1.本发明属于功能材料技术领域,尤其涉及一种单氰基乙烯的衍生物及其晶体和晶体的应用。
背景技术:2.智能材料是能够感知环境刺激,对之进行分析、判断,并采取一定措施进行适度响应的新型功能材料,如在压力、温度、光照等外界刺激条件下会产生光色、亮度、机械性能等物理化学变化,因此智能材料在生物探针、光电显示和光学存储等领域有着巨大的应用前景。
3.在有机发光领域中,压致变色材料是一种刺激响应变色材料,通过研磨、压缩、剪切等机械力作用能够可逆地改变颜色或发光,同样在受到静水压力等外部力刺激时,其自身发光性能会发生改变,在压敏元件、信息的存储显示、压致传感等领域具有广阔的应用前景。压致变色效应的本质是高压诱导分子聚集态结构或分子构象改变,从而造成电子能级的扰动、相变、晶格缺陷或分子结构异构化等现象的发生,最终影响材料的电子吸收和发射光谱的谱峰形状和位置。开发更多种类的压致变色材料,是目前需要解决的问题。
技术实现要素:4.本发明提供了一种单氰基乙烯的衍生物及其晶体和晶体的应用,能够压制变色,且在高压下的荧光发射峰位与外界压力的大小呈现线性关系,可通过荧光发射峰位的变化定量检测外界压力。
5.本发明提供了一种单氰基乙烯的衍生物,具有式i的结构:
[0006][0007]
优选的,所述r为单键、奈、蒽或芘。
[0008]
本发明提供了一种上述方案所述的单氰基乙烯的衍生物的制备方法,包括如下步骤:
[0009]
将含甲醛的化合物、2-萘乙腈、有机溶剂和弱碱混合,在保护气氛中进行克脑文格尔反应,得到单氰基乙烯的衍生物;
[0010]
所述克脑文格尔反应的温度为40~50℃,时间为45~55min。
[0011]
优选的,所述弱碱为四丁基氢氧化铵溶液;所述有机溶剂为四氢呋喃和叔丁醇。
[0012]
优选的,得到单氰基乙烯的衍生物后还包括固液分离,并将得到的固体分离物进行柱层析色谱分离,得到纯化的单氰基乙烯的衍生物。
[0013]
本发明提供了一种单氰基乙烯的衍生物晶体,由上述方案所述的单氰基乙烯的衍生物制备得到。
[0014]
优选的,包括如下步骤:
[0015]
将具有式i所示结构的单氰基乙烯的衍生物溶解于良溶剂中,得到单氰基乙烯的衍生物溶液;
[0016]
向所述单氰基乙烯的衍生物溶液中加入不良溶剂进行静置结晶,得到单氰基乙烯的衍生物晶体。
[0017]
优选的,所述良溶剂包括二氯甲烷、三氯甲烷或四氢呋喃,所述不良溶剂包括甲醇或乙醇。
[0018]
优选的,所述静置的时间为7天;所述结晶在室温和避光条件下进行。
[0019]
上述方案所述的单氰基乙烯的衍生物晶体或上述任意一项方法制备得到的单氰基乙烯的衍生物晶体在压致荧光变色材料中的应用。
[0020]
与现有技术相比,本发明的优点和积极效果在于:
[0021]
本发明提供的含单氰基乙烯的衍生物,在不改变分子结构的情况下,可通过响应诸如压力、研磨或剪切等外部机械刺激而显示出明显的荧光变化,能够作为压制荧光变色材料。同时该衍生物制备简单、快速,且产率高,适合工业化应用,可用于光电器件、变形检测、传感器和防伪纸领域。
[0022]
进一步的,将本发明提供的含单氰基乙烯的衍生物结晶得到晶体,该晶体在高压下的荧光发射峰位与外界压力的大小呈现线性关系,基于此可通过荧光发射峰位的变化定量检测外界压力。
附图说明
[0023]
图1为实施例2制备的绿色ancn晶体在原位加压和原位释压的荧光光谱图及荧光照片;
[0024]
图2为实施例4制备的蓝色cnn晶体在原位加压和原位释压的荧光光谱图及荧光照片;
[0025]
图3为实施例2制备的绿色ancn晶体和实施例4制备的蓝色cnn晶体的压力-波长曲线图;
[0026]
图4为实施例2制备的绿色ancn晶体和实施例4制备的蓝色cnn晶体在常压下的荧光发射光谱图和荧光寿命图。
具体实施方式
[0027]
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0028]
本发明提供了一种含有单氰基乙烯的衍生物,具有式i的结构:
[0029][0030]
在本发明中,所述r优选为单键、奈、蒽或芘,更优选为奈或蒽。
[0031]
本发明提供的单氰基乙烯的衍生物具有氰基扭转结构,作为压制变色材料在不改变分子结构的情况下通过响应诸如压力、研磨或剪切等外部机械刺激即可显示出明显的荧光变化。
[0032]
本发明提供了一种上述方案所述的单氰基乙烯的衍生物的制备方法,包括如下步骤:
[0033]
将含甲醛的化合物、2-萘乙腈、有机溶剂和弱碱混合,在保护气氛中进行克脑文格尔反应,得到单氰基乙烯的衍生物;
[0034]
所述克脑文格尔反应的温度为40~50℃,时间为45~55min。
[0035]
在本发明中,所述含甲醛的化合物优选为2-奈甲醛、9-蒽甲醛或1-芘甲醛。在本发明中,所述弱碱优选为四丁基氢氧化铵溶液。在本发明中,所述四丁基氢氧化铵溶液的质量浓度优选为30%。在本发明中,所述四丁基氢氧化铵的作用是提供碱性反应条件。在本发明中,所述有机溶剂优选为四氢呋喃和叔丁醇。在本发明中,所述有机溶剂作为溶剂,对其用量没有特殊限定,保证克脑文格尔反应顺利进行即可。在本发明中,所述克脑文格尔反应优选在搅拌条件下进行。本发明对所述搅拌速率没有特殊限定,保证克脑文格尔反应顺利进行即可。在本发明中,优选在油浴条件下进行克脑文格尔反应。本发明对提供保护气氛的保护气体种类没有特殊限定,采用本领域技术人员熟知的保护气体即可,具体如氮气。
[0036]
在本发明中,得到单氰基乙烯的衍生物后,优选还包括固液分离,并将得到的固体分离物进行柱层析色谱分离,得到纯化的单氰基乙烯的衍生物。在本发明中,所述固液分离的方式优选为过滤。在本发明中,所述柱层析色谱分离时采用的展开剂优选为石油醚与二氯甲烷的混合物;所述石油醚与二氯甲烷的体积比优选为1:1。
[0037]
本发明提供了一种单氰基乙烯的衍生物晶体,由上述方案所述的单氰基乙烯的衍生物制备得到。
[0038]
本发明还提供了一种上述方案所述的单氰基乙烯的衍生物晶体的制备方法,包括如下步骤:
[0039]
将具有式i所示结构的单氰基乙烯的衍生物溶解于良溶剂中,得到单氰基乙烯的衍生物溶液;
[0040]
向所述单氰基乙烯的衍生物溶液中加入不良溶剂进行结晶,得到单氰基乙烯的衍生物晶体。
[0041]
在本发明中,所述良溶剂优选包括二氯甲烷、三氯甲烷或四氢呋喃,所述不良溶剂优选包括甲醇或乙醇。
[0042]
在本发明中,所述静置的时间优选为7天;所述结晶优选在室温和避光条件下进行。在本发明中,所述室温优选为25℃。
mark ii型单反相机拍摄单氰基乙烯的衍生物晶体原位高清照片。
[0056]
在本发明中,为保证测试结果的稳定性和可靠性,选取的单氰基乙烯的衍生物晶体形状尽量保持一致,并严格控制光斑在晶体的照射区域能保持完全一致。
[0057]
为了进一步说明本发明,下面结合实施例对本发明提供的技术方案进行详细地描述,但不能将它们理解为对本发明保护范围的限定。
[0058]
实施例1
[0059]
制备单氰基乙烯的衍生物(ancn)粉末
[0060]
向圆底烧瓶中加入1.56g的9-蒽甲醛(cas号:66-99-9)、1.67g的2-萘乙腈(cas号:7498-57-9)和15ml无水四氢呋喃与15ml叔丁醇,然后在46℃的油浴与氮气保护条件下,逐滴加入2ml含质量分数为30%四丁基氢氧化铵溶液,然后将所得反应体系在冰水浴(46℃)中搅拌反应50min;反应完成后,过滤圆底烧瓶中的混合物,将所得滤饼进行柱层析色谱分离(按体积比计,展开剂为石油醚:二氯甲烷=1:1),得到2.84g绿色粉末,产率为80%。具体反应式如下所示:
[0061][0062]
实施例2
[0063]
制备绿色ancn晶体
[0064]
将5mg实施例1制备的ancn绿色粉末溶解在4ml二氯甲烷中,将所得ancn溶液置于干净干燥的10ml试管中,在所述ancn溶液上方缓慢加入1ml甲醇,然后用一小团棉花紧紧堵住试管口,再将试管放入500ml干净广口瓶中,并在广口瓶中加入100ml甲醇,同时在广口瓶外侧包裹锡纸来避光,在室温(25℃)环境中静置7天,得到若干稳定的方块状绿色ancn。
[0065]
实施例3
[0066]
制备单氰基乙烯的衍生物(cnn)粉末
[0067]
向圆底烧瓶中加入2.06g的2-奈甲醛(cas号:642-31-9)、1.67g的2-萘乙腈cas号:(7498-57-9)和15ml无水四氢呋喃与15ml叔丁醇,然后在46℃的油浴与氮气保护条件下,逐滴加入2ml含质量分数为30%四丁基氢氧化铵溶液,然后将所得反应体系在冰水浴(46℃)中搅拌反应50min;反应完成后,过滤圆底烧瓶中的混合物,将所得滤饼进行柱层析色谱分离(按体积比计,展开剂为石油醚:二氯甲烷=1:1),得到2.74gcnn白色粉末,产率为90%。
[0068]
[0069]
实施例4
[0070]
制备蓝色cnn晶体
[0071]
将5mg实施例3制备的cnn绿色粉末溶解在4ml二氯甲烷中,将所得cnn溶液置于干净干燥的10ml试管中,在所述cnn溶液上方缓慢加入1ml甲醇,然后用一小团棉花紧紧堵住试管口,再将试管放入500ml干净广口瓶中,并在广口瓶中加入100ml甲醇,同时在广口瓶外侧包裹锡纸来避光,在室温(25℃)环境中静置7天,得到若干稳定的方块状蓝色cnn晶体。
[0072]
性能测试
[0073]
1.对实施例2和4制备得到的单氰基乙烯的衍生物晶体的结构参数进行测定,具体结果如表1所示:
[0074]
表1单氰基乙烯的衍生物晶体的结构参数
[0075][0076]
[0077]
2.测试实施例2制备的绿色ancn晶体在高压下的物相变化,包括以下步骤:
[0078]
取绿色ancn晶体,要求肉眼观察透明,纯度大于99.9%,晶粒大小为0.3mm
×
0.3mm
×
0.2mm;
[0079]
采用mao式dac压机装置预压t301钢片,得到压痕;其中,mao式dac压机装置中金刚石砧面直径为400μm,t301钢片的厚度为0.24mm,压痕的厚度为50μm;
[0080]
在压痕中心钻孔,孔洞直径为140μm,将绿色ancn晶体置于孔洞中央,并将直径为10μm的红宝石微球置于绿色ancn晶体旁边,作为压力探测器;
[0081]
对绿色ancn晶体加压,压力范围为0~8.35gpa,直至检测不到荧光光谱;利用355nm dpss激光器作为激发光源,使用海洋光学qe65000型光谱仪连接nikon ti-u型倒置荧光显微镜作为检测系统,并在每测量完一个压力点就通过canon eos 6d mark ii型单反相机拍摄绿色ancn晶体原位高清照片;
[0082]
为保证测试结果的稳定性和可靠性,选取的绿色ancn晶体形状尽量保持一致,并严格控制光斑在晶体的照射区域能保持完全一致。ancn晶体在原位加压和原位释压的荧光光谱图及荧光照片如图1所示。其中图1中的a为绿色ancn晶体的原位加压荧光光谱,b为绿色ancn晶体的原位释压荧光光谱,c为绿色ancn晶体原位加压和原位释压的荧光照片,激发波长均为355nm。由图1可知,在加压过程中对绿色ancn晶体进行测试的结果显示,将绿色ancn晶体加压至2.52gpa,光谱从491nm直接突变到550nm并且荧光强度淬灭一半,同时,从荧光显微镜下的照片发现晶体形状发生了突变。在原位释压过程中,压力恢复到0.00gpa时光谱强度可恢复到初始状态,荧光发射峰为491nm。在此加压及释压过程中并未发现晶相变化。
[0083]
按照上述方法测试实施例4制备的蓝色cnn晶体在高压下的物相变化,不同之处仅在于选取蓝色cnn晶体的晶粒大小为0.3mm
×
0.2mm
×
0.2mm,压力范围为0~14.09gpa。
[0084]
cnn晶体在原位加压和原位释压的荧光光谱图及荧光照片如图2所示,其中图2中的a为蓝色cnn晶体的原位加压荧光光谱,b为蓝色cnn晶体的原位释压荧光光谱,c为蓝色cnn晶体原位加压和原位释压的荧光照片,激发波长均为355nm。由图2可知,对蓝色cnn晶体进行测试的结果显示,在0.00gpa时,发射峰位在443nm,小压力(《4.05gpa)下蓝色cnn晶体的荧光光谱变化较小;当压力继续升高到14.09gpa以上,荧光光谱上仍能采集到信号,光谱红移能达到最大峰位为559nm左右。原位释压过程时,蓝色cnn晶体的荧光强度缓慢恢复到初始的荧光强度,但其荧光发射峰位一致。在蓝色cnn晶体中,原位加压和原位释压整个过程可逆,且在此加压及释压过程中也并未发现晶相变化。
[0085]
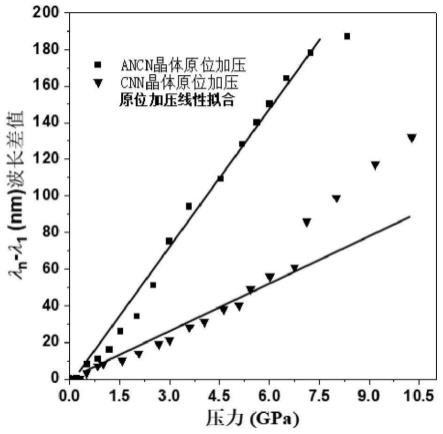
绘制实施例2中的绿色ancn晶体和实施例4中的蓝色cnn晶体的压力-波长曲线图,具体结果如图3所示。
[0086]
由图3可知,绿色ancn晶体的荧光峰位伴随着压力增加呈现出了良好的线性变化,而蓝色cnn晶体的荧光峰位伴随着压力增加也呈现出了良好的线性变化,但对比发现绿色ancn晶体对于压力变化的灵敏度更高,表现出了更大的荧光发射波长变化。
[0087]
对实施例2中的绿色ancn晶体和实施例4中的蓝色cnn晶体在常压下的荧光发射光谱和荧光寿命进行检测(荧光寿命衰减由英国爱丁堡公司的爱丁堡仪器fls920测试,并通过单光子计数技术完成。该激光器为epl皮秒脉冲激光器,激发波长为375nm),具体结果如图4所示,其中图4中的a为两种晶体的荧光发射光谱图,b为两种晶体的荧光寿命图。由图4
可知,绿色ancn晶体荧光发射峰位为491nm,蓝色cnn晶体发射峰位为443nm,而两种晶体ancn与cnn的荧光寿命并没有太大差异。
[0088]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。