实现蟾蜍内酯类化合物的大量制备,都不具有实用性和经济性。
技术实现要素:
5.本发明的目的在于提供一种能够大规模合成天然蟾毒基内酯类化合物的制备方法。
6.本发明的第一方面,提供一种式i所示的蟾毒基内酯类化合物的制备方法,所述制
备方法包括以下步骤:式ii所示的蟾蜍内酯类化合物通过化学半合成完成六元二烯内酯环的上载,再经过氧化态修饰获得式i所示的蟾毒基内酯类化合物,
[0007][0008]
式中,r1、r2和r4各自独立地为-h或-oh;
[0009]
r3为-ch3或-cho;
[0010]
r5和r6各自独立地为-h、-oh或酮基;
[0011]
r7为-h、oh或氧乙酰基;
[0012]
ra为h、羟基或叔丁基二甲基硅氧基。
[0013]
在另一优选例中,ra为羟基或叔丁基二甲基硅氧基。
[0014]
在另一优选例中,r1、r2、r4、r5、r6、r7都为h,r3为甲基。
[0015]
在另一优选例中,r1、r2、r4、r5、r6、r7都为h,r3为甲基,ra为叔丁基二甲基硅氧基时,所述制备方法还包括式ii所示的蟾蜍内酯类化合物,即式6化合物的合成步骤,包括:步骤4或步骤a~4,
[0016][0017]
式中tbs为叔丁基二甲基硅基,
[0018]
其中:步骤a:化合物2经甾体3β还原酶(3βhsd)、甾体5β还原酶和甾体c14α羟化酶生物催化得到化合物5;
[0019]
步骤4:化合物5在有机溶剂中与叔丁基二甲基氯硅烷(tbscl)或叔丁基二甲硅基三氟磺酸酯(tbsotf)和有机碱反应得到化合物6;所述有机溶剂为二氯甲烷或n,n-二甲基甲酰胺;所述有机碱为吡啶或咪唑。
[0020]
本发明中,对于甾体3β还原酶(3βhsd)、甾体5β还原酶和甾体c14α羟化酶生物催化,酶的使用顺序没有特别的要求,并非按照上述顺序进行生物催化,酶的使用顺序可变换,此外,可以分别单独使用,也可以混合使用,也就是说,所述的生物催化,包括但不仅限于纯酶分步催化、一锅法酶催化、粗酶液催化、多细胞共转化和整细胞转化。
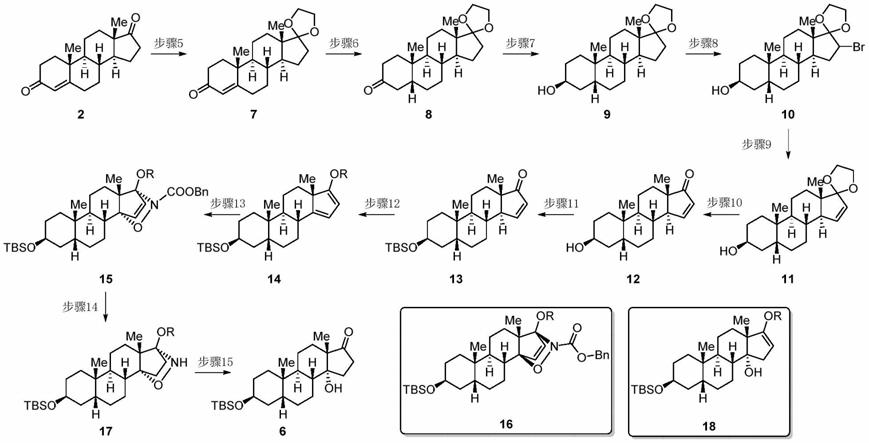
[0021]
在另一优选例中,步骤a包括:步骤3~4或步骤2~3~4或步骤1~2~3~4,
[0022][0023]
式中tbs为叔丁基二甲基硅基,
[0024]
其中:
[0025]
步骤1:化合物2经甾体3β还原酶(3βhsd)生物催化得到化合物3,不需要分离纯化;
[0026]
步骤2:化合物3经甾体5β还原酶生物催化还原得到的化合物经催化加氢得到化合物4,不需要分离纯化;
[0027]
步骤3:化合物4经甾体c14α羟化酶生物催化氧化得到化合物5;
[0028]
步骤4:化合物5在有机溶剂中与叔丁基二甲基氯硅烷(tbscl)或叔丁基二甲硅基三氟磺酸酯(tbsotf)和有机碱反应得到化合物6;所述有机溶剂为二氯甲烷或n,n-二甲基甲酰胺;所述有机碱为吡啶或咪唑。
[0029]
在另一优选例中,所述步骤1、步骤2和步骤3的反应顺序可变换,或者同时进行,例如,在完成步骤1后,可以使用甾体5β还原酶和甾体c14α羟化酶的混合酶对化合物3进行生物催化得到化合物5。
[0030]
在另一优选例中,先用甾体3β还原酶(3βhsd)的大肠杆菌工程菌粗酶液催化4-ad转化成3β-羟基-4-雄烯-17-酮,再用甾体5β还原酶和c14α羟化酶的酵母工程菌细胞转化3β-羟基-4-雄烯-17-酮生成3β,14α-二羟基-5β-雄甾烷-17-酮。
[0031]
本发明中,所述大肠杆菌工程菌粗酶液为大肠杆菌工程菌经超声或高压均质仪破碎,离心取上清部分。
[0032]
在另一优选例中,所述的甾体3β还原酶选自:来源于digitalis lanata的甾体3β还原酶dlhsd1和来源于erysimum crepidifolium的甾体3β还原酶echsd2,其氨基酸序列分别如seq id no:2、seq id no:4所示。
[0033]
在另一优选例中,所述的甾体5β还原酶选自:来源于arabidopsis thaliana的甾体5β还原酶atst5βr1、atst5βr2和来源于amborella trichopoda的甾体5β还原酶amtst5βr2,其氨基酸序列如seq id no:6、seq id no:8和seq id no:10所示。
[0034]
在另一优选例中,所述的c14α羟化酶为复合酶,包括以下两种酶:(a)来源于cochliobolus lunatus的细胞色素p450酶,其氨基酸序列如seq id no:12所示;和(b)来源于cochliobolus lunatus的细胞色素p450还原酶cpr,其氨基酸序列如seq id no:14所示。
[0035]
在另一优选例中,r1、r2、r4、r5、r6、r7都为h,r3为甲基,ra为叔丁基二甲基硅氧基时,所述制备方法还包括式ii所示的蟾蜍内酯类化合物的合成步骤,包括:
[0036]
步骤15或步骤14~15或步骤13~14~15或步骤12~13~14~15或步骤11~12~13~14~15或步骤10~11~12~13~14~15或步骤9~10~11~12~13~14~15或步骤8~9~10~11~12~13~14~15或步骤7~8~9~10~11~12~13~14~15或步骤6~7~8~9~10~11~12~13~14~15或步骤5~6~7~8~9~10~11~12~13~14~15,
[0037][0038]
式中tbs为叔丁基二甲基硅基,r为乙酰基,丙酰基或丁酰基,bn为苄基,
[0039]
其中:
[0040]
步骤5:化合物2在酸性条件下与乙二醇反应得到化合物7;酸性条件使用的酸为对甲基苯磺酸、苯磺酸、樟脑磺酸、柠檬酸、丙二酸、对硝基苯酚、质量分数50%~98%硫酸或质量分数30%~36.5%盐酸中的一种或两种以上的混合酸;
[0041]
步骤6:在氢化催化剂作用下,化合物7在有机溶剂中经催化加氢得到化合物8;所述的氢化催化剂为二氧化铂、钯碳、氢氧化钯、兰尼镍或铑/三氧化铝;所述的有机溶剂为吡啶、4-甲基吡啶、四氢呋喃、甲醇或乙醇中的一种或两种以上的混合溶剂;
[0042]
步骤7:化合物8在有机溶剂中通过还原剂还原得到化合物9;所述还原剂为三仲丁基硼氢化锂、三仲丁基硼氢化钾或二异丁基氢化铝;所述有机溶剂为四氢呋喃,乙醚,1,4-二氧六环,乙二醇二甲醚、苯或甲苯中的一种或两种以上的混合溶剂;
[0043]
步骤8:化合物9在有机溶剂中与溴代试剂反应得到化合物10;所述溴代试剂为苯基三甲基三溴化铵,吡啶鎓三溴化物或液溴;所述有机溶剂为四氢呋喃,乙醚,1,4-二氧六环,乙二醇二甲醚、二氯甲烷、1,2-二氯乙烷或四氯化碳中的一种或两种以上的混合溶剂;
[0044]
步骤9:化合物10与碱在有机溶剂中加热反应得到化合物11;所述有机溶剂为四氢呋喃,乙醚、1,4-二氧六环或二甲亚砜;所述的碱为叔丁醇钾或叔丁醇钠;所述反应温度为20~80℃;
[0045]
步骤10:化合物11与酸在溶剂中反应得到化合物12;所述的酸为对甲基苯磺酸、吡啶对甲苯磺酸盐(ppts)、苯磺酸、樟脑磺酸、柠檬酸、丙二酸、对硝基苯酚、高氯酸、高碘酸、质量分数2%~10%稀硫酸或质量分数2~5%稀盐酸;所述溶剂为丙酮、水、四氢呋喃、1,4-二氧六环的一种或两种以上混合溶剂;
[0046]
步骤11:化合物12与叔丁基二甲基氯硅烷(tbscl)或叔丁基二甲硅基三氟磺酸酯(tbsotf)和咪唑或吡啶反应得到化合物13;反应溶剂为二氯甲烷或n,n-二甲基甲酰胺;
[0047]
步骤12:化合物13在酸作用下与酸酐反应得到化合物14;所述的酸为对甲基苯磺酸或硫酸;所述酸酐为乙酸酐、丙酸酐或丁酸酐;
[0048]
步骤13:化合物14与n-(苄氧羰基)羟胺和氧化剂反应得到化合物15和16的混合物;将该混合物溶于溶剂,加热反应,得到化合物15;所述氧化剂为四正丁基高氯酸铵或四
正丁基高碘酸铵;所述的溶剂为苯、甲苯、二氯甲烷、四氢呋喃、n,n-二甲基甲酰胺中的一种或两种以上的混合溶剂;所述反应温度为20~80℃;
[0049]
步骤14:在氢化催化剂作用下,化合物15在有机溶剂中经催化加氢得到化合物17;所述氢化催化剂为钯碳、氢氧化钯、兰尼镍或碳酸钙钯;所述有机溶剂为甲醇,乙醇,四氢呋喃,1,4-二氧六环或水中的一种或两种以上的混合溶剂;
[0050]
步骤15:化合物17与二水合氯化铜反应得到化合物6和18;将化合物18或化合物6和18的混合物溶于溶剂,加入碱反应得到单一化合物6;所述的溶剂为甲醇、乙醇、水、四氢呋喃或1,4-二氧六环中的一种或两种以上的混合溶剂;所述的碱为碳酸钠、碳酸钾、氢氧化钾、氢氧化钠、氢化钠、三乙胺中的一种或两种以上的混合物。
[0051]
在另一优选例中,r1、r2、r4、r5、r6、r7都为h,r3为甲基,ra为叔丁基二甲基硅氧基时,所述制备方法包括如下步骤:
[0052]
步骤21或步骤20~21或步骤19~20~21或步骤18~19~20~21或步骤17~18~19~20~21或步骤16~17~18~19~20~21;
[0053][0054]
式中tbs为叔丁基二甲基硅基,
[0055]
其中,
[0056]
步骤16:化合物6的有机溶液中加入水合肼和碱反应,粗品中再加入单质碘和碱反应得到化合物19;所述的有机溶剂为四氢呋喃、乙醇或甲醇的一种或两种以上的混合物;所述的碱为二异丙基乙基胺、三乙胺或4-二甲基氨基吡啶;
[0057]
步骤17:化合物19和化合物20中加入过渡金属催化剂和碱反应得到化合物21;所述的过渡金属催化剂为[1,1'-双(二苯基膦基)二茂铁]二氯化钯、四(三苯基膦)钯、醋酸钯或二氯双(三苯基膦)钯;所述碱为碳酸钾、磷酸钾、磷酸氢钾、叔丁醇钾或氢氧化钾等;反应溶剂为四氢呋喃、甲醇、水、n,n-二甲基甲酰胺的一种或两种以上的混合溶剂;
[0058]
步骤18:化合物21的有机溶液中加入氢化均相催化剂,经催化加氢反应得到化合物22;所述氢化均相催化剂为三苯基膦氯化铑、六氟磷酸(三环已基膦)(1,5-环辛二烯)(吡啶)合铱或(1,5-环辛二烯)氯铑(i)二聚体;采用的有机溶剂为二氯甲烷,苯或甲苯;
[0059]
步骤19:化合物22与脱水剂反应得到化合物23;所述的脱水剂为n-(三乙胺基硫酰)氨基甲酸甲酯、双[a,a-双(三氟甲基)苯甲醇]-二苯基硫、二氯亚砜或甲磺酰氯和有机碱的组合;所述有机碱为三乙胺、二异丙基乙基胺、吡啶或2,6-二甲基吡啶;
[0060]
步骤20:化合物23进行脱硅醚保护反应得到化合物24;所述脱硅醚保护试剂为四丁基氟化铵的四氢呋喃溶液、氢氟酸吡啶溶液、氢氟酸水溶液或氢氟酸乙腈溶液;
[0061]
步骤21:化合物24的有机溶液中加入卤化试剂和高氯酸水溶液反应,粗品不经纯化直接溶于吡啶中,在50~70℃之间加热反应0.5~2.0小时得到目标化合物1;采用的有机溶剂为1,4-二氧六环、四氢呋喃、乙醚、丙酮或水的一种或两种以上的混合溶剂;所述卤化试剂为n-溴代琥珀酰亚胺(nbs),n-碘代琥珀酰亚胺(nis)或n-氯代琥珀酰亚胺(ncs);所述高氯酸水溶液的质量分数为1%~70%。
[0062]
在另一优选例中,步骤5中的酸为对甲基苯磺酸、苯磺酸、樟脑磺酸或质量分数90%-98%浓硫酸。
[0063]
在另一优选例中,步骤6中的氢化催化剂为氢氧化钯、质量分数5%的钯碳或10%的钯碳,所述有机溶剂为吡啶或4-甲基吡啶。
[0064]
在另一优选例中,步骤7中的还原剂为三仲丁基硼氢化钾或二异丁基氢化铝,反应温度为-78~0℃,有机溶剂为四氢呋喃或甲苯。
[0065]
在另一优选例中,步骤8中的溴代试剂为吡啶鎓三溴化物或液溴,有机溶剂为四氢呋喃或四氯化碳。
[0066]
在另一优选例中,步骤9中有机溶剂为二甲亚砜或1,4-二氧六环。
[0067]
在另一优选例中,步骤10中的酸为对甲基苯磺酸、吡啶对甲苯磺酸盐(ppts)、苯磺酸、樟脑磺酸、质量分数2~15%的硫酸或质量分数2~10%的盐酸;所述溶剂为丙酮与水的混合溶剂,其体积比例为2:1~6:1。
[0068]
在另一优选例中,步骤12中酸为对甲苯磺酸或硫酸,酸酐为乙酸酐。
[0069]
在另一优选例中,步骤13中所述氧化剂为四正丁基高碘酸铵;所述的溶剂为甲苯或二氯甲烷。
[0070]
在另一优选例中,步骤14中所述氢化催化剂为碳酸钙钯或质量分数5%钯碳;所述的有机溶剂为乙醇、四氢呋喃、甲醇或水的一种或两种以上的混合溶剂。
[0071]
在另一优选例中,步骤15中所述的溶剂为水、四氢呋喃、乙醇或1,4-二氧六环的一种或两种以上的混合溶剂;所述的碱为碳酸钾或氢氧化钠。
[0072]
在另一优选例中,步骤16中所述的有机溶剂为乙醇或四氢呋喃的一种或两种的混合溶剂;所述的碱为二异丙基乙基胺或三乙胺。
[0073]
在另一优选例中,步骤17中所述的过渡金属催化剂为[1,1'-双(二苯基膦基)二茂铁]二氯化钯或四三苯基膦钯;所述碱为磷酸钾或碳酸钾;所述溶剂为n,n-二甲基甲酰胺或四氢呋喃。
[0074]
在另一优选例中,步骤18中所述氢化均相催化剂为六氟磷酸(三环已基膦)(1,5-环辛二烯)(吡啶)合铱或三苯基膦氯化铑,所述有机溶剂为二氯甲烷或甲苯。
[0075]
在另一优选例中,步骤19中所述的脱水剂为二氯亚砜或甲基磺酰氯和吡啶的组合或n-(三乙胺基硫酰)氨基甲酸甲酯。
[0076]
在另一优选例中,步骤20中所述的脱硅醚保护试剂为氢氟酸吡啶溶液或四丁基氟化铵的四氢呋喃溶液。
[0077]
在另一优选例中,步骤21中所述的有机溶剂为1,4-二氧六环与水或丙酮与水的混合溶剂,其体积比例为12:1~5:1;所述卤化试剂为n-溴代琥珀酰亚胺(nbs)或n-碘代琥珀
酰亚胺(nis)。
[0078]
本发明的第二方面,提供一种蟾毒基内酯类化合物中间体,具有下式:
[0079][0080]
式中,r1、r2和r4各自独立地为-h或-oh;
[0081]
r3为-ch3或-cho;
[0082]
r5和r6各自独立地为-oh或酮基;
[0083]
r7为oh或氧乙酰基。
[0084]
本发明提供的制备方法操作简单,收率较高,可用于大规模制备该类天然产物,具有广泛的应用前景。
[0085]
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。说明书中所揭示的各个特征,可以被任何提供相同、均等或相似目的的替代性特征取代。限于篇幅,在此不再一一累述。
附图说明
[0086]
图1中a、b、c、d分别示出了雄烯二酮(4-ad)转化为5β-雄甾烷-3,17-二酮、3β-羟基-4-雄烯-17-酮、14α-羟基-4-雄烯-3,17-二酮、3β,14α-二羟基-5β-雄甾烷-17-酮的反应方程式。
[0087]
图2示出了上述酶蛋白的sds-page电泳图。
[0088]
图3示出了甾体3β还原酶dlhsd1的大肠杆菌工程菌粗酶液催化底物雄烯二酮(4-ad)转化成3β-羟基-4-雄烯-17-酮的hplc谱图。
[0089]
图4示出了甾体5β还原酶atst5βr2的大肠杆菌工程菌细胞催化底物雄烯二酮(4-ad)转化成5β-雄甾烷-3,17-二酮的gc谱图。
[0090]
图5示出了甾体c14α羟化酶催化4-ad转化生成14α-羟基-4-雄烯-3,17-二酮0h、72h的lc-ms,提取目标分子量图谱。
具体实施方式
[0091]
本技术的发明人经过广泛而深入的研究,提供一种14,15β-环氧基-3β-羟基-5β,14β-蟾毒基-20,22-二烯内酯的制备方法,以价廉易得的甾体化合物雄烯二酮(4-ad)或3β-羟基-5β-雄甾烷-17-酮为原料,通过生物-化学接力合成或化学合成天然蟾蜍内酯类化合物,用以促进该类化合物在医药领域的广泛应用,可以缓解蟾酥中活性物质药理研究的物料稀缺压力。
[0092]
制备方法
[0093]
本发明提供了一种甾体母核3β羟基、ab环十氢萘环顺式构型、c14α构型羟基的甾体化合物,所述甾体化合物具有以下结构式所示的结构,用于作为化学半合成蟾蜍内酯类
化合物的中间体。
[0094][0095]
式中,r1、r2和r4为-h或-oh;r3为-ch3或-cho;r5和r6为-oh或酮基;r7为oh或氧乙酰基。
[0096]
在另一优选例中,所述甾体化合物r1、r2、r4-r7基团都为h,r3为甲基,即3β,14α-二羟基-5β-雄甾烷-17-酮。
[0097]
本发明提供了一种生物催化获得蟾蜍内酯化合物化学合成中间体3β,14α-二羟基-5β-雄甾烷-17-酮的方法,以雄烯二酮(4-ad)或3β-羟基-5β-雄甾烷-17-酮为原料,采用甾体3β还原酶(3βhsd)、5β还原酶和c14α羟化酶分别生物催化还原3位酮基成β构型羟基、还原甾体母核c4-c5位双键形成ab环十氢萘环顺式构型骨架,以及c14α构型羟基化,如图1所示。
[0098]
本发明中,所述甾体3β还原、5β还原和c14α羟化的反应顺序可相互变换或同时进行。
[0099]
所述生物催化方法,包括但不仅限于纯酶分步催化、一锅法酶催化、粗酶液催化、多细胞共转化和整细胞转化。
[0100]
本发明提供了两种将甾体母核c3位酮基还原成β构型羟基的酶dlhsd1、echsd2,所述酶的氨基酸序列分别如seq id no:2、seq id no:4所示。
[0101]
本发明提供了三种将甾体母核c4-c5位双键还原形成ab环十氢萘环顺式构型骨架的甾体5β还原酶atst5βr1、atst5βr2、amtst5βr2,所述酶的氨基酸序列分别如seq id no:6、seq id no:8、seq id no:10所示。
[0102]
本发明提供了一种将甾体母核c14位羟基化成α构型羟基的细胞色素p450酶及相关p450还原酶(cytochrome p450 reductase,cpr),所述酶的氨基酸序列分别如seq id no:12、seq id no:14所示。
[0103]
本发明对甾体3β还原酶、5β还原酶的dna序列进行大肠杆菌和毕赤酵母密码子偏爱序列优化,适合大肠杆菌和毕赤酵母高效表达,dlhsd1、echsd2、atst5βr1、atst5βr2、amtst5βr2密码子优化后的dna序列分别如seq id no:1、seq id no:3、seq id no:5、seq id no:7、seq id no:9所示。
[0104]
本发明对甾体母核c14位α羟基化酶及相关的氧化还原酶(cpr)的dna序列进行毕赤酵母密码子偏爱序列优化,适合毕赤酵母高效表达,密码子优化后的dna序列分别如seq id no:11、seq id no:13所示。
[0105]
本发明提供了一种生物催化高效制备合成中间体3β,14α-二羟基-5β-雄甾烷-17-酮的方法。先用甾体3β还原酶(3βhsd)的大肠杆菌工程菌粗酶液催化4-ad转化成3β-羟基-4-雄烯-17-酮,再用甾体5β还原酶和c14α羟化酶的酵母工程菌细胞转化3β-羟基-4-雄烯-17-酮生成3β,14α-二羟基-5β-雄甾烷-17-酮。
[0106]
本发明中,获得的3β,14α-二羟基-5β-雄甾烷-17-酮经羟基保护后,可以通过以下步骤化学合成式1所示的蟾毒基内酯类化合物:
[0107][0108]
本发明中,还提供通过化学合成获得式1所示的蟾毒基内酯类化合物的制备方法,包括以下步骤:
[0109][0110][0111]
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条
件,例如sambrook等人,分子克隆:实验室手册(new york:cold spring harbor laboratory press,1989)中所述的条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数是重量百分比和重量份数。
[0112]
序列:
[0113][0114]
实施例1
[0115]
纯酶atst5βr1、atst5βr2、dlhsd1、echsd2的制备
[0116]
合成密码子优化后的甾体5β还原酶基因atst5βr1(seq id no:5)和atst5βr2(seq id no:7)和amtst5βr2(seq id no:9),甾体3β还原酶基因dlhsd1(seq id no:1)和echsd2(seq id no:3),分别插入大肠杆菌蛋白表达常规载体pcdfduet-1和pet28a(+)载体(购自novagen)的多克隆位点,获得大肠杆菌表达重组质粒pcdfduet-1-atst5βr1、pcdfduet-1-atst5βr2、pcdfduet-1-amtst5βr2、pet28a(+)-dlhsd1、pet28a(+)-echsd2。按常规方法转化大肠杆菌bl21(de3),获得相应蛋白的表达菌株。
[0117]
含pet28a(+)-dlhsd1、pet28a(+)-echsd2大肠杆菌加入0.1mm iptg,18℃,220rpm,诱导表达18~20h;含pcdfduet-1-atst5βr1、pcdfduet-1-atst5βr2的大肠杆菌培养物加入0.5mm iptg,30℃,220rpm,诱导表达16~18h。
[0118]
常规蛋白ni株纯化方法纯化蛋白,sds-page凝胶电泳检测催化到的蛋白,结果表明,表达的蛋白atst5βr1、atst5βr2、dlhsd1、echsd2的分子量与预测值相符。上述纯化好的蛋白sds-page凝胶电泳图如图2所示。
[0119]
实施例2甾体3β还原酶(3βhsd)、5β还原酶纯酶活性检测。
[0120]
在50mm hepes钠-hcl缓冲液中(含150mm nacl、1mm dtt、10%甘油,ph=7.0),加入底物4-ad、葡萄糖、nadp+、葡萄糖脱氢酶,再分别加入纯酶atst5βr2、atst5βr2、dlhsd1和echsd2,30℃,反应3h。其中,atst5βr2和atst5βr2能催化甾体母核c4-c5位双键还原,dlhsd1和echsd2能催化3位酮基还原,atst5βr2和dlhsd1催化甾体母核c4-c5位双键还原和3位酮基还原的活性更优。
[0121]
实施例3甾体5β还原酶atst5βr2大肠杆菌工程菌细胞转化底物4-ad。
[0122]
10ml含抗生素的lb培养基中,接种重组大肠杆菌单菌落,37℃,220rpm,过夜培养。1l含有抗生素的lb培养基,按1%接种重组的大肠杆菌过夜培养物,37℃、220rpm培养2~3h。测od
600
值,到达0.6~0.8时,加入iptg,加入底物4-ad,220rpm,30℃,转化60h,取200μl菌液,用等体积乙酸乙酯萃取两次,氮吹,200μl乙酸乙酯复溶,shimadzu2020gcms检测,表达甾体5β还原酶atst5βr2的工程菌可以细胞催化底物4-ad转化成5β-雄甾烷-3,17-二酮。
[0123]
实施例4甾体5β还原酶atst5βr2的大肠杆菌工程菌细胞转化底物4-ad条件的优化。
[0124]
具体条件包括c源、助溶剂和转速,其中,最优c源为糊精(浓度为1mg/ml)、最优助溶剂为吐温80(浓度为0.2%)、最优转速220rpm。在该优化的条件下,每1l的甾体5β还原酶atst5βr2的大肠杆菌工程菌细胞培养物中加入100mg 4-ad,30℃,220rpm,60h,转化生产5β-雄甾烷-3,17-二酮,转化率为88%,反应结果如图3所示,分离纯化得到化合物5β-雄甾烷-3,17-二酮。
[0125]
实施例5甾体3β还原酶dlhsd1的粗酶液催化底物4-ad转化为3β-羟基-4-雄烯-17-酮。
[0126]
大量表达甾体3β还原酶dlhsd1大肠杆菌工程菌5l(0.1mm iptg诱导,18℃,诱导20h),收集得菌体约25g,用250ml 50mm hepes钠-hcl buffer(ph=6.5)重悬菌体,均质机高压破菌,4℃,12000rpm离心30min,收集破碎液。150ml 50mm hepes钠-hcl buffer(ph=6.5)中加入1.35g葡萄糖(终浓度为7.5mm)、78.4mg nadp+钠盐(终浓度为0.1mm),再缓慢加入500mg 4-ad(用10%dmso或乙腈溶解终浓度为1.75mm,),混匀后缓慢加入250ml上述破碎液上清,混匀,最后加入60μl浓度为150mg/ml的葡萄糖脱氢酶gdh,利用葡萄糖脱氢酶的体外循环体系再生辅助因子nadph,反应总体积为500ml。25℃,100rpm,反应24h,取200μl菌液,用等体积乙酸乙酯萃取两次,氮吹,200μl甲醇复溶,用shimadzu 8040lcms检测,反应结果如图4所示,分离纯化得到化合物3β-羟基-4-雄烯-17-酮。每1l表达甾体3β还原酶dlhsd1的大肠杆菌工程菌破碎液催化100mg4-ad转化成3β-羟基-4-雄烯-17-酮,转化率为81.8%。
[0127]
实施例6甾体3β还原酶、5β还原酶及c14位羟基化酶酵母工程菌构建
[0128]
对目的基因dlhsd1、echsd2、atst5βr1、atst5βr2、amtst5βr2于相应的大肠杆菌表达载体中进行pcr克隆,用ecori、xhoi酶切质粒插入到酵母蛋白表达常规载体ppicza(购自金斯瑞公司),构建酵母表达载体ppicza-dlhsd1、ppicza-echsd2、ppicza-atst5βr1、ppicza-atst5βr2、ppicza-amtst5βr2。
[0129]
构建酵母蛋白表达载体ppicza-duet:以ppicza为模板,以gaccttcgtttgtgcggcggccgcgatctaacatccaaagacgaaag、ttaacactagtcatatgggatccatggtgatggtgatgatgacccatgg为引物克隆出dna片段fragment-1;以atatgactagtgttaacctgcagtgagttttagccttagacatgac、gctatggtgtgtggggggtctcacttaatcttctgtac为引物克隆出dna片段fragment-2,用bamh i酶切ppicza,最后用诺唯赞公司的重组酶重组构建质粒ppicza-duet。
[0130]
合成密码子优化后的来源于cochliobolus lunatus的细胞色素p450(seq id no:11)和细胞色素p450还原酶(cytochrome p450 reductase,cpr)(seq id no:11)基因。先用bamhi和spei酶切酵母表达载体ppicza-duet,构建重组载体ppicza-duet-cpr,再用kpni和xhoi酶切重组载体ppicza-duet-cpr,构建酵母表达载体ppicza-duet-p450-cpr。
[0131]
将上述质粒ppicza-dlhsd1、ppicza-echsd2、ppicza-atst5βr1、ppicza-atst5β
r2、ppicza-amtst5βr2和ppicza-duet-p450-cpr分别按照常规电转方法导入毕赤酵母km71h,获得表达相应酶的工程菌。
[0132]
实施例7酵母工程菌阳性克隆的筛选及细胞转化实验。
[0133]
挑5~10个酵母工程菌单克隆于5ml ypd液体培养基中(含100μg/ml的zeocin),30℃,260rpm,培养24小时。将0.5ml培养物接入5ml mgyh甘油培养基,30℃,260rpm,培养24h。室温,2500g离心10min,在超净台中更换至5ml mmh甲醇培养基(含甲醇0.5%),加入底物4-ad,30℃,260rpm,每24小时加0.5%甲醇,总共摇了3天,取200μl菌液用等体积乙酸乙酯萃取两次,氮吹,用200μl甲醇复溶,用shimadzu 8040lcms检测,甾体3β还原酶的酵母工程菌不能细胞催化4-ad生产相应的产物3β羟基-4-雄烯-17-二酮;甾体5β还原酶的酵母工程菌能细胞催化4-ad生产相应的产物5β-雄甾烷-3,17-二酮;c14α羟化酶的酵母工程菌能细胞催化4-ad生产相应的产物14α-羟基-4雄烯-3,17-二酮(见图5)。
[0134]
实施例9化合物5的生物催化法制备
[0135]
分别采用如实施例2纯酶催化、实施例4大肠杆菌细胞转化、实施例5粗酶液催化、实施例6酵母细胞转化的方法对进行3β还原酶、5β还原酶和c14α羟化酶反应顺序的排列组合,以及两步或三步反应同时进行尝试,获得最优制备方法组合,包含2个步骤,步骤(1)粗酶液催化;步骤(2)酵母细胞转化。具体如下:
[0136]
(1)甾体3β还原酶dlhsd1粗酶液催化4-ad转化成3β-羟基-4-雄烯-17-酮。
[0137]
甾体3β还原酶dlhsd1在大肠杆菌中大量表达,粗酶液上清中,加入底物4-ad制备3β-羟基-4-雄烯-17-酮,具体方法详见实施例5。
[0138]
(2)5β还原酶和c14α羟化酶同步催化3β-羟基-4-雄烯-17-酮转化生成中间体3β,14α-二羟基-5β-雄甾烷-17-酮。
[0139]
分别挑ppicza-atst5βr2、ppicza-duet-p450-cpr阳性单克隆于10ml ypd液体培养基中(含100μg/ml的zeocin),30℃,260rpm,培养24h。将ppicza-atst5βr2、ppicza-duet-p450-cpr培养24h的菌液各10ml转接至1l mgyh甘油培养基,30℃,260rpm,培养24h。室温、2500g离心10min,在超净台中更换成1l mmh甲醇培养基(含甲醇0.5%),加入底物3β-羟基-4-雄烯-17-酮,终浓度为80mg/l,30℃,260rpm,培养72h,获得化合物5。
[0140]
甾体3β还原酶大肠杆菌细胞、酵母细胞不能直接催化4-ad转化生成3β羟基-4-雄烯-17-二酮,其大肠杆菌粗酶液很容易得到,催化反应简单,并且效率较高;然而,14α-羟化酶只能在酵母中直接细胞催化底物得到相应产物;甾体5β还原酶的蛋白表达量低,粗酶液催化反应效率较低,其大肠杆菌、酵母工程菌都能细胞催化4-ad生成相应的产物5β-雄甾烷-3,17-二酮,效率高于粗酶液催化。因此采用甾体3β还原酶dlhsd1粗酶液催化再进行5β还原酶和c14α羟化酶酵母工程菌同步催化,缩短了三步反应时间,这种组合是最优制备方法组合。
[0141]
实施例10化合物3β-叔丁基二甲基硅醚-14α-羟基-5β-雄甾烷-17-酮的化学合成方法
[0142]
步骤4:合成化合物6
[0143]
化合物5(200mg,0.653mmol)、叔丁基二甲基氯硅烷(128mg,0.849mmol)及咪唑(223mg,3.27mmol)溶于无水dmf(1ml),室温搅拌6小时。加入乙酸乙酯(10ml)稀释后,饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=15:1)得到
化合物13白色固体(0.246mg,收率90%)。1h nmr(600mhz,cdcl3):δ=4.07
–
3.99(m,1h),2.43
–
2.31(m,2h),1.99
–
1.71(m,8h),1.59
–
1.45(m,3h),1.45
–
1.32(m,5h),1.31
–
1.18(m,3h),0.98(s,3h),0.97(s,3h),0.88(s,9h),0.01(s,6h)ppm。
[0144]
步骤5:合成化合物7
[0145]
雄烯二酮化合物2(30.00g,104.7mmol)及分子筛(60g)悬浮于乙二醇(350ml),加入对甲苯磺酸一水合物(19.92g,104.7mmol)。室温搅拌3小时后,将反应液倒入饱和碳酸氢钠的冰水溶液(200ml)中,硅藻土抽滤,乙酸乙酯洗涤滤饼,滤液加入乙酸乙酯(3
×
150ml)萃取。合并有机相,饱和氯化钠水溶液洗涤,无水硫酸钠干燥。浓缩后得到粗品7直接投下一步反应。1h nmr(600mhz,cdcl3):δ=5.71(s,1h),3.92
–
3.88(m,2h),3.87
–
3.81(m,2h),2.46
–
2.24(m,4h),2.04
–
1.95(m,2h),1.88
–
1.83(m,1h),1.82
–
1.77(m,1h),1.74
–
1.66(m,2h),1.61
–
1.50(m,3h),1.47
–
1.38(m,3h),1.33
–
1.24(m,1h),1.18(s,3h),1.08
–
0.99(m,1h),0.98
–
0.94(m,1h),0.88(s,3h)ppm。
[0146]
步骤6:合成化合物8
[0147]
化合物7(15.00g,45.39mmol)溶于4-甲基吡啶(90ml),加入质量分数10%pd/c(6.66g,含水量55%)。氢气氛围下反应24小时后,加入硅藻土抽滤,并用400ml乙酸乙酯淋洗稀释。滤液依次用质量分数10%柠檬酸水溶液(4
×
150ml),饱和碳酸氢钠溶液(200ml)和饱和氯化钠水溶液洗涤。有机相用无水硫酸钠干燥。浓缩后得到粗品8直接投下一步反应。1hnmr(600mhz,cdcl3):δ=3.94
–
3.86(m,2h),3.85
–
3.79(m,2h),2.64(t,j=13.8hz,1h),2.30(td,j=15.0,5.4hz,1h),2.16
–
2.10(m,1h),2.05
–
1.91(m,3h),1.88
–
1.73(m,3h),1.69
–
1.63(m,1h),1.58(td,j=13.2,4.2hz,1h),1.52
–
1.40(m,6h),1.37
–
1.28(m,2h),1.26
–
1.19(m,2h),1.43
–
1.04(m,1h),0.99(s,3h),0.83(s,3h)ppm。
[0148]
步骤7:合成化合物9
[0149]
化合物8粗品(12.00g,36.09mmol)溶于无水四氢呋喃(120ml),0℃下加入三仲丁基硼氢化锂溶液(54ml,54.0mmol,1.0m in thf)。冰水浴下反应1小时后,缓慢滴加3mnaoh(aq.,30ml),质量分数30%h2o2(aq.,30ml)淬灭反应。反应液加入二氯甲烷(3
×
50ml)萃取,合并有机相后饱和硫代硫酸钠水溶洗涤,饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=10:1)得到化合物9白色固体(9.9g,三步总收率67%)。1h nmr(600mhz,cdcl3):δ=4.09(s,1h),3.96
–
3.80(m,4h),2.01
–
1.82(m,3h),1.81
–
1.59(m,3h),1.58
–
1.29(m,13h),1.25
–
1.12(m,3h),1.11
–
1.01(m,1h),0.95(s,3h),0.83(s,3h)ppm。
[0150]
步骤8:合成化合物10
[0151]
化合物9(18.00g,53.81mmol)溶于无水四氢呋喃(180ml),0℃下加入三溴吡啶(21.03g,59.19mmol)的四氢呋喃溶液(60ml)。室温搅拌3小时后,冰水浴下加入饱和硫代硫酸钠水溶液(30ml)和饱和碳酸氢钠水溶液(30ml)淬灭反应。反应体系加入乙酸乙酯(3
×
60ml)萃取,合并有机相后饱和氯化钠水溶液洗涤,无水硫酸钠干燥。浓缩后加入石油醚/etoac(20:1,80ml)的混合溶液稀释,放入冰箱静置4小时后抽滤,得化合物10白色固体(18.3g,收率82%),直接投下一步反应。1h nmr(600mhz,cdcl3):δ=4.48(dd,j=10.8,4.2hz,1h),4.25
–
4.19(m,1h),4.14
–
4.06(m,2h),3.94
–
3.86(m,2h),2.11
–
2.01(m,1h),2.00
–
1.89(m,2h),1.87
–
1.80(m,1h),1.78
–
1.67(m,2h),1.63
–
1.55(m,1h),1.55
–
1.44(m,
4h),1.41
–
1.27(m,7h),1.27
–
1.18(m,1h),1.18
–
1.12(m,1h),1.10
–
1.01(m,1h),0.93(s,3h),0.85(s,3h)ppm。
[0152]
步骤9:合成化合物11
[0153]
化合物10(19.00g,45.96mmol)溶于无水二甲亚砜(300ml),加入叔丁醇钾(15.47g,137.9mmol)后室温搅拌过夜。将反应液倒入饱和氯化铵的冰水溶液(500ml),二氯甲烷(3
×
200ml)萃取。合并有机相后饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=8:1)得到化合物11白色固体(14.8g,收率73%)。1h nmr(600mhz,cdcl3):δ=6.13(dd,j=6.0,0.6hz,1h),5.67(dd,j=6.0,3.6hz,1h),4.11
–
4.07(m,1h),3.98
–
3.90(m,3h),3.84
–
3.77(m,1h),2.27
–
2.20(m,1h),2.00
–
1.87(m,2h),1.80
–
1.68(m,2h),1.66
–
1.58(m,2h),1.56
–
1.29(m,10h),1.23
–
1.11(m,2h),0.98(s,3h),0.90(s,3h)ppm。
[0154]
步骤10:合成化合物12
[0155]
化合物11(5.00g,15.0mmol)溶于丙酮(125ml)和水(25ml)的混合溶液,加入吡啶对甲苯磺酸盐(756mg,3.01mmol)后室温搅拌8小时。加入饱和nahco3水溶液(30ml)淬灭反应,加水稀释后乙酸乙酯(3
×
60ml)萃取。合并有机相后饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=5:1)得到化合物12白色固体(4.1g,收率94%)。1h nmr(600mhz,cdcl3):δ=7.53
–
7.45(m,1h),5.98(dd,j=6.0,3.0hz,1h),4.12
–
4.06(m,1h),2.31(dt,j=11.4,2.4hz,1h),1.99
–
1.90(m,2h),1.87
–
1.75(m,3h),1.75
–
1.66(m,2h),1.58
–
1.40(m,8h),1.39
–
1.32(m,1h),1.28
–
1.17(m,2h),1.02(s,3h),0.99(s,3h)ppm。
[0156]
步骤11:合成化合物13
[0157]
化合物12(3.00g,10.4mmol)、叔丁基二甲基氯硅烷(2.04g,13.5mmol)及咪唑(2.12g,31.2mmol)溶于无水n,n-二甲基甲酰胺(15ml),室温搅拌6小时。加入乙酸乙酯(150ml)稀释后,饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=20:1)得到化合物13白色固体(3.6g,收率86%)。1h nmr(600mhz,cdcl3):δ=7.54
–
7.49(m,1h),6.01(dd,j=6.0,3.0hz,1h),4.06
–
4.02(m,1h),2.36
–
2.31(m,1h),1.99
–
1.91(m,1h),1.90
–
1.79(m,4h),1.75
–
1.69(m,1h),1.65
–
1.49(m,4h),1.48
–
1.36(m,5h),1.27
–
1.20(m,3h),1.05(s,3h),1.00(s,3h),0.88(s,9h),0.02(s,6h)ppm。
[0158]
步骤12:合成化合物14
[0159]
化合物13(1.00g,2.49mmol)、对甲苯磺酸(214mg,1.25mmol)溶于乙酸酐(25ml),室温搅拌2小时,浓缩后粗品经柱层析分离纯化(石油醚/etoac=100:1)得到化合物14白色固体(780mg,收率71%)。1h nmr(600mhz,cdcl3):δ=6.09(d,j=2.4hz,1h),5.75(t,j=2.4hz,1h),4.00(s,1h),2.20(s,3h),2.02
–
1.95(m,1h),1.93
–
1.89(m,1h),1.85
–
1.79(m,2h),1.68
–
1.48(m,5h),1.45
–
1.19(m,8h),1.05(s,3h),1.02(s,3h),0.88(s,9h),0.01(s,3h),0.01(s,3h)ppm。
[0160]
步骤13:合成化合物15
[0161]
化合物14(600mg,1.35mmol)、n-(苄氧羰基)羟胺(293mg,1.76mmol)溶于二氯甲烷(30ml),0℃下加入四正丁基高碘酸铵(761mg,1.76mmol)的二氯甲烷溶液(30ml)。加料完毕后,室温搅拌30分钟。冰水浴下加入饱和硫代硫酸钠水溶液(20ml)淬灭反应,二氯甲烷(3
×
20ml)萃取,饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品溶于甲苯(30ml),加热至110℃反应4小时。停止反应后浓缩反应液,粗品经柱层析分离纯化(石油醚/etoac=20:1)得到化合物15白色固体(445mg,收率54%)。1h nmr(600mhz,cdcl3):δ=7.39
–
7.30(m,5h),6.85(d,j=6.0hz,1h),6.23(d,j=6.0hz,1h),5.16(d,j=12.0hz,1h),5.05(d,j=12.0hz,1h),4.04(s,1h),2.33
–
2.25(m,1h),2.08
–
1.94(m,2h),1.92(s,3h),1.93
–
1.84(m,1h),1.84
–
1.77(m,1h),1.73
–
1.61(m,1h),1.60
–
1.50(m,3h),1.46
–
1.12(m,8h),0.92(s,3h),0.90(s,3h),0.88(s,9h),0.02(s,6h)ppm。同时还得到化合物21白色固体(173mg,收率21%)。
[0162]
步骤14:合成化合物17
[0163]
化合物15(300mg,0.492mmol)溶于乙醇(9ml)和四氢呋喃(9ml)的混合溶液,加入质量分数5%pd/caco3(90mg,30%w/w)。抽换氢气,室温搅拌5小时。硅藻土抽滤,乙酸乙酯洗涤滤饼。浓缩滤液后粗品经柱层析分离纯化(石油醚/etoac=20:1)得到化合物17白色固体(180mg,收率84%)。1h nmr(600mhz,cdcl3):δ=6.29(s,1h),4.03
–
3.99(m,1h),2.61
–
2.53(m,1h),2.11
–
2.07(m,1h),2.07(s,3h),2.06
–
2.02(m,1h),1.92
–
1.80(m,2h),1.79
–
1.72(m,2h),1.62
–
1.45(m,7h),1.45
–
1.25(m,5h),1.22
–
1.12(m,3h),1.02(s,3h),0.91(s,3h),0.87(s,9h),0.01(s,3h),0.00(s,3h)ppm。
[0164]
步骤15:合成化合物6
[0165]
将化合物17(200mg,0.419mmol)溶于四氢呋喃(7.5ml)和水(1.5ml)的混合溶液,加入cucl2·
2h2o(143mg,0.838mmol)。室温搅拌12小时,加水(8ml)稀释后乙酸乙酯(3
×
8ml)萃取。饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=15:1)得到化合物6白色固体(95mg,收率54%)。1h nmr(600mhz,cdcl3):δ=4.07
–
3.99(m,1h),2.43
–
2.31(m,2h),1.99
–
1.71(m,8h),1.59
–
1.45(m,3h),1.45
–
1.32(m,5h),1.31
–
1.18(m,3h),0.98(s,3h),0.97(s,3h),0.88(s,9h),0.01(s,6h)ppm。同时还得到化合物18白色固体(50mg,收率26%)
[0166]
化合物18向化合物6的转化:
[0167]
将化合物18(50mg,0.108mmol)溶于甲醇(2ml),加入碳酸钾(30mg,0.216mmol)。室温搅拌5小时后,加水(2ml)稀释后乙酸乙酯(3
×
2ml)萃取。饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=5:1)得到化合物6白色固体(38mg,收率85%)。
[0168]
实施例11目标化合物14,15β-环氧基-3β-羟基-5β,14β-蟾毒基-20,22-二烯内酯的化学合成方法
[0169]
步骤16:合成化合物19
[0170]
氮气保护下将化合物6(100mg,0.238mmol)溶于乙醇(24ml),加入三乙胺(0.66ml,4.76mmol)和水合肼(0.29ml,4.76mmol)。加热至50℃反应过夜,原料转化完全。减压浓缩除去反应溶剂后,粗品直接投下一步反应。氮气保护下将粗品腙化合物溶于四氢呋喃(20ml),加入三乙胺(0.66ml,4.76mmol)。冰水浴下滴加碘(181mg,0.714mmol)的四氢呋喃溶液(4ml)。室温搅拌4小时后,加入饱和硫代硫酸钠水溶液(10ml)和饱和碳酸氢钠水溶液(10ml)淬灭反应。乙酸乙酯(3
×
20ml)萃取后饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=30:1)得到化合物19白色固体(95mg,收率75%)。1h nmr(600mhz,cdcl3):δ=6.22
–
6.13(m,1h),4.07
–
4.01(m,1h),2.41
–
2.32(m,1h),2.21(dd,
j=15.6,3.0hz,1h),2.13(td,j=12.0,5.4hz,1h),1.98
–
1.74(m,5h),1.62
–
1.54(m,1h),1.53
–
1.44(m,3h),1.42
–
1.25(m,5h),1.21
–
1.15(m,2h),0.97(s,3h),0.88(s,9h),0.87(s,3h),0.01(s,6h)ppm。
[0171]
步骤17:合成化合物21
[0172]
氮气保护下将化合物19(80mg,0.151mmol)、硼酸酯化合物20(40mg,0.181mmol)、[1,1'-双(二苯基膦基)二茂铁]二氯化钯(11mg,0.0151mmol)和磷酸钾(119mg,0.559mmol)溶于n,n-二甲基甲酰胺(3ml),加热至60℃反应4小时。加水(10ml)稀释后,乙酸乙酯(3
×
5ml)萃取,饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=8:1)得到化合物21白色固体(53mg,收率71%)。1h nmr(600mhz,cdcl3):δ=7.55
–
7.50(m,1h),7.43(dd,j=9.6,2.4hz,1h),6.33(dd,j=9.6,0.6hz,1h),5.89
–
5.84(m,1h),4.06
–
4.01(m,1h),2.43
–
2.36(m,1h),2.32
–
2.26(m,1h),2.18
–
2.10(m,2h),1.98
–
1.86(m,3h),1.83
–
1.75(m,1h),1.63
–
1.53(m,2h),1.52
–
1.48(m,2h),1.44
–
1.31(m,4h),1.26
–
1.17(m,3h),1.04(s,3h),0.98(s,3h),0.88(s,9h),0.01(s,6h)ppm。
[0173]
步骤18:合成化合物22
[0174]
将化合物21(50mg,0.1mmol)溶于二氯甲烷(2ml),加入六氟磷酸(三环已基膦)(1,5-环辛二烯)(吡啶)合铱(24mg,0.03mmol)。抽换氢气,0℃下反应6小时。硅藻土抽滤,二氯甲烷洗涤滤饼。浓缩滤液后粗品经柱层析分离纯化(石油醚/etoac=5:1)得到化合物22白色固体(35mg,收率70%)。1h nmr(600mhz,cdcl3):δ=7.30
–
7.24(m,2h),6.30
–
6.24(m,1h),4.07
–
4.01(m,1h),3.16
–
3.10(m,1h),2.04
–
1.97(m,1h),1.95
–
1.83(m,3h),1.83
–
1.74(m,2h),1.73
–
1.65(m,2h),1.62
–
1.56(m,1h),1.53
–
1.46(m,2h),1.44
–
1.31(m,5h),1.28
–
1.17(m,4h),0.95(s,3h),0.87(s,9h),0.63(s,3h),0.01(s,6h)ppm。
[0175]
步骤19:合成化合物23
[0176]
氮气保护下将化合物22(25mg,0.05mmol)溶于二氯甲烷(2.5ml),加入吡啶(32μl,0.4mmol)。-20℃下加入氯化亚砜(11μl,0.15mmol),反应40分钟。加水(5ml)淬灭,二氯甲烷(5
×
2ml)萃取,饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=15:1)得到化合物23白色固体(20mg,收率83%)。1h nmr(600mhz,cdcl3):δ=7.32(dd,j=9.6,2.4hz,1h),7.30(s,1h),6.30(d,j=9.6hz,1h),5.25
–
5.20(m,1h),4.04
–
4.00(m,1h),2.78
–
2.70(m,1h),2.43
–
2.36(m,2h),2.09
–
2.00(m,1h),1.97
–
1.90(m,1h),1.86
–
1.79(m,3h),1.69
–
1.50(m,4h),1.48
–
1.35(m,5h),1.34
–
1.18(m,4h),0.94(s,3h),0.88(s,9h),0.71(s,3h),0.01(s,6h)ppm。
[0177]
步骤20:合成化合物24
[0178]
将化合物23(8mg,0.017mmol)溶于四氢呋喃(0.8ml),加入质量分数70%氢氟酸吡啶(0.8ml)。室温搅拌20分钟。缓慢滴加饱和碳酸氢钠水溶液(2ml)淬灭,乙酸乙酯(3
×
2ml)萃取,饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=5:1)得到化合物24(4mg,收率67%)。1h nmr(600mhz,meoh-d4):δ=7.59(dd,j=9.6,2.4hz,1h),7.57
–
7.54(m,1h),6.33(d,j=9.0hz,1h),5.31
–
5.27(m,1h),4.05
–
4.02(m,1h),2.86
–
2.78(m,1h),2.58
–
2.49(m,1h),2.42
–
2.33(m,1h),2.25
–
2.17(m,2h),2.14
–
1.90(m,5h),1.88
–
1.84(m,1h),1.82
–
1.76(m,1h),1.71
–
1.66(m,1h),1.62
–
1.45(m,7h),0.99(s,3h),0.76(s,3h)ppm。
[0179]
步骤21:合成化合物1
[0180]
将化合物24(2.9mg,0.0079mmol)溶于丙酮(0.36ml)和水(40μl)的混合溶液中,加入n-溴代琥珀酰亚胺(1.8mg,0.010mmol)和质量分数1%高氯酸水溶液(5μl)。室温搅拌1小时。缓慢滴加饱和碳酸氢钠水溶液(0.5ml)和饱和硫代硫酸钠水溶液(0.5ml)淬灭,乙酸乙酯(3
×
3ml)萃取,饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品不经纯化,直接溶于吡啶(0.4ml),在60℃下反应2小时。浓缩后粗品经柱层析分离纯化(石油醚/etoac=2:1)得到化合物1白色固体(1.6mg,收率53%)。核磁氢谱与标准谱图对比,结果一致。
[0181]
实施例12:
[0182]
合成化合物7:
[0183]
将原料雄烯二酮化合物2(10g,34.97mmol)溶于100ml甲苯中,加入乙二醇(7ml,123mmol)。升温回流1小时后,加入160μl浓硫酸(3mmol),tlc监测反应,反应完全后,冷却至室温,用饱和碳酸氢钠水溶液(50ml)淬灭。硅藻土抽滤,乙酸乙酯洗涤滤饼,滤液加入乙酸乙酯(3
×
50ml)萃取。合并有机相,饱和氯化钠水溶液洗涤,无水硫酸钠干燥,浓缩后柱层析分离(石油醚/etoac=8:1)得到化合物7白色固体(8.7g,收率75%)。
[0184]
合成化合物8:
[0185]
化合物7(15g,45.39mmol)溶于吡啶(90ml),在氮气保护下加入质量分数10%氢氧化钯(1.5g,10%w/w)。用氢气置换3次后,反应12小时。反应毕,用硅藻土抽滤,并用乙酸乙酯(400ml)洗涤滤饼。滤液用10%柠檬酸水溶液(4
×
150ml)洗涤,后用饱和碳酸氢钠溶液(200ml)洗涤,无水硫酸钠干燥。浓缩后得到粗品8直接用于下一步反应。
[0186]
合成化合物9:
[0187]
化合物8(12g,36.09mmol)溶于无水甲苯(120ml),-78℃下缓慢滴加二异丁基氢化铝(36ml,54.08mmol,1.5m in toluene)。加毕,在该温度下反应6小时后,用饱和氯化铵溶液(50ml)淬灭反应,将反应升至室温后搅拌过夜。抽滤,用乙酸乙酯洗涤沉淀。滤液用乙酸乙酯(3
×
50ml)萃取,合并有机相后用无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=10:1)得到化合物9白色固体(8.2g,两步总收率54%)。
[0188]
合成化合物10:
[0189]
化合物9(18g,53.81mmol)溶于无水四氯化碳(180ml),0℃下使用恒压滴液漏斗缓慢滴加液溴(5.5ml,107.62mmol)的四氯化碳溶液(20ml)。室温搅拌3小时后,冰水浴下加入饱和硫代硫酸钠水溶液(30ml)和饱和碳酸氢钠水溶液(30ml)淬灭反应。分液后,水相用乙酸乙酯(3
×
100ml)萃取。合并有机相后饱和氯化钠水溶液洗涤,无水硫酸钠干燥,浓缩后使用柱层析分离(石油醚/etoac=10/1)得化合物10白色固体(13.8g,收率62%),直接投下一步反应。
[0190]
合成化合物11:
[0191]
化合物10(19g,45.96mmol)溶于无水1,4-二氧六环(300ml),加入叔丁醇钾(15.47g,137.88mmol)后室温搅拌过夜。反应完成后使用硅藻土抽滤,加入饱和氯化铵溶液(500ml),二氯甲烷(3
×
200ml)萃取。合并有机相后饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=8:1)得到化合物11白色固体(14.8g,收率73%)。
[0192]
合成化合物12:
[0193]
化合物11(5g,15.04mmol)溶于二氧六环(150ml),加入质量分数5%硫酸水溶液10ml后室温搅拌12小时。反应毕,加入饱和nahco3水溶液(30ml)淬灭反应,加水稀释后乙酸乙酯(3
×
60ml)萃取。合并有机相后饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=5:1)得到化合物12白色固体(4.1g,收率94%)。
[0194]
合成化合物13:
[0195]
化合物12(3g,10.40mmol)溶于吡啶(15ml)中,冰水浴下滴加叔丁基二甲硅基三氟磺酸酯(4.8ml,20.80mmol),并在该温度下持续搅拌1小时。反应完成后,加入乙酸乙酯(150ml)稀释,饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=20:1)得到化合物13白色固体(3.8g,收率91%)。
[0196]
合成化合物14:
[0197]
化合物13(1g,2.49mmol)、硫酸(27μl,0.5mmol)溶于乙酸酐(25ml),室温搅拌2小时。冰水浴下缓慢加入饱和碳酸氢钠水溶液(50ml)淬灭反应,乙酸乙酯(3
×
30ml)萃取,饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=100:1)得到化合物14白色固体(969mg,收率63%)。
[0198]
合成化合物17:
[0199]
化合物15(300mg,0.492mmol)溶于甲醇(14ml)和水(1ml)的混合溶液,加入质量分数5%pd/c(30mg,10%w/w)。抽换氢气,室温搅拌1小时。硅藻土抽滤,乙酸乙酯洗涤滤饼。浓缩滤液后粗品经柱层析分离纯化(石油醚/etoac=20:1)得到化合物167白色固体(199mg,收率85%)。
[0200]
合成化合物6:
[0201]
将化合物17(200mg,0.419mmol)溶于乙醇(3ml)、二氧六环(3ml)和水(3ml)的混合溶液,加入二水合氯化铜(143mg,0.838mmol)。室温搅拌24小时,反应完成后浓缩,粗品经柱层析分离纯化(石油醚/etoac=15:1)得到化合物6白色固体(76mg,收率43%),以及化合物18白色固体(67mg,收率38%)。
[0202]
实施例13:
[0203]
合成化合物6:
[0204]
将化合物18(50mg,0.108mmol)溶于乙醇(2ml)和水(0.5ml)的混合溶剂中,加入氢氧化钠(8.6mg,0.216mmol)。室温搅拌1小时后,加水(2ml)稀释后乙酸乙酯(3
×
2ml)萃取。饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=5:1)得到化合物6白色固体(40mg,收率73%)。
[0205]
实施例14:
[0206]
合成化合物19:
[0207]
氮气保护下将化合物6(100mg,0.238mmol)溶于乙醇(24ml),加入n,n-二异丙基乙胺(207μl,1.19mmol)和水合肼(58μl,1.19mmol)。在50℃下反应12小时。减压浓缩除去反应溶剂后,粗品直接投下一步反应。氮气保护下将上步所得化合物溶于四氢呋喃(6ml)和乙醇(10ml)的混合溶剂中,加入三乙胺(207μl,1.19mmol)。冰水浴下滴加碘(181mg,0.714mmol)的四氢呋喃溶液(4ml)。加毕,室温搅拌4小时,加入饱和硫代硫酸钠水溶液(10ml)和饱和碳酸氢钠水溶液(10ml)淬灭反应。乙酸乙酯(3
×
20ml)萃取后饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=30:1)得到化合物19白色固体(95mg,收
率75%)。
[0208]
合成化合物21:
[0209]
氮气保护下将化合物19(80mg,0.151mmol)、硼酸酯化合物20(40mg,0.181mmol)、pd(pph3)4(17mg,0.015mmol)和碳酸钾(77mg,0.559mmol)溶于四氢呋喃(3ml)和水(0.3ml)的混合溶剂中,加热至40℃反应12小时。反应完成后,加水(10ml)稀释,乙酸乙酯(3
×
5ml)萃取,饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=8:1)得到化合物21白色固体(65mg,收率87%)。
[0210]
合成化合物22:
[0211]
将化合物21(20mg,0.04mmol)溶于二氯甲烷(0.8ml),加入三苯基膦氯化铑(2mg,10%w/w)。抽换氢气,室温下反应12小时。反应完成后,用硅藻土抽滤,二氯甲烷洗涤滤饼。取滤液浓缩后粗品经柱层析分离纯化(石油醚/etoac=5:1)得到化合物22白色固体(16mg,收率80%)。
[0212]
合成化合物23:
[0213]
氮气保护下将化合物22(10mg,0.02mmol)溶于二氯甲烷(1ml),加入吡啶(10μl,0.16mmol)。冰浴下加入甲基磺酰氯(5μl,0.09mmol),反应10分钟。加水(2ml)淬灭,二氯甲烷(3
×
2ml)萃取,饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=15:1)得到化合物23白色固体(6.9mg,收率71%)。
[0214]
合成化合物24:
[0215]
将化合物23(4mg,0.0083mmol)溶于四氢呋喃(0.4ml),加入四丁基氟化铵的四氢呋喃溶液(0.4ml,1.0m in thf)。室温搅拌2分钟。反应完成后用水淬灭,乙酸乙酯(3
×
2ml)萃取后用水反复洗涤(5
×
5ml),取有机相用无水硫酸钠干燥。浓缩后粗品经柱层析分离纯化(石油醚/etoac=5:1)得到化合物24(2mg,收率67%)。
[0216]
合成化合物1:
[0217]
将化合物24(2.9mg,0.0079mmol)溶于丙酮(0.36ml)和水(40μl)的混合溶液中,加入n-碘代琥珀酰亚胺(2.2mg,0.010mmol)和质量分数1%高氯酸水溶液(5μl)。室温搅拌1小时。缓慢滴加饱和碳酸氢钠水溶液(0.5ml)和饱和硫代硫酸钠水溶液(0.5ml)淬灭,乙酸乙酯(3
×
3ml)萃取,饱和食盐水洗涤,无水硫酸钠干燥。浓缩后粗品不经纯化,直接溶于吡啶(0.4ml),在60℃下反应2小时。浓缩后粗品经柱层析分离纯化(石油醚/etoac=2:1)得到化合物1白色固体(1.6mg,收率53%)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1