Ash1p作为负调控因子在提高宿主细胞中蛋白表达中的应用
ash1p作为负调控因子在提高宿主细胞中蛋白表达中的应用
技术领域
1.本发明属于分子生物学和生物工程学领域,涉及真核基因表达的转录调控因子的应用,特别是涉及一种组成型启动子pgap的转录调控因子ash1p的应用。
背景技术:
2.启动子是调控基因表达的最重要元件之一,p
aox1
启动子(诱导型)和pgap启动子(组成型)是毕赤酵母外源蛋白表达中最具代表性的启动子。
3.甲醇诱导毕赤酵母表达系统是目前用于表达大多数异源蛋白常用的表达系统,然而并不是所有的外源蛋白均适合甲醇诱导表达而且甲醇在大规模生产时存在极大的安全隐患。相对甲醇诱导,组成型毕赤酵母表达系统表达异源蛋白时不使用甲醇诱导,但是大多数的组成型启动子强度相对较弱,无法获得较高的蛋白产量,致使其在应用上受限制。
4.针对甲醇诱导的弊端,近年来对毕赤酵母表达系统的优化改造成为了研究热点。在启动子的改造研究上,目前主要是通过各种方法构建新的启动子文库。其中,有不少研究通过在p
aox1
上删除或插入顺式作用元件、对5'utr或核心启动子区域进行点突变等来改造p
aox1
从而导致p
aox1
的激活,但是这些改造似乎不能很好地消除由高水平葡萄糖和甘油等替代碳源引起的抑制,且远达不到工业应用的水平。对于pgap文库的构建,唯一的研究是qin等通过易错pcr进行随机突变构建gap启动子文库,但是方法随机,无法解释pgap的调控。随着转录组数据分析的应用,启动子开发工作得到很大的提升。关于调控和提高p
aox1
的表达强度相关研究已有很多报道,目前研究表明,甲醇可通过调控多个反式作用元件(主要集中于p
aox1
启动子的转录调控因子或者碳源阻遏相关转录因子等)的活性或亚细胞定位,在转录水平调控p
aox1
,从而影响甲醇代谢通路相关基因的表达。尽管如此,参与p
aox1
调控的机制复杂,部分碳源的去阻遏,在蛋白表达上并未能超越传统的甲醇诱导。目前在启动子调控的研究上主要集中于paox1,而对于探索pgap的转录调控的研究,目前只有ozge ata等,通过有针对性地删除或复制转录因子结合位点(tfbs),构建启动子变异体的高表达rhgh菌株,而在公开文献中报道通过对pgap启动子的转录调控的改进,来增强pgap启动子表达目的蛋白产量的报道几乎是空白的。
5.根据文献报道,ash1p在酿酒酵母中定位于细胞核时,可以关闭ho基因的转录,影响酿酒酵母交配型的转换,通过比对,在毕赤酵母中ash1基因推定为pas_chr1-1_0414(序列号为:nc_012963.1),而目前尚未有报道ash1p对于转录调控的功能。
技术实现要素:
6.本发明的主要目的是针对宿主细胞中异源蛋白表达存在的问题和不足,提供了一种调控宿主细胞中异源蛋白表达的转录因子ash1p,能通过减少阻遏作用来增强毕赤酵母表达系统中组成型启动子pgap转录调控,进而提高目的蛋白的表达效率和产量。
7.本发明的第一个方面,提供了ash1p作为负调控因子在提高宿主细胞中蛋白表达中的应用,所述ash1p的氨基酸序列是由核苷酸序列为seq id no:1的ash1基因所编码的;
所述应用为通过敲除ash1基因从而提高宿主细胞中蛋白的表达。
8.根据本发明所述的应用,所述宿主细胞是选自:毕赤酵母、酿酒酵母、产甘油假丝酵母。
9.根据已有文献报道,毕赤酵母、酿酒酵母、产甘油假丝酵母等均可以接受以pgap为启动子的酵母表达载体,用以表达异源蛋白,因此可作为本发明的宿主细胞。
10.本发明的第二个方面,提供了一种调控宿主细胞中异源蛋白表达的基因表达盒,以pgap为启动子并敲除ash1基因。
11.本发明的第三个方面,提供了一种载体,其含有本发明所述的基因表达盒。
12.本发明的第四个方面,提供了一种宿主细胞,其含有本发明所述的基因表达盒,或者含有本发明所述的载体。
13.本发明的第五个方面,提供了一种增强宿主细胞中蛋白表达的方法,包括:提供如本发明所述的宿主细胞,在合适所述异源蛋白表达的条件下培养所述宿主细胞,从培养基中分离所表达的蛋白。
14.本发明通过实验揭示了:在具有以pgap为启动子的异源蛋白合成通路中对ash1基因进行敲除,该序列翻译后形成转录因子ash1p(ash1蛋白)参与pgap启动子的反向调控,去除其对以pgap为启动子的后续外源基因的阻遏表达,从而实现目的蛋白的非诱导大量累积。
15.本发明因此可以构建一种以pgap为启动子并敲除ash1基因的基因表达盒,ash1转录抑制因子的敲除能够增强毕赤酵母组成型启动子pgap启动子的转录从而使基因表达盒中的外源基因在毕赤酵母(或其他可以接受以pgap为启动子的酵母)中高效表达,可以显著提高外源基因的表达强度。
附图说明
16.图1是ash1基因敲除表达盒图谱;a图为kan基因替换敲除表达盒;b图为完全敲除表达盒。
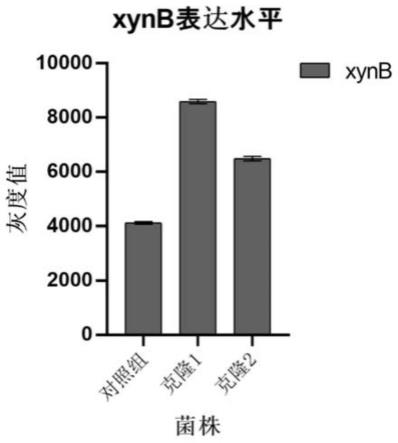
17.图2是本发明菌株(毕赤酵母)表达木聚糖酶xynb的增长率,图中对照组为未敲除菌株;克隆1和克隆2为ash1基因敲除菌株。
具体实施方式
18.以下将结合具体实施例对本发明的技术方案做更进一步的详细说明。应当理解,下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
19.除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。下列实施例所采用的分子克隆技术参见j.萨母布鲁克等编的《分子克隆实验指南》中所述的条件,或按照制造厂商所建议的条件。
20.本文中,ash1p表示ash1基因表达的蛋白。
21.实施例1:kan基因替换转录抑制因子ash1基因敲除表达盒的构建1、上游同源臂的扩增
22.本发明以毕赤酵母的基因组序列作为参考,采用软件dnaman 8设计并合成两条寡
核苷酸引物,通过pcr法扩增ash1(seq id no:1)上游同源臂序列。
23.两条pcr引物如下:
24.1-f:acttacgagctcgacaggcaaatcattca(seq id no:4)
25.1-r:acttacgcggatccgcgcgtatttagctacaatc(seq id no:5)
26.下划线碱基分别为saci、bamhi限制性内切酶酶切位点。
27.pcr反应体系如下表1。
28.表1:
29.组分体积(μl)2
×
q5 master mix12.5primer f(10μm)1.25primer r(10μm)1.25模板dna1(100ng)ddh2o9ul总体积25ul
30.pcr程序设定如下表2。
31.表2:
[0032][0033]
pcr扩增产物经1%的琼脂糖凝胶电泳后,用dna回收试剂盒进行胶回收,得到大约729bp的片段。
[0034]
2、下游同源臂的扩增
[0035]
本发明以毕赤酵母的基因组序列作为参考,采用软件dnaman 8设计并合成两条寡核苷酸引物,通过pcr法扩增ash1下游同源臂序列。
[0036]
两条pcr引物如下:
[0037]
2-f:acttaccggaattccggtcactaactgtatact(seq id no:6)
[0038]
2-r:acttatttgcggccgctttaacaatcaagaggacatactt(seq id no:7)
[0039]
下划线碱基分别为ecori、noti限制性内切酶酶切位点。
[0040]
pcr反应体系与pcr程序设定如上所述的上游同源臂的扩增方法。
[0041]
pcr扩增产物经1%的琼脂糖凝胶电泳后,用dna回收试剂盒进行胶回收,得到大约745bp的片段。
[0042]
3、kan基因的扩增
[0043]
本发明以ppic3.5k质粒(来源于invitrogen公司)的kan基因(卡那霉素抗性基因,
该基因在真核生物中表达可以使细胞表现出g418抗性)作为参考,采用软件dnaman 8设计并合成两条寡核苷酸引物,通过pcr法扩增kan基因。
[0044]
两条pcr引物如下:
[0045]
3-f:acttacgcggatccgcgatgagccatattcaac(seq id no:8)
[0046]
3-r:acttaccggaattccggttagaaaaactcatcgag(seq id no:9)
[0047]
下划线碱基分别为bamhi、ecori限制性内切酶酶切位点。
[0048]
pcr反应体系与pcr程序设定如上述。
[0049]
pcr扩增产物经1%的琼脂糖凝胶电泳后,用dna回收试剂盒进行胶回收,得到大约816bp的片段。
[0050]
4、ppic3.5k载体与kan片段的连接
[0051]
将ppic3.5k质粒与kan基因pcr产物分别用限制性内切酶ecori、bamhi于37℃酶切20min,酶切条件如下表3。
[0052]
表3:
[0053]
组分体积(μl)10
×
cutsmart缓冲液1plasmid1(约200ng)ecori、bamhi0.2ddh2o7.6总体积10
[0054]
酶切产物经1%的琼脂糖凝胶电泳后分别回收两个目的片段,用t4dna连接酶连接,连接体系如下表4。
[0055]
表4:
[0056][0057][0058]
用连接酶16℃连接12h,连接产物转化dh5a感受态细胞扩增后用质粒抽提试剂盒抽提质粒,用ecori和bamhi双酶切后跑电泳结果显示有9kb和816bp两条带,表明连接成功,通过dna测序,确定为kan基因。
[0059]
5、上游同源臂片段与ppi3.5k-kan载体连接
[0060]
上游同源臂片段与ppi3.5k-kan载体分别用限制性内切酶saci、bamhi内切酶双酶切并纯化回收获得。
[0061]
上游同源臂片段与ppi3.5k-kan载体连接方法同步骤4所示,用saci、bamhi双酶切后跑电泳结果显示有9kb和750bp两条带,表明连接成功,通过dna测序,确定为上游同源臂
片段(序列为seq id no:2)。
[0062]
6、下游同源臂片段与ppic3.5k-(上游同源臂)-kan载体连接
[0063]
下游同源臂片段与ppic3.5k-(上游同源臂)-kan载体分别用限制性内切酶ecori、noti内切酶双酶切并纯化回收获得。
[0064]
下游同源臂片段与ppic3.5k-(上游同源臂)-kan载体连接方法同步骤4所示,用ecori、noti双酶切后跑电泳结果显示有8kb和750bp两条带,表明连接成功,通过dna测序,确定为下游同源臂片段(序列为seq id no:3)。
[0065]
由此kan基因替换转录抑制因子ash1基因敲除表达盒构建成功(如图1的a图所示)。
[0066]
实施例2:转录抑制因子ash1基因完全敲除表达盒的构建
[0067]
如实施例1中的方法,使用引物1-f、1-r以及2-f、2-r扩增ash1基因的上下的同源臂,并将上下游同源臂片段通过上述酶切连接的方法与ppic3.5k载体连接,构建ppic3.5k-(上游同源臂)-(下游同源臂)敲除表达盒。
[0068]
实施例3:毕赤酵母基因组ash1敲除
[0069]
为了提高单拷贝表达盒在毕赤酵母染色体上的整合效率,用限制性内切酶saci、noti将敲除表达盒线性化并用试剂盒纯化回收。本实验的受体菌为毕赤酵母smd1168(已含在pgap之后插入木聚糖酶xynb基因的重组菌,ex6),实施例1敲除表达盒电转化后使用含0.3mg/ml的g418平板进行筛选,并提基因组pcr鉴定。实施例2敲除表达盒电转化后使用ypg平板进行筛选,并提基因组pcr鉴定。
[0070]
通过pcr产物测序结果显示筛选到的菌株为成功敲除了ash1的阳性克隆。
[0071]
实施例4:ash1敲除菌株中组成型启动子pgap所驱动的异源蛋白表达测定
[0072]
将实施例1筛选到的阳性克隆1和实施例2筛选到的阳性克隆2进行发酵,发酵条件为28℃,200rpm,培养72h。同时以含有xynb基因的毕赤酵母(ex6)作为对照,培养72h后取上清进行sds-page电泳检测,分析木聚糖酶xynb表达水平。
[0073]
结果如图2所示,敲除ash1p转录因子后所挑取的阳性克隆1相比于对照组的表达量提高了109%,阳性克隆2相比于对照组的表达量提高了58%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1