一种耐高温的聚酰亚胺复合材料及其制备方法与应用
1.本发明涉及高分子材料技术领域,尤其涉及一种耐高温的聚酰亚胺复合材料及其制备方法与应用。
背景技术:
2.近年来,树脂基复合材料由于其质轻高强和优异的耐腐蚀性而被广泛用于航空航天领域,在飞机、导弹和火箭等制造中占比逐年增大。在耐热性聚合物领域中,热固性聚酰亚胺具有十分突出的热稳定性和机械性能,成为制备耐高温复合材料中最受欢迎的树脂基体。常规聚酰亚胺的热稳定性与分子本体结构密切相关,热分解性能随主链结构差别较大,主链结构键能越大,热稳定性等级越高,常规苯乙炔封端的聚酰亚胺的热失重5%温度均在500℃以上,但由于受到有机键能的制约,热分解温度很难大于600℃;玻璃化转变温度是热固性材料温度评价的一项重要参数,通常为服役最高温度,其同样受分子结构影响,主链刚性、取代基团的空间位阻、侧链柔性、分子间作用力(侧链极性、氢键)、共聚、交联、分子量(超过一定分子量后,增加不明显)等均可提升pi(聚酰亚胺树脂)树脂体系的tg(转变温度),常规聚酰亚胺的玻璃化转变温度可达300~400℃,后固化处理后其tg可进一步增大;树脂基体的性能也影响着力学性能,单向碳纤维增强聚酰亚胺复合材料的弯曲强度约为1400mpa、弯曲模量可达120gpa的量级。由于聚酰亚胺体系固化温度较高,复合材料成型多采用热模压成型或热压罐成型,体系粘度直接影响着其工艺成型制度,加工窗口一般在300~350℃,粘度为10~2000pa
·
s。相对于其他树脂基体而言,pi熔体粘度太大,以致于成型较困难,加工条件苛刻,工艺性严重限制了其在各大科学技术中的应用。
3.目前,含腈基聚酰亚胺逐渐得到学者研究,通过将腈基(cn)引入到聚酰亚胺主链苯环上,增大分子间相互作用力,从而使得聚酰亚胺体系的耐热性进一步提升。研究发现将cn接在聚酰亚胺主链联苯上,会提高分子链内部旋转能量壁垒,使聚合物骨架刚性增强,链运动性大幅受限,tg升高,热重分析测试氮气条件时,由于氮含量的增加使800℃时残炭率有所升高,利于制成阻燃性和热稳定性强的终端材料;调整侧链腈基引入位点还可以一定程度上打破链的规整性并形成非共平面的结构,有助于改善溶解性能,而聚合物中cn间的强相互作用,也有可能对于pi材料的机械强度有明显的加强效果。imre treufeld和david h.wang分别制备了含3个cn的新型二胺单体,与二酐共聚将cn作为主链苯环的侧基引入到聚酰亚胺体系中,每个重复单元存在3个相邻的cn来增加偶极矩密度可以增加整体高温介电常数,同时可保持介电损耗相对较低,最终制得的3cn-pi薄膜均具有优异的电性能和热性能。目前,含腈基聚酰亚胺的研究大多基于热塑性聚酰亚胺主链的研究,制备成为高介电耐热的pi薄膜并加以应用。官能团cn的引入只起到了功能性基团的作用,未有学者将其作为反应性基团,探究其对热固性聚酰亚胺及其复合材料的性能影响。
4.现在,聚酰亚胺复合材料已经发展到第四代,大量研究表明,引入含硅或硼等无机组分可显著改善聚酰亚胺的热性能,通过分子结构设计也能改善一定的工艺性。但是,将硼或硅引入到pi体系的反应过程比较繁琐,合成成本较高。
5.因此,如何将腈基官能团引入到聚酰亚胺树脂体系中,开发出一种新型含腈基官能团的聚酰亚胺固化交联剂,并将其应用于聚酰亚胺树脂体系中成为了本领域技术人员亟需解决的问题。
技术实现要素:
6.有鉴于此,本发明提供了一种耐高温的聚酰亚胺复合材料及其制备方法与应用,该方法合成了含腈基和苯乙炔基三官能度的酰亚胺低聚物单体,并将其引入到聚酰亚胺体系中共混改性,得到了一种热稳定性十分优异的聚酰亚胺复合材料,有效解决耐高温聚酰亚胺复合材料长期服役温度低于420℃的现状,更满足了航空航天领域中耐高温结构部件的性能要求。
7.为了达到上述目的,本发明采用如下技术方案:
8.一种耐高温的聚酰亚胺复合材料的制备方法,包括以下步骤:
9.将酰亚胺低聚物单体和聚酰亚胺预聚物依次进行热亚胺化反应、固化交联反应即得耐高温的聚酰亚胺复合材料。
10.进一步的,所述热亚胺化反应的反应温度为240~260℃,反应时间为1.5~2.5h;所述固化交联反应的反应温度为370~390℃,反应时间为1.5~2.5h。
11.进一步的,所述酰亚胺低聚物单体的添加质量为聚酰亚胺预聚物质量的1~20%。
12.进一步的,所述酰亚胺低聚物单体的结构式如下式i~x所示:
[0013][0014]
其制备方法包括以下步骤:
[0015]
先将氰基硝基苯与sncl2溶液进行第一反应得到氰基苯胺,将氰基苯胺与4-苯乙炔苯酐溶液进行第二反应即得酰亚胺低聚物单体。
[0016]
进一步的,所述第一反应的反应时间为7~10h;
[0017]
所述第二反应为化学亚胺化反应或热亚胺化反应,所述化学亚胺化反应的反应温度为50~80℃,反应时间为10~15h;所述热亚胺化反应的反应温度为150~200℃,反应时间为15~50h。
[0018]
进一步的,所述sncl2溶液的溶剂为无水乙醇或乙酸乙酯,4-苯乙炔苯酐溶液的溶剂为非质子型高沸点溶剂;
[0019]
所用氰基硝基苯与sncl2的摩尔比为1:12~14;
[0020]
所用氰基苯胺与4-苯乙炔苯酐的摩尔比为1:1~2。
[0021]
进一步的,所述聚酰亚胺预聚物的结构式如下:
[0022][0023]
其中,n为0~5之间的整数,ar1和ar2独立的为芳香杂环结构;
[0024]
所述聚酰亚胺预聚物的制备方法包括以下步骤:
[0025]
将芳香二酐单体和4-苯乙炔苯酐加入到乙醇中进行第一反应得到第一混合物,将第一混合物与芳香二胺单体进行第二反应即得聚酰亚胺预聚物。
[0026]
进一步的,所述第一反应的反应温度为70~90℃,反应时间为4~8h;第二反应的反应时间为12~16h;
[0027]
所述芳香二酐单体和4-苯乙炔苯酐的摩尔比为1~5:2,所述芳香二酐单体与芳香二胺单体的摩尔比为1~5:2~6。
[0028]
本发明提供了上述耐高温的聚酰亚胺复合材料的制备方法所制备的耐高温的聚酰亚胺复合材料。
[0029]
本发明还提供了上述耐高温的聚酰亚胺复合材料在航空航天领域中的应用。
[0030]
经由上述的技术方案可知,与现有技术相比,本发明的有益效果如下:
[0031]
本发明是将含腈基和苯乙炔基三官能度的酰亚胺低聚物单体作为第三组分引入到优选的聚酰亚胺复配体系中,实现物理共混改性。低聚物单体中2个cn和1个苯乙炔基均可作为反应性基团,在高温下发生交联固化,并与pi主链上的苯乙炔基缠结交联形成复杂的网络结构。苯乙炔基的交联反应温度高,从而使得pi具有更宽的加工窗口,交联反应活性高,其本身的芳香性也使得固化物热性能得到保证。cn交联固化后形成芳杂环为主的网络结构,其本身强极性使得其热稳定性十分突出,2个cn的存在增加了体系中的交联位点,使得单元交联密度得以显著增加,热性能得以提高。设计的含腈基单体为非对称性低聚物,有助于促进体系形成非共平面结构从而降低熔体粘度。交联过程中cn具有的强极性和电负性使得分子间相互作用力增强,pi的机械性能也可明显增加。
[0032]
本发明所制备的耐高温的聚酰亚胺复合材料,均是通过物理共混的形式将含双腈基和单苯乙炔基的酰亚胺单体引入到聚酰亚胺树脂树脂体系中,一方面可以在维持原有基础主链良好耐热性的基础上进一步增加交联位点,提升交联密度,形成更为复杂且牢固的交联网络;另一方面在体系中引入了小分子量的非对称性结构以及极性基团,可促使体系形成非共平面结构,降低了熔体粘度,且随着分子间作用力的不断增大,体系的热稳定性和耐热氧化性也得到了提升,愈发满足航空航天领域中耐高温结构部件的制备要求。
附图说明
[0033]
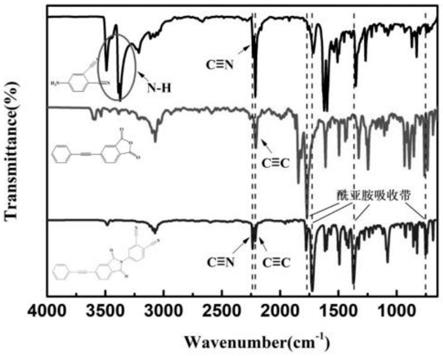
图1为实施例1的原料4-apn、4-pepa以及产物apn-pepa的红外光谱对比图;
[0034]
图2为实施例1产物apn-pepa的1h-nmr图;
[0035]
图3为实施例1产物apn-pepa的质谱esi图;
[0036]
图4为实施例1产物apn-pepa的在不同升温速率下的dsc曲线;
[0037]
图5为实施例1产物apn-pepa分别在n2和空气气氛下的tg曲线;
[0038]
图6为pi(对应a图)、pi-apn5%(对应b图)、pi-apn10%(对应c图)、pi-apn15%(对应d图)和pi-apn20%(对应e图)聚酰亚胺复合材料在不同升温速率下的dsc曲线;
[0039]
图7为pi、pi-apn5%、pi-apn10%、pi-apn15%和pi-apn20%聚酰亚胺复合材料的粘温曲线;
[0040]
图8为pi、pi-apn5%、pi-apn10%、pi-apn15%和pi-apn20%聚酰亚胺复合材料分别在n2(对应a图)和空气气氛下(对应b图)的tg曲线;
[0041]
图9为pi、pi-apn5%聚酰亚胺复合材料的红外光谱对比图;
[0042]
图10为pi、pi-apn5%、pi-apn10%、pi-apn15%和pi-apn20%聚酰亚胺测试材料的dma曲线图;
[0043]
图11为pi、pi-apn5%、pi-apn10%、pi-apn15%和pi-apn20%聚酰亚胺测试材料的室温力学性能对比图;
[0044]
图12为制备碳纤维增强聚酰亚胺测试材料的温度-压力-时间程序图。
具体实施方式
[0045]
本发明提供了一种耐高温的聚酰亚胺复合材料的制备方法,包括以下步骤:
[0046]
将酰亚胺低聚物单体和聚酰亚胺预聚物依次进行热亚胺化反应、固化交联反应即得耐高温的聚酰亚胺复合材料。
[0047]
在本发明中,所述热亚胺化反应的反应温度为240~260℃,优选为245~255℃,进一步优选为250℃。反应时间为1.5~2.5h,优选为1.8~2.2h,进一步优选为2h。
[0048]
所述固化交联反应的反应温度为370~390℃,优选为375~385℃,进一步优选为380℃。反应时间为1.5~2.5h,优选为1.8~2.2h,进一步优选为2h。
[0049]
所述制备聚酰亚胺复合材料的具体操作步骤为:
[0050]
将酰亚胺低聚物单体用二氯甲烷搅拌溶解,按照质量分数分别为0wt%、5wt%、10wt%、15wt%、20wt%加入到聚酰亚胺预聚物中,共混搅拌2~4h,得到灰褐色澄清聚酰胺酸树脂共混溶液,5种体系分别命名为pi、pi-apn5%、pi-apn10%、pi-apn15%和pi-apn20%。
[0051]
将聚酰胺酸树脂共混溶液,通过真空浓缩、旋转蒸发,加热进行热亚胺化,得到5种不同含量apn-pepa改性的聚酰亚胺预聚物。
[0052]
在真空马弗炉中,按照如下程序加热上述5种apn-pepa改性的聚酰亚胺预聚物:60min内温度从室温升到310~330℃,保温15min;而后20min内继续升温至370~390℃,保温120min;最后随炉自然冷却,得到5种不同含量apn-pepa改性的聚酰亚胺树脂固化物,即聚酰亚胺复合材料。
[0053]
作为优选,真空马弗炉的参数设定为:60min内温度从室温升到320℃,保温15min;而后20min内继续升温至380℃,保温120min;最后随炉自然冷却,即得产物。
[0054]
所述酰亚胺低聚物单体的结构式如下式i~x所示:
[0055][0056]
作为优选,所述酰亚胺低聚物单体的结构式为式i~ⅲ、
ⅹ
所示,进一步优选为式x所示的结构。
[0057]
其制备方法包括以下步骤:
[0058]
先将氰基硝基苯与sncl2溶液进行第一反应得到氰基苯胺,将氰基苯胺与4-苯乙炔苯酐溶液进行第二反应即得酰亚胺低聚物单体。
[0059]
在本发明中,所述第一反应的反应时间为7~10h;优选为7.8~9h,进一步优选为8h。
[0060]
所述第二反应为化学亚胺化反应或热亚胺化反应,所述化学亚胺化反应的反应温度为50~80℃,优选为60~70℃,进一步优选为65℃。反应时间为10~15h,优选为12~14h,进一步优选为13h。
[0061]
所述化学亚胺化反应的具体操作步骤为:
[0062]
按照氰基硝基苯与sncl2的摩尔比为1:12~14在室温下发生第一反应得到氰基苯胺,按照氰基苯胺与4-苯乙炔苯酐的摩尔比为1:1~2室温下搅拌2~4h;再按照氰基苯胺:
乙酸酐:吡啶=1:2~4:2~4的摩尔比,于60℃下搅拌12h,即得酰亚胺低聚物单体。
[0063]
所述热亚胺化反应的反应温度为150~200℃,反应时间为15~50h;优选为160~180℃,反应时间为20~40h;进一步优选温度为170℃,时间为30h。
[0064]
所述热亚胺化反应的具体操作步骤为:
[0065]
按照氰基硝基苯与sncl2的摩尔比为1:12~14在室温下发生第一反应得到氰基苯胺,按照氰基苯胺与4-苯乙炔苯酐的摩尔比为1:1~2室温下搅拌2~4h;再按照氰基苯胺:甲苯=1:4~6的摩尔比,于180℃下反应18h,即得酰亚胺低聚物单体。
[0066]
所述sncl2溶液的溶剂为无水乙醇或乙酸乙酯,优选为无水乙醇。所述4-苯乙炔苯酐溶液的溶剂为非质子型高沸点溶剂;优选为二甲基亚砜、n-甲基吡咯烷酮、二甲基乙酰胺、n,n-二甲基甲酰胺中的任意一种;进一步优选为二甲基亚砜。
[0067]
所用氰基硝基苯与sncl2的摩尔比为1:12~14;优选为1:13。
[0068]
所用氰基苯胺与4-苯乙炔苯酐的摩尔比为1:1~2;优选为1:1.5。
[0069]
所述聚酰亚胺预聚物的结构式如下:
[0070][0071]
其中,n为0~5之间的整数,优选为1~3之间的整数,进一步优选为n=2。
[0072]
ar1和ar2独立的为芳香杂环结构,优选为以下结构:
[0073][0074]
所述聚酰亚胺预聚物的制备方法包括以下步骤:
[0075]
将芳香二酐单体和4-苯乙炔苯酐加入到乙醇中进行第一反应得到第一混合物,将第一混合物与芳香二胺单体进行第二反应即得聚酰亚胺预聚物。
[0076]
在本发明中,所述第一反应的反应温度为70~90℃,优选为75~85℃,进一步优选为80℃。反应时间为4~8h,优选为5~7h;进一步优选为6h。
[0077]
所述第二反应的反应时间为12~16h,优选为13~14h,进一步优选为13.5h。
[0078]
所述芳香二酐单体和4-苯乙炔苯酐的摩尔比为1~5:2,优选为2~3:2,进一步优选为1:1。
[0079]
所述芳香二酐单体与芳香二胺单体的摩尔比为1~5:2~6,优选为2~4:3~5,进一步优选为3:4。
[0080]
本发明提供了上述耐高温的聚酰亚胺复合材料的制备方法所制备的耐高温的聚酰亚胺复合材料。
[0081]
本发明还提供了上述耐高温的聚酰亚胺复合材料在航空航天领域中的应用。
[0082]
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0083]
以下实施例中涉及到的化合物单体均采用缩写,2,3,3',4-联苯四甲酸二酐对应缩写为α-bpda、4-苯乙炔苯酐对应缩写为pepa、4,4'-二氨基-2,2
’‑
双三氟甲基联苯对应缩写为tfmb和4,4'-二氨基联苯对应缩写为bz。
[0084]
实施例1
[0085]
本实施例为酰亚胺低聚物单体的制备,本实施例所用的原料为4-硝基邻苯二甲腈(apn),具体操作步骤如下:
[0086]
在装有机械搅拌三口烧瓶中称取45.13g(200mmol)sncl
2`
2h2o于装有机械搅拌的250ml三口烧瓶中,加入50ml无水乙醇搅拌溶解,待溶液搅拌均一后,再加入8.66g(50mmol)4-硝基邻苯二甲腈,分批次缓慢加入,防止冲料,室温搅拌10min后升温至80℃冷凝回流,反应3~4h,采用tcl法时刻监控反应程度。反应结束后冷却至室温,旋蒸掉大部分乙醇溶剂,倒入500ml烧杯,加入150ml去离子水和150ml乙酸乙酯,先后加入固体naoh、nahco3将溶液ph调至9,溶液出现分层,上层为有机层,下层为锡盐层。将上述分层溶液进行抽滤,不断用乙酸乙酯清洗析出的固体锡盐,分液取有机层,加入适量无水mgso4静置脱水1h,抽滤得有机溶液,旋蒸,70℃真空干燥12h,得淡黄色固体粉末6.74g,即4-氨基邻苯二甲腈,产率为94.2%。
[0087]
于装有磁力搅拌以及分水器装置的250ml三口烧瓶中加入上述制得的4-氨基邻苯二甲腈2.86g(20mmol)和4-苯乙炔苯酐4.96g(20mmol),取10ml dmso加入到烧瓶中溶解,室温搅拌2h,得到均一透明的淡黄色的paa溶液。滴加乙酸酐4.08g(40mmol)和吡啶3.16g(40mmol)的混合溶液,升温至60℃搅拌,可采用tcl法时刻监控反应程度,反应12h,溶液变成棕褐色液体。反应结束后将溶液冷却至室温,倒入装有250ml去离子水的500ml烧杯中,搅拌生成棕色沉淀,并用50ml去离子水分别润洗2次,抽滤得棕色粗产物,将粗产物倒入装有200ml乙酸乙酯的500ml烧杯中,充分搅拌,抽滤,70℃真空干燥12h,得淡黄色固体粉末,即为目标产物酰亚胺低聚物单体(apn-pepa),产物质量为6.48g,产率为86.8%。
[0088]
实施例2
[0089]
本实施例为酰亚胺低聚物单体的制备,本实施例所用的原料为4-硝基邻苯二甲腈,具体操作步骤如下:
[0090]
在装有机械搅拌三口烧瓶中称取45.13g(200mmol)sncl
2`
2h2o于装有机械搅拌的250ml三口烧瓶中,加入50ml无水乙醇搅拌溶解,待溶液搅拌均一后,再加入8.66g(50mmol)4-硝基邻苯二甲腈,分批次缓慢加入,防止冲料,室温搅拌10min后升温至80℃冷凝回流,反应3~4h,采用tcl法时刻监控反应程度。反应结束后冷却至室温,旋蒸掉大部分乙醇溶剂,倒入500ml烧杯,加入150ml去离子水和150ml乙酸乙酯,先后加入固体naoh、nahco3将溶液ph调至9,溶液出现分层,上层为有机层,下层为锡盐层。将上述分层溶液进行抽滤,不断用乙酸乙酯清洗析出的固体锡盐,分液取有机层,加入适量无水mgso4静置脱水1h,抽滤得有机溶液,旋蒸,70℃真空干燥12h,得淡黄色固体粉末6.74g,即4-氨基邻苯二甲腈,产率为94.2%。
[0091]
于装有磁力搅拌以及分水器装置的250ml三口烧瓶中加入上述制得的4-氨基邻苯二甲腈2.86g(20mmol)和4-苯乙炔苯酐4.96g(20mmol),取50mlnmp加入到烧瓶中溶解,室温搅拌12h,得到均一的橙黄色的paa溶液。加入4ml无水甲苯,搅拌升温至180℃,而后每隔1h加入2ml无水甲苯,直至180℃反应进行10h后,可以观察分水器中陆续有水被蒸出,同时也采用tcl法时刻监控反应程度,反应16h,溶液变成深褐色液体。反应结束后将溶液冷却至室温,旋蒸掉甲苯溶剂,倒入装有250ml去离子水的500ml烧杯中,搅拌生成棕色沉淀,抽滤得棕色粗产物,将粗产物倒入装有200ml乙酸乙酯的500ml烧杯中,充分搅拌,抽滤,70℃真空干燥12h,得淡黄色固体粉末,即为目标产物酰亚胺低聚物单体(apn-pepa),产物质量为
6.18g,产率为82.8%。
[0092]
实例1和实例2中合成方法区别在于形成酰亚胺环的方式不同,实例1为化学亚胺化,实例2为热亚胺化。将两种方法最终得到的产物apn-pepa通过红外光谱分析(ir)、核磁共振氢谱(1h-nmr)、质谱(esi)进行化学结构表征,所得测试结果与各化合物结构吻合。
[0093]
由图1、图2和图3可知:4-氨基邻苯二甲腈:ft-ir(kbr,cm-1
):1366(c-n的弯曲振动),2231(c≡n的伸缩振动),3496、3374(n-h的伸缩振动);1h-nmr(400mhz,cdcl3):δ=4.0(2h,n-h),δ=6.88(1h,苯环c-h),δ=6.97(1h,苯环c-h),δ=7.28(1h,苯环c-h)。酰亚胺低聚物单体:ft-ir(kbr,cm-1
):752(c=o的变角振动),1366(c-n的弯曲振动),1726(c=o的对称伸缩振动),1780(c=o的不对称伸缩振动),2206(c≡c的伸缩振动)2231(c≡n的伸缩振动);1h-nmr(400mhz,dmso-d6):δ=7.44-7.56(3h,苯环c-h),δ=7.61-7.72(2h,苯环c-h),δ=8.06(1h,苯环c-h),δ=8.08-8.13(2h,苯环c-h),δ=8.18(1h,苯环c-h),δ=8.27(1h,苯环c-h),δ=8.30-8.38(1h,苯环c-h);esi:m=373.3g/mol。
[0094]
产物apn-pepa通过热分析dsc测试表征单体的物理性能,并在不同升温速率下进行热流扫描外推得到低聚物的固化工艺制度,通过热重tg测试表征单体的热稳定性。
[0095]
由图4和图5可知,单体熔点为200℃,由于分子内同时存在腈基和苯乙炔基,可发生高温交联固化,初始固化温度为270℃,固化峰值温度为370℃,固化终止温度为410℃。同时,单体固化后产物也具有十分优异的稳定性。热分解温度高达500℃以上,800℃残碳率达73.4%。
[0096]
实施例3
[0097]
本实施例为聚酰亚胺预聚物的制备,具体操作步骤如下:
[0098]
称取4.85g(16.5mmol)的α-bpda、4.09g(16.5mmol)的pepa加入到装有磁力搅拌的250ml三口烧瓶中,加入25ml无水乙醇搅拌溶解,在80℃下冷凝回流2h,待溶液呈无色澄清透明时,冷却至室温,加入4.56g(24.7mmol)的bz,室温搅拌12h,得到苯乙炔封端的bz/α-bpda聚酰亚胺酸溶液。
[0099]
bz/α-bpda聚酰亚胺的结构式如下:
[0100][0101]
其中,n=2。
[0102]
实施例4
[0103]
本实施例为聚酰亚胺复合材料的制备,具体操作步骤如下:
[0104]
(1)取10ml二氯甲烷溶解实施例1所制备的酰亚胺低聚物单体apn-pepa,按照质量分数分别为0wt%、5wt%、10wt%、15wt%、20wt%加入实施例3所制得bz/α-bpda聚酰亚胺酸溶液中,共混搅拌3h,得到灰褐色澄清聚酰胺酸树脂共混溶液,5种体系分别命名为pi、
pi-apn5%、pi-apn10%、pi-apn15%和pi-apn20%;
[0105]
(2)将聚酰胺酸树脂共混溶液,通过真空浓缩、旋转蒸发,加热至250℃进行热亚胺化,反应2h,得到5种不同含量apn-pepa改性的聚酰亚胺预聚物;
[0106]
(3)在真空马弗炉中,按照如下程序加热上述5种apn-pepa改性的聚酰亚胺预聚物:60min内温度从室温升到320℃,保温15min;而后20min内继续升温至380℃,保温120min;最后随炉自然冷却,即得5种不同含量apn-pepa改性的聚酰亚胺复合材料。
[0107]
将上述所制得的5种聚酰亚胺复合材料通过dsc测试后外推得到各自体系的固化特征温度,在各自对应的固化峰值温度加热保温固化2h,即得到5种聚酰亚胺树脂固化物。
[0108]
分别对5个不同改性体系的聚酰亚胺复合材料进行不同升温速率下的dsc扫描测试,见图6。由图6可知各体系升温速率为0时的固化工艺温度参数,如表1所示。
[0109]
表1:5种不同改性体系的聚酰亚胺复合材料的固化工艺温度参数
[0110]
样品命名t
i0
(℃)t
p0
(℃)t
t0
(℃)pi346393445pi-apn5%343380425pi-apn10%342379440pi-apn15%340379441pi-apn20%336381438
[0111]
其中:t
i0
表示预固化温度;t
p0
表示恒温固化温度;t
t0
表示后终止温度。
[0112]
对5个不同体系的聚酰亚胺复合材料分别进行高温流变测试,得到各体系的粘温曲线(见图7),由图7可知,随着apn-pepa组分含量的增加,共混pi体系的最低粘度逐渐降低,工艺性得到了显著改善。各体系最低粘度复合材料以及其对应的温度如表2所示。
[0113]
表2:5种不同体系的聚酰亚胺复合材料及其对应的温度
[0114][0115]
对5个不同体系的聚酰亚胺复合材料分别在氮气和空气气氛下进行tg测试(见图8),由图8可知,树脂固化物均具有较高的热分解温度和残碳率,热稳定性十分优异。各体系热分解参数如表3所示。
[0116]
表3:5种不同体系的聚酰亚胺复合材料的热分解参数
[0117][0118]
将pi和pi-apn5%体系聚酰亚胺复合材料进行红外光谱测试(见图9),由图9可知,二者吸收曲线均未出现2206cm-1
(c≡c的伸缩振动)以及2231cm-1
(c≡n的伸缩振动)的吸收峰,表明苯乙炔基和腈基已发生固化交联,形成芳杂环类的交联完固结构,树脂得到了比较充分的固化。
[0119]
性能表征
[0120]
将实施例4所制备的5种聚酰亚胺复合材料制成测试材料,具体步骤如下:
[0121]
(1)预浸料的制备:将7块大小约为95mm
×
95mm的碳纤维布平铺于大小合适的白色离型膜上,用无水乙醇提前润好的软刷将聚酰胺酸溶液均匀涂覆于每一块纤维布上,涂覆一遍后可静置20min以确保浸润良好,反复涂刷,直至5种聚酰亚胺复合材料的溶液完全浸渍碳纤维布。将涂覆完成的碳纤维布连同离型膜放入真空干燥烘箱中,设置温度程序,起始温度为40℃,每隔1h升温10℃,直至90℃,并在90℃保温10h,使得溶剂尽可能完全挥发。随炉降温至室温,即得到聚酰亚胺预浸料。
[0122]
(2)模压成型制备碳纤维/聚酰亚胺测试材料:将烘干的预浸料装入模具,压机的温度程序设定为8段,20min内加热到150℃,保温30min,除去预浸料中残余的溶剂;20min内升温到260℃,保温120min,让预聚体充分热亚胺化脱水形成聚酰亚胺树脂;20min内继续升温至330℃,保温20min,让pi树脂熔融从而与碳纤维实现充分浸润,待压机温度加热到330℃后保温5min,启动压力程序,施加2mpa,并保压至板材冷却结束;最后在20min内升温至380℃,保温120min,让复合材料能够充分固化;随后随热压机自然冷却,消除残余应力,卸载压力,即得到5个不同体系的碳纤维增强聚酰亚胺测试材料。
[0123]
(3)测试材料后固化处理:将所得的测试材料板材去除约10mm边缘余料区域,随后用砂轮切割机裁剪出至少2个大小为8mm
×
40mm的测试材料样条,放入真空马弗炉中进行后固化,温度程序设定为:60min内从室温加热到450℃,保温2h,然后随炉自然冷却,即得测试材料后固化dma测试样条。
[0124]
(4)测试材料室温力学性能试样制备:将所得的测试材料板材去除约10mm边缘余料区域,随后用砂轮切割机裁出至少5个大小为12.5mm
×
80mm的测试材料样条用于弯曲性能测试,裁出至少5个大小为10mm
×
20mm的测试材料样条用于剪切性能测试。
[0125]
将得到的大小为8mm
×
40mm的测试材料标准样条分别进行dma测试(见图10),由图10可知:5种测试材料体系初步固化后tg均在470℃以上,后固化进一步发生固化交联,tg值又得到了显著提高,均大于500℃,并随着apn-pepa组分含量的增大而增大,即表明,pi基础体系的耐热性已经十分优异,引入含腈基组分后,其耐温性又得到了进一步提升。各体系玻璃化转变温度如表4所示。
[0126]
表4:5种测试材料的玻璃化转变温度
[0127][0128][0129]
各测试材料标准样条在室温下分别进行弯曲及剪切两项力学性能测试(见图11),由图11可知,测试材料力学较为优异,且引入含腈基组分后,共混pi体系的力学性能优于纯pi体系,各体系力学性能具体数值如表5所示。
[0130]
表5:5种测试材料的力学性能测试数据
[0131]
体系弯曲强度(mpa)弯曲模量(gpa)剪切强度(mpa)pi118510066pi-apn5%129410381pi-apn10%13219777pi-apn15%14889672pi-apn20%12709066
[0132]
本发明涉及到的仪器与测试方法如下:
[0133]
非等温热分析测试(dsc):使用日本岛津生产的dsc-60测定,采用5℃/min、10℃/min、15℃/min、20℃/min四个升温速率分别对目标物进行固化行为扫描测试,通入n2保护,测试温度范围为rt-470℃。
[0134]
热重测试(tg):使用日本岛津生产的tg-60测定,升温速率为15℃/min,气氛为
air,测试温度范围为rt-800℃。
[0135]
红外分析(ir):使用nicolet ft-ir 6700型红外光谱仪测试,采用kbr压片法进行红外表征,测试范围为500cm-1
~4000cm-1
。
[0136]
核磁分析(1h-nmr):使用bruker 400mhz核磁共振波谱仪进行1h-nmr测试,采用cdcl3和cd3socd3为溶剂。
[0137]
流变分析:使用美国ta公司生产的dhr-2型流变仪进行测定聚合物流变行为,测试温度范围为200~400℃,升温速率为4℃/min。
[0138]
质谱分析(esi):使用bruker 7.0tapex iv型傅里叶变换高分辨率质谱仪对高分子单体进行测试。
[0139]
动态热机械性能分析(dma):使用mettlertoledo dma 1动态分析仪测试,标准样品大小为40mm
×
8mm
×
2mm,选用三点弯曲模式,升温速率为5℃/min,测试温度范围为rt-500℃。本发明选取损耗因子(tanδ)测试曲线的峰值温度作为聚酰亚胺复合材料的玻璃化转变温度(tg)。
[0140]
室温机械性能分析:使用instron model 5565型万能测验机测试,按照国标切割成标准样条。弯曲性能按照国标《gb/t3356-1999》单向纤维增强塑料弯曲性能试验方法,测试样品大小为80mm
×
15mm
×
2mm,拉伸速率为2.0mm/min,室温下测试5个及以上的样条,求取平均值得最终试样的弯曲强度和弯曲模量;剪切性能按照国标《jct 773-2010》纤维增强塑料短梁法,测试样品大小20mm
×
10mm
×
2mm,拉伸速率为1.0mm/min,室温下测试5个及以上的样条,求取平均值得最终试样的剪切强度。
[0141]
本说明书中各个实施例采用递进的方式描述,每个实施例重点说明的都是与其他实施例的不同之处,各个实施例之间相同相似部分互相参见即可。
[0142]
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1