季铵盐类化合物或其盐及其用途,区分2位羟基取代脂肪酸和3位羟基取代脂肪酸的方法
1.本发明涉及一种季铵盐类化合物或其通过氨基与酸配对形成的盐及其用途,特别涉及将该季铵盐类化合物用作衍生化试剂,利用质谱分析区分2位羟基取代脂肪酸和3位羟基取代脂肪酸的方法。
背景技术:
2.脂肪酸(fa)因其在很多生理过程中的关键作用而一直吸引着科学家的兴趣。近年来,有报道显示多不饱和脂肪酸(pufa)和含羟基脂肪酸(ohfa) 也是重要的生物活性分子,它们在许多疾病中显示出显著的生物活性,包括炎症、糖尿病、肝损伤,还有一些癌症等。随着链长、双键或羟基的数量和位置不同,不同的pufa、ohfa可以发挥完全不同的活性。花生四烯酸(aa,c20:4) 等n-6pufa是生物系统中重要的免疫和促炎因子;具有更高不饱和度的二十碳五烯酸(epa,c20:5)和二十二碳六烯酸(dha,c22:6)都属于n-3pufa,是大脑功能的重要参与者;由于epa和dha可以抑制n-6pufas的体内生物合成,它们也具有抗炎活性。
3.2位羟基取代脂肪酸(以下也记做2-ohfa)和3位羟基取代脂肪酸(以下也记做3-ohfa)是两类较为特殊的含羟基脂肪酸,它们与脂肪酸α-氧化和β-氧化密切相关,这也表明2/3-ohfa在体内能量代谢过程中可能发挥重要作用。
4.更重要的是,有报道显示(r)-2-ohfa以及不饱和的2-ohfa对肿瘤细胞生长有很好的抑制作用(参见非专利文献:sun,l.;yang,x.;huang,x.;yao,y.; wei,x.;yang,s.;zhou,d.;zhang,w.;long,z.;xu,x.;zhu,x.;he,s.;su,x. cancer res 2021,81,289-302.)。
5.然而,如何区分和鉴别2-ohfa和3-ohfa至今仍然缺乏有效的手段,两者在主要红外、核磁共振、紫外等主要波谱学数据方面都表现得比较相似,难以区分。这一难题,在2-ohfa和3-ohfa中存在位置未知的不饱和键时(即不饱和的2-ohfa和3-ohfa,以下也分别写做2-ohufa和3-ohufa)更加难以解决,虽然近些年来,液相色谱-质谱(lc-ms)联用技术因其良好的灵敏度、能够准确提供化合物的质荷比、离子碎片和保留时间等诊断信息的能力而被广泛用于各种类型化合物的结构鉴定。然而,因为2/3-ohufa标准品合成难度大以及质谱上电离效率较低等问题,很难通过lc-ms建立相应的标准品数据库,从而区分和鉴定2/3-ohufa;此外,通过lc-ms直接判断 2/3-ohufa中oh基团的位置也很困难,因为2/3-ohufa在质谱上得到的离子碎片不能提供有效的官能团信息。不仅如此,生物体内含羟基脂肪酸的含量远远低于其他脂肪酸,这也给含羟基脂肪酸的鉴定增加了很多难度。这对于深入研究2/3-ohfa和2/3-ohufa在生物体内的生理活性带来了较大困难,由于缺乏有效的分析方法,2/3-ohufa的生物学活性研究也一直进展缓慢。
6.近年来,科学家们已经发展出了很多方法来尝试解决2/3-ohfa(包括 2/3-ohufa)的分析问题。由于上述的那些问题,通过lc-ms直接分析 2/3-ohfa受到了很大的限制;迄今为止,对ohfa进行快速、全面的分析仍然是一个巨大的挑战。
技术实现要素:
7.本发明中,发明人发现了一种衍生化试剂,其为一类季铵盐类化合物,可以通过使其与脂肪酸样品反应,将衍生化产物进行质谱检测,通过碎裂产生的特征离子碎片识别2/3-oh的位置,从而快速地区分该样品是2-ohfa还是 3-ohfa。本发明的衍生化试剂还可以用于从其他异构体中识别出2-ohfa或者3-ohfa。此外,同位素标记的本发明的季铵盐类化合物(例如d3标记),可以用于快速区分复杂样本中的低含量2/3-ohfas。
8.具体而言,本发明提供一种式(1)所示的季铵盐类化合物或其通过氨基与酸配对形成的盐,
[0009][0010]
其中,
[0011]
r1、r2各自独立地为饱和或不饱和的c1~c6的烷基、饱和或部分不饱和的c3-6环烃基、饱和或部分不饱和的3~10元杂环基或者c6~10芳基;
[0012]
x-表示抗衡阴离子;
[0013]
a和b各自独立地为直链或支链的c3~c10的亚烷基链,其中所述直链或支链的c3~c10的亚烷基链可选地被一或多个选自-o-、-co-、-c(=o)o-、-conh-、-nhco-、-nhconh-、-nh-、-nr
3-、-c(r3)
2-、-s-、亚磺酰基、磺酰基、亚磺酰氧基、磺酰氧基、-氨基磺酰基氨基-、亚炔基、亚烯基、亚环烷基、或它们的任意组合中的二价基团中断一或多次,其中,r3为h、c1~c6 烷基、饱和或部分不饱和的c
3-6
环烃基、饱和或部分不饱和的3-10元杂环基或者c6-10芳基。
[0014]
在本发明优选的实施方式中,式(i)所示的化合物为式(ii)所示的季铵盐类化合物,
[0015][0016]
其中,
[0017]
r1、r2各自独立地为c1-c6的烷基;
[0018]
x-表示抗衡阴离子;
[0019]
m和n各自独立的表示1~6的整数。
[0020]
在本发明优选的实施方式中,其为式(i)的化合物通过氨基与酸配对形成的盐,所述酸为选自氢氟酸、盐酸、氢溴酸、氢碘酸、磷酸、甲酸、乙酸、三氟乙酸、草酸、硫酸、硝酸、甲磺酸、三氟甲磺酸、胺基磺酸中的无机酸,或者选自马来酸、柠檬酸、乳酸、酒石酸、琥珀酸中
的有机酸,x-中的x为卤素原子。
[0021]
在本发明优选的实施方式中,其中,r1、r2各自独立地为c1-c3的烷基,
[0022]
进一步优选地,r1、r2各自独立地为甲基或者乙基,
[0023]
更进一步优选地r1、r2都为甲基。
[0024]
在本发明优选的实施方式中,x为氯、溴或碘。
[0025]
在本发明优选的实施方式中,m=1且n=1,或者m=2且n=2。
[0026]
本发明的另一个方面,提供本发明所述的季铵盐类化合物或其通过氨基与酸配对形成的盐作为脂肪酸衍生化试剂用于在质谱分析中区分2位羟基取代脂肪酸和3位羟基取代脂肪酸的用途。
[0027]
本发明的另一个方面,提供本发明所述的季铵盐类化合物或其通过氨基与酸配对形成的盐,作为脂肪酸衍生化试剂用于在质谱分析中鉴别2位羟基取代脂肪酸或3位羟基取代脂肪酸的用途。
[0028]
本发明的另一个方面,提供一种基于质谱分析区分2位羟基取代脂肪酸和 3位羟基取代脂肪酸的方法,其使用本发明所述的季铵盐类化合物或其通过氨基与酸配对形成的盐,作为脂肪酸衍生化试剂,通过质谱检测的碎片的m/z值,区分2位羟基取代脂肪酸和3位羟基取代脂肪酸。
[0029]
本发明的优选实施方式中,区分2位羟基取代脂肪酸和3位羟基取代脂肪酸的方法包含以下工序:
[0030]
s1衍生化工序:在碱性环境下,在多肽缩合剂的存在下,使待测样品与权利要求1所述所述的季铵盐类化合物或其通过氨基与酸配对形成的盐发生缩合反应,得到衍生化产物;
[0031]
s2质谱检测工序:将s1中获得的衍生化产物,进行质谱检测;
[0032]
s3质谱数据分析工序:在二级质谱的离子碎片中,寻找特征碎片,当存在[式(i)的分子量-x的分子量+26]的m/z值的二级碎片时判断为存在2位羟基取代脂肪酸,当存在[式(i)的分子量-x的分子量+43]的m/z值的二级碎片时判断为存在3位羟基取代脂肪酸。
[0033]
经由本发明的化合物进行衍生化的2-ohfa或3-ohfa在较高的碰撞能量诱导下能够分别产生特征离子碎片;这两个特征离子能够特异性区分脂肪酸是 2位取代的羟基还是3位取代的羟基,由此实现了完全不依赖标准品的 2/3-ohfa快速且全面的鉴定,本发明化合物和鉴别方法同样适合2/3羟基取代不饱和脂肪酸的区分。
附图说明
[0034]
图1为表示化合物1和化合物2的结构式的图;
[0035]
图2为表示实施例1中的衍生化产物的质谱分析结果的示意图;
[0036]
图3为表示实施例1中的衍生化产物的质谱分析结果的说明图;
[0037]
图4为将化合物1用于其他羟基位置取代的脂肪酸时的质谱分析的结果的图;
[0038]
图5为比较例2的质谱分析结果的图;
[0039]
图6为小鼠肿瘤转移组与空白对照组衍生化之后的质谱分析结果图。
具体实施方式
[0040]
以下更加详细的说明本发明。
[0041]
本发明提供一种式(1)所示的季铵盐类化合物或其通过氨基与酸配对形成的盐,
[0042][0043]
其中,
[0044]
r1、r2各自独立地为饱和或不饱和的c1~c6的烷基、饱和或部分不饱和的c3-6环烃基、饱和或部分不饱和的3~10元杂环基或者c6~10芳基;
[0045]
x-表示抗衡阴离子;
[0046]
a和b各自独立地为直链或支链的c3~c10的亚烷基链,其中所述直链或支链的c3~c10的亚烷基链可选地被一或多个选自-o-、-co-、-c(=o)o-、-conh-、-nhco-、-nhconh-、-nh-、-nr
3-、-c(r3)
2-、-s-、亚磺酰基、磺酰基、亚磺酰氧基、磺酰氧基、-氨基磺酰基氨基-、亚炔基、亚烯基、亚环烷基、或它们的任意组合中的二价基团中断一或多次,其中,r3为h、c1~c6 烷基、饱和或部分不饱和的c
3-6
环烃基、饱和或部分不饱和的3-10元杂环基或者c6-10芳基。
[0047]
本发明中,ca~cb的表达方式代表该基团具有的碳原子数为a~b,除非特殊说明,一般而言该碳原子数不包括取代基的碳原子数。本发明中,对于化学元素的表述,若无特别说明,通常包含化学性质相同的同位素的概念,例如“氢”的表述,也包括化学性质相同的“氘”、“氚”的概念。
[0048]
因此,所谓的c1~c6的烷基,例如可举出甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、2-甲基丁基、正戊基、仲戊基、新戊基、正己基、新己基、正庚基等,但是并不限于这些其中,优选为c1~c5的烷基,作为优选的基团可举出甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、2-甲基丁基、正戊基、仲戊基、新戊基等。
[0049]
所谓的亚烷基,是指上述的烷基再去除掉一个氢原子而成的二价基团,例如可举出,亚甲基、亚乙基、亚丙基、亚丁基等。
[0050]
所谓的c6~c10的芳基,可以举出苯基、萘基、蒽基、菲基等,其中优选苯基。
[0051]
所谓的c6~c10的亚芳基,可举出上述c6~c10的芳基再去除掉一个氢原子而成的二价基团,其中优选亚苯基、亚萘基。
[0052]
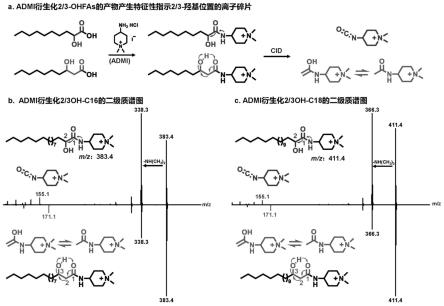
本发明的发明人发现,具有环状结构的季铵盐结构在质谱裂解中的稳定性较好,将其与脂肪酸通过氨基形成肽键,在二级质谱中的裂解规律有迹可循,尤其是当脂肪酸的2位取代羟基,或者3位取代羟基时,其与具有环状结构的季铵盐结构衍生化试剂连接后,在以标准品2-羟基十六酸(2-oh-c16)为例,相应的利用本发明化合物进行衍生化的衍生产物在专属性二级质谱(ms2)中能够产生m/z[式(i)的分子量-x的分子量+26]的离子碎片,这是由oh附着的碳原子与临近羰基碳原子之间的键断裂产生的,因此能够指向2-oh基团的位置;利用本发明的化合物进行衍生化的3-羟基十六酸(3-oh-c16)在ms2 中能够产生m/z[式
(i)的分子量-x的分子量+43]的离子碎片。因此能够指区分羟基具体是取代在脂肪酸的2位还是3位。
[0053]
如果详细说明,当使用合成例1中合成的4-氨基-1,1-二甲基哌啶碘化物 (本发明中也称之为admi),以标准品2-羟基十六酸(2-oh-c16)为例,相应的admi衍生产物在专属性二级质谱(ms2)中能够产生m/z 155.1的离子碎片,这是由oh附着的碳原子与临近羰基碳原子之间的键断裂产生的,因此能够指向2-oh基团的位置(可以参照图2);admi衍生化的3-羟基十六酸 (3-oh-c16)在ms2中能够产生m/z 171.1的离子碎片,这是由oh附着的碳原子与靠近羧基的邻近碳原子之间键断裂产生的,因此能够指向3-oh基团的位置(具体的裂解方式分析,可以参照图2)。
[0054]
本发明的化合物中季铵盐阳离子用于在质谱碎片中方便的最终的所需的阳离子,与季铵盐中的n连接的r1、r2可以为饱和或不饱和的c1~c6的烷基、饱和或部分不饱和的c
3-6
环烃基、饱和或部分不饱和的3~10元杂环基或者c6~10芳基,但是从合成的容易程度,以及从裂解中的稳定性好的观点出发,优选的是r1、r2为饱和的c1~c6的烷基,
[0055]
式(i)的化合物的环状结构至关重要。发明人也合成了如下的非环状结构的季铵盐,其同样具备氨基和季铵盐阳离子部分。
[0056][0057]
但是利用与本发明同样的方法对2/3羟基取代的脂肪酸进行区分和鉴别时,无法形成特征的离子碎片,无法进行区分和鉴别。由此可推知环状结构对于衍生物产生特征性的裂解行为的重要性。通过实施例的几种环状结构产生特征性二级质谱离子碎片的规律,可以确定环结构是本发明中必须的。
[0058]
x-表示抗衡阴离子,可列举质子酸的共轭碱。作为质子酸的共轭碱的具体例子,可列举卤离子、硫酸根离子、硝酸根离子、碳酸根离子、高氯酸根离子、四氟硼酸根离子、四(五氟苯基)硼酸根离子、六氟磷酸根离子、甲烷磺酸根离子以及三氟乙酸根离子。优选的是卤离子,即x-中的x为卤素原子。
[0059]
氨基可以与各种酸配对形成盐的形式保存,所谓的酸只要是在进行衍生化时不影响衍生化反应即可,例如所述酸为选自氢氟酸、盐酸、氢溴酸、氢碘酸、磷酸、甲酸、乙酸、三氟乙酸、草酸、硫酸、硝酸、甲磺酸、三氟甲磺酸、胺基磺酸中的无机酸,或者选自马来酸、柠檬酸、乳酸、酒石酸、琥珀酸中的有机酸。
[0060]
氨基用于与脂肪酸的羧基缩合从而完成衍生化。氨基与脂肪酸的缩合,等同于多肽合成过程中,肽键合成工序。该工序一般在碱性条件下,利用多肽缩合剂催化来实现。本发明的季铵盐化合物与脂肪酸的衍生化反应是公知技术,本领域人员可以根据需要选择脂肪酸与本发明的季铵盐化合的缩合反应方式。
[0061]
所谓的多肽缩合剂,没有特别的限定,可以使用公知的多肽缩合剂,例如可举出碳二亚胺型、磷正离子型和脲正离子型。作为碳二亚胺型缩合剂,例如可举出n,n'-二环己基碳二亚胺(dcc)、n,n'-二异丙基碳二亚胺(dic)、 dcc-hobt复合缩合剂、dcc-hoat复合缩合剂等,其中的hobt,hoat是缩合活化剂。作为磷正离子型缩合剂,例如可举出卡特缩合剂bop、pybop、 aop、pyaop、brop、pyclop、pybrop等。作为脲正离子型缩合剂,例如可举出
tbtu、hbpyu、hatu、totu、pyciu、tffh、btffh、cip等。本发明中,优选使用反应速度快,产物消旋小、收率高的脲正离子型缩合剂,进一步优选使用hatu(化合物名为:o-(7-氮杂苯并三唑-1-基)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯)。
[0062]
缩合反应一般在弱碱环境使用,可以使用有机碱,例如dipea、三乙胺, 也可以使用碳酸氢钠。溶剂可以使用任意溶剂,例如dmf、二氯甲烷、乙腈,从直接用于质谱检测的便利性角度出发,优选使用乙腈作为溶剂。
[0063]
式(i)的化合物中的a和b是形成环的连接基团,其各自独立地为直链或支链的c3~c10的亚烷基链,其中所述直链或支链的c3~c10的亚烷基链可选地被一或多个选自-o-、-co-、-c(=o)o-、-conh-、-nhco-、-nhconh-、
ꢀ‑
nh-、-nr
3-、-c(r3)
2-、-s-、亚磺酰基、磺酰基、亚磺酰氧基、磺酰氧基、
‑ꢀ
氨基磺酰基氨基-、亚炔基、亚烯基、亚环烷基、或它们的任意组合中的二价基团中断一或多次,其中,r3为h、c1~c6烷基、饱和或部分不饱和的c
3-6
环烃基、饱和或部分不饱和的3-10元杂环基或者c6-10芳基。其中优选为饱和的亚烷基链c1~c3的亚烷基链,a和b可以相同也可以不同。
[0064]
作为进一步优选的本发明的化合物,式(i)所示的化合物为式(ii)所示的季铵盐类化合物,
[0065][0066]
其中,
[0067]
r1、r2各自独立地为c1-c6的烷基;
[0068]
x-表示抗衡阴离子;
[0069]
m和n各自独立的表示1~6的整数。
[0070]
在本发明优选的实施方式中,其中,r1、r2各自独立地为c1-c3的烷基,
[0071]
进一步优选地,r1、r2各自独立地为甲基或者乙基,
[0072]
更进一步优选地r1、r2都为甲基。
[0073]
在本发明优选的实施方式中,x为氯、溴或碘。
[0074]
在本发明优选的实施方式中,m=1且n=1,或者m=2且n=2。
[0075]
从合成容易性,通用性角度出发,作为本发明最优选的化合物的例子,可以举出以下结构的化合物,但本发明不限于以下的化合物。
[0076][0077]
本发明所述的季铵盐类化合物或其通过氨基与酸配对形成的盐作为脂肪酸衍生化试剂用于在质谱分析中区分2位羟基取代脂肪酸和3位羟基取代脂肪酸的用途。
[0078]
另外,本发明也可以用于复杂混合物样品中,检测2位羟基取代脂肪酸或 3位羟基
取代脂肪酸的有无。通过本发明所述的季铵盐类化合物或其通过氨基与酸配对形成的盐作为脂肪酸衍生化试剂,使其与复杂样品反应,将产物直接进入质谱,或者利用lc-ms进行检测,如果检测到m/z[式(i)的分子量-x 的分子量+26]的离子碎片,或者[式(i)的分子量-x的分子量+43]的离子碎片,则可以证明复杂混合物样品中2位羟基取代脂肪酸和3位羟基取代脂肪酸的有无。
[0079]
综上所述,本发明能够提供一种基于质谱分析区分2位羟基取代脂肪酸和 3位羟基取代脂肪酸的方法,其使用本发明所述的季铵盐类化合物或其通过氨基与酸配对形成的盐,作为脂肪酸衍生化试剂,通过质谱检测的碎片的m/z值,区分2位羟基取代脂肪酸和3位羟基取代脂肪酸。本发明区分2位羟基取代脂肪酸和3位羟基取代脂肪酸的方法可以包含以下工序,但是并不限于以下的工序:
[0080]
s1衍生化工序:在碱性环境下,在多肽缩合剂的存在下,使待测样品与权利要求1所述所述的季铵盐类化合物或其通过氨基与酸配对形成的盐发生缩合反应,得到衍生化产物;
[0081]
s2质谱检测工序:将s1中获得的衍生化产物,进行质谱检测;
[0082]
s3质谱数据分析工序:在二级质谱的离子碎片中,寻找特征碎片,当存在[式(i)的分子量-x的分子量+26]的m/z值的二级碎片时判断为存在2位羟基取代脂肪酸,当存在[式(i)的分子量-x的分子量+43]的m/z值的二级碎片时判断为存在3位羟基取代脂肪酸。
[0083]
本发明的所谓的质谱检测,只要是能进行二级质谱检测的仪器检测手段都能使用。由于得到的衍生化产物结构中含有正电荷,因此衍生化产物可通过各种商业上常用的电离方式实现离子化,包括电子轰击电离(ei)、化学电离(ci)、解吸电离(di)、喷雾电离(si)等。本发明中,优选的质谱分析仪器可以是超高性能液相色谱-四极杆串联飞行时间(uplc-q/tof)质谱系统,但是并不限于该系统,只要能进行二级质谱检测的质谱系统均可使用。本发明的衍生化产物能够产生非常明确的指示2位或3位羟基位置的特征裂解碎片,具有很强专属性,因此可以通过直接比较离子碎片确定未知含羟基脂肪酸的羟基位置,而不一定需要通过液相色谱比较相应标准品的保留时间。因此在本发明设计的分析流程中,可以直接进样,也可以经由高效液相色谱之后进样。
[0084]
上述的碱性环境以及多肽缩合剂等的发明要素,在上述本发明的化合物的说明中详述,在此不赘述。
[0085]
实施例
[0086]
以下通过具体的实施例说明本发明的制备方法,以下实施例用于说明本发明,但不用来限制本发明的范围。提供本发明的实施例是为了向本领域技术人员更充分地描述本发明,并且以下实施例可以修改为各种其他配置,并且本发明的范围如下但是,本发明不限于该实施例。相反,提供这些实施例是为了使本公开彻底和完整,并且将本发明的精神完全传达给本领域技术人员。术语“和/或”包括一个或多个所列项目的任何和所有组合。在以下附图中,清楚的方便起见,厚度或尺寸可能被夸大描述。
[0087]
仪器和试剂:所有实验均在超高性能液相色谱-四极杆串联飞行时间 (uplc-q/tof)质谱系统(h-class uplc、synapt g2 si ms,waters,usa) 上进行。采用waters acquity uplc beh c8柱(2.1
×
100mm,1.8μm)进行分离;柱温保持在45℃,自动进样器温度设置为15℃,流速为0.4ml/min。流动相a和b分别为含0.1%甲酸的水和含0.1%甲酸的乙腈
(acn),梯度设置如下:0-8分钟,5%-95%b;8-10分钟,95%b;10-12分钟,5%b。质谱的设置如下:负离子模式的电喷雾离子源电压为-2800v,正离子模式的电喷雾离子源电压为3300v,源温度为120℃,去溶剂气体流速为750l/h。
[0088]
实施例中所使用的试剂购自aldrich chemical company、上海毕得医药科技有限公司、上海国药试剂有限公司、百灵威试剂有限公司、韶远科技(上海) 有限公司或艾柏科技有限公司等公司。
[0089]
衍生试剂的合成
[0090]
合成例1:4-氨基-1,1-二甲基哌啶-1-碘化铵盐酸盐(admi)的合成
[0091][0092]
将(1-甲基哌啶-4-基)氨基甲酸叔丁酯(395mg,1.8mmol)溶于二氯甲烷(4ml)中,然后加入碘甲烷(0.135ml,2.16mmol),并室温反应过夜。减压蒸干得黄色胶状物,即为4-((叔丁氧羰基)氨基)-1,1-二甲基哌啶-1-碘化铵。
[0093]
将上述产物溶于甲醇(4ml),加入浓盐酸(0.3ml),然后于50℃搅拌反应3h,通过lc-ms监测反应进程。反应完成后,减压蒸干溶剂,粗产品以无水乙醇重结晶得370mg淡黄色固体,即为目标化合物例4(admi)。1h nmr (600mhz,dmso-d6)δ7.91(s,3h,-nh2hcl),3.49(m,2h),3.43(m,2h),3.31 (m,1h),3.14(s,3h),3.04(s,3h),2.04(m,2h),1.97(m,2h);
13
c nmr(151 mhz,meod)δ60.03,54.91,46.91,44.75,23.95;ms(esi):m/z=129.3[m-i
-
- hcl]
+
。
[0094]
合成例2化合物2的合成
[0095][0096]
化合物2合成方法与化合物相似,只是用n,n-二甲基三甲基二胺取代 1-甲基-4-哌啶胺。1h nmr(600mhz,dmso-d6)δ3.2(s,9h,),3.5(m,2h),2.19 (m,2h),3.06(td,2h,j=7.6,1.6);
13
c nmr(151mhz,meod)δ53.9,64.4,22.6, 37.7;ms(esi):m/z=117.1[m-i
-
-hcl]
+
。
[0097]
化合物1和化合物2两种衍生化试剂的结构展示于图1。
[0098]
合成例3:4-氨基-1,1-二甲基哌啶-1-溴化铵氢溴酸盐的合成
[0099][0100]
将4-氨基-1-甲基哌啶(1g,8.77mmol)溶于二氯甲烷(10ml)中,加入二碳酸二叔丁酯(2.32ml,10.10mmol)和三乙胺(1.58ml,11.40mmol),于常温搅拌反应8h。用tlc监测至原料反应完毕后,加入饱和碳酸氢钠水溶液淬灭反应,分出二氯甲烷相,以饱和氯化钠水溶液清洗三次,无水硫酸镁干燥,随后减压浓缩,通过柱色谱法(二氯甲烷/甲醇=10/1)纯化,得到淡黄色固体 1.2g,即为(1-甲基哌啶-4-基)氨基甲酸叔丁酯。1h nmr(500mhz, chloroform-d)δ4.44(s,1h),3.44(m,1h),277(m,2h),2.27(s,3h),2.06(t,2h, j=11.4),
1.92(m,2h),1.47(m,2h),1.43(s,9h);ms(esi):m/z=215.2[m+h]
+
。
[0101][0102]
将(1-甲基哌啶-4-基)氨基甲酸叔丁酯(200mg,0.93mmol)溶于二氯甲烷(4ml)中,置于-20℃搅拌15分钟,然后将溴甲烷(0.16ml,5.61mmol) 以注射器加入到反应体系中,并于-20℃搅拌反应过夜。有白色固体析出,过滤,收集滤饼,用少量二氯甲烷冲洗,40℃真空干燥3h得到白色固体226mg,即为4-((叔丁氧羰基)氨基)-1,1-二甲基哌啶-1-溴化铵。1h nmr(500mhz, methanol-d4)δ3.67(m,1h),3.58
–
3.40(m,4h),3.18(s,3h),3.15(s,3h),2.16
–ꢀ
2.02(m,2h),1.98
–
1.84(m,2h),1.45(s,9h);ms(esi):m/z=229.2[m-br
-
]
+
。
[0103][0104]
将4-((叔丁氧羰基)氨基)-1,1-二甲基哌啶-1-溴化铵(100mg,0.44mmol) 溶于甲醇(4ml)中,加入氢溴酸(0.5ml),于50℃搅拌反应2h,通过lc-ms 监测反应进程。反应完成后,旋干得到白色固体93mg,即为目标化合物例1。1h nmr(500mhz,dmso-d6)δ8.13(s,3h,-nh2hbr),3.54(m,2h),3.48(m, 2h),3.36(m,1h),3.14(s,3h),3.08(s,3h),2.10(m,4h);ms(esi):m/z=129.3[m-br
-
-hbr]
+
。
[0105]
合成例4:4-氨基-1-乙基-1-甲基哌啶-1-碘化铵盐酸盐的合成
[0106][0107]
将原料(1-甲基哌啶-4-基)氨基甲酸叔丁酯(415mg,1.94mmol)溶于二氯甲烷(8ml)中,加入碘乙烷(0.64ml,8.53mmol),于室温搅拌过夜,通过lc-ms监测反应进程。反应完成后,旋干得到黄色固体710mg,即为4-((叔丁氧羰基)氨基)-1-乙基-1-甲基哌啶-1-碘化铵。1h nmr(500mhz,methanol-d4) δ3.71(m,1h),3.52(m,4h),3.43(m,2h),3.09/3.09(2
×
s,3h),2.11(m,2h), 1.95(m,2h),1.47(s,9h),1.42/1.39(2
×
t,3h,j=7.0);ms(esi):m/z=243.3[m-i -
]
+
。
[0108][0109]
将4-((叔丁氧羰基)氨基)-1-乙基-1-甲基哌啶-1-碘化铵(300mg,1.24mmol) 溶于无水甲醇(4ml)中,加入1ml浓度为2m的氯化氢/1,4-二氧六环溶液,于室温搅拌反应9h,通过lc-ms监测反应进程。反应完毕后,旋干得到粗产物,将粗产物用无水乙醇打浆,然后于50℃真空干燥,得到124mg淡黄色固体,即为目标化合物例2。1h nmr(500mhz,dmso-d6)δ8.51(s,3h,-nh2hcl), 3.57(m,2h),3.49
–
3.28(m,5h),3.00(s,3h),2.25
–
1.97(m,4h),1.26/1.23(2
×
t, 3h,j=7.2);ms(esi):m/z=143.2[m-i
-
-hcl]
+
。
[0110]
合成例5:3-氨基-1,1-二甲基氮杂环丁烷-1-碘化铵盐酸盐
[0111][0112]
将原料氮杂环丁烷-3-氨基甲酸叔丁酯(100mg,0.58mmol)溶于四氢呋喃 (5ml)中,加入碳酸钾(321g,2.32mmol)和碘甲烷(0.145ml,2.32mmol),于室温反应3h,通过lc-ms监测反应进程。反应完毕后,过滤除去不溶物,旋干得粗产物。然后用甲基叔丁基醚和乙腈混合溶液打浆,并于50℃真空干燥,得到淡黄色固体27mg,即为3-((叔丁氧羰基)氨基)-1,1-二甲基氮杂环丁烷-1-碘化铵.1h nmr(500mhz,dmso-d6)δ7.64(d,j=7.7hz,1h),4.63(m, 1h),4.45(m,2h),4.21(dd,j=11.4,7.5hz,2h),3.17(s,3h),3.16(s,3h),1.39 (s,9h);ms(esi):m/z=201.2[m-i
-
]
+
。
[0113][0114]
将3-((叔丁氧羰基)氨基)-1,1-二甲基氮杂环丁烷-1-碘化铵(150mg, 0.75mmol)溶于甲醇(3ml)中,加入1ml浓度为2m的氯化氢/1,4-二氧六环溶液,于室温搅拌反应3h,通过lc-ms监测反应进程。反应完毕后,旋干得到粗产物,将粗产物用无水乙醇打浆并真空干燥,得淡黄色固体56mg,即为目标化合物例3。1h nmr(500mhz,dmso-d6)δ9.06(s,3h,-nh2hcl),4.67 (m,2h),4.43(m,3h),3.33(s,3h),3.23(s,3h);ms(esi):m/z=101.2[m-i
-
- hcl]
+
。
[0115][0116]
合成例6:4-氨基-1-甲基-1-(甲基-d3)哌啶-1-碘化铵盐酸盐(dadmi)
[0117][0118]
制备方法同合成例1,除了以cd3i代替碘甲烷为原料,得目标化合物例5 为淡黄色固体。1h nmr(600mhz,dmso-d6)δ9.09(s,3h,-nh2hcl),3.48(m, 2h),3.41(m,2h),3.30(m,1h),3.12/3.04(2
×
s,3h),2.04(m,2h),1.98(m,2h); ms(esi):m/z=132.2[m-i
-
-hcl]
+
。
[0119]
实施例1基于本发明代表化合物admi的脂肪酸衍生化物质谱分析实验利用admi缩合反应:将吹干的2-羟基十六酸标准品溶于200μl乙腈(acn)、10μl hatu(50μm,溶于acn)和10μl碳酸氢钠(nahco3,20μm,溶于h2o)的混合溶液中,并在室温下振摇反应5分钟;然后,向反应液中加入20μl 1:1混合的admi衍生化试剂[50μm,溶解于acn/h2o(8:2, v/v)],然后在37℃下反应30分钟。反应结束时,向反应液中加入200μl乙酸乙酯(ea)和200μl去离子水,将产物萃取到有机层中,并且也可去除对下一步氧化反应有较大干扰的nahco3;萃取结束后转移上层有机层,然后重复萃取步骤两次;最后合并ea层并用氮气吹干;吹干样品经meoh复溶上样 uplc-q/tof ms分析。
[0120]
质谱结果分析:
[0121]
admi衍生化鉴定2/3-oh位置:衍生策略有助于提高脂肪酸的质谱响应。在本篇文章中,季铵盐试剂admi,可以同时提高2/3-ohfa的质谱响应和液相色谱分离效率。更重要的是,admi衍生化2/3-ohfa的产物经较高的碰撞能量诱导可以碎裂出特征离子碎片,能够用
于鉴定2/3-oh基团的位置(图2a)。以标准品2-羟基十六酸(2-oh-c16)为例,相应的admi衍生产物在专属性二级质谱(ms2)中能够产生m/z 155.1的离子碎片,这是由oh附着的碳原子与临近羰基碳原子之间的键断裂产生的,因此能够指向2-oh基团的位置(图 2);admi衍生化的3-羟基十六酸(3-oh-c16)在ms2中能够产生m/z 171.1 的离子碎片,这是由oh附着的碳原子与靠近羧基的邻近碳原子之间键断裂产生的,因此能够指向3-oh基团的位置(图2)。
[0122]
实施例2admi衍生化应用于其他c8-c18碳链长度的饱和2/3-ohfa标准品
[0123]
为了进一步确定这两个诊断离子的产生不受碳链长度的影响,将admi 衍生化应用于其他c8-c18碳链长度的饱和2/3-ohfa标准品(图3),并都得到了预期的结果。
[0124]
如果是液质联用设备,由于不同碳链长度的ohfa同分异构体,经admi 衍生化后在液相色谱上都能取得很好的色谱分离效果。还可以将脂肪酸的同分异构体衍生化产物的分子式、质荷比以及保留时间的信息一同分析,结合精确质荷比、155.1/171.1的碎片离子片段和保留时间等信息,能建立起对饱和 2/3-ohfas的快速、有效并且全面的分析方法。
[0125]
实施例3基于4-氨基-1-乙基-1-甲基哌啶-1-碘化铵盐酸盐(合成例4的化合物)的脂肪酸衍生化物质谱分析实验
[0126]
实施例4基于3-氨基-1,1-二甲基氮杂环丁烷-1-碘化铵盐酸盐(合成例5 的化合物)的脂肪酸衍生化物质谱分析实验
[0127]
比较例1
[0128]
需要说明的是,发明人也尝试了使用admi对于羟基位于其他位置的 ohfa同分异构体,admi衍生化产物没能产生类似的可以指出oh位置的离子碎片(图4);这可能是由于admi衍生化产物中羟基官能团和新形成的酰胺键之间较长的碳链限制了类似的离子碎片的形成。(图4)
[0129]
比较例2
[0130]
发明人采用与实施例同样的方法,利用化合物2对2-羟基十六酸标准品进行衍生化,结果并没有没能产生类似的可以指出oh位置的离子碎片。利用化合物2衍生化2/3-ohfa产物的质谱图中没有发现能够指向2/3-oh位置的离子碎片(图5).
[0131]
实施例5复杂组织提取液中2/3-oh脂肪酸有无的鉴别
[0132]
小鼠黑色素瘤肺转移模型构建和衍生化:本研究中使用的黑色素瘤细胞系 b16f10购自美国模式培养物集存库,细胞培养在含10%牛血清蛋白、100u/ml 青霉素和100μg/ml链霉素(gibco)的dmem培养基中;细胞在37℃、含 5%co2的增湿空气环境中培养。本研究使用18只雄性c57bl/6j小鼠。三周龄雄性小鼠购自上海slac实验动物公司。小鼠随机分为肿瘤组及空白对照组,分别尾静脉注射100μl含1x106个肿瘤细胞的培养液或空白dmem培养液。两周后处死小鼠,并采集肺组织;实验前采集的组织样本储存在-80℃冰箱中。首先,称取200mg小鼠组织,于2ml去离子水中匀浆;然后将匀浆液超声处理30分钟;超声完毕后,取50μl样品匀浆液,转移到1.5ml离心管中;然后向离心管中加入10μl磷酸盐缓冲盐水(dpbs),并使用0.1%的hcl 酸化样品;然后向样品中加入100μl乙酸乙酯,并涡旋40s;涡旋完毕后转移出上层有机层,并再次加入100μl乙酸乙酯,重复萃取程序两次。最后,合并全部ea层并用氮气吹干。
[0133]
将吹干的小鼠组织液溶于200μl乙腈/四氢呋喃(acn/thf,1:1)、10μ l hatu(50μm,溶于acn)和10μl碳酸氢钠(nahco3,20μm,溶于h2o)的混合溶液中,并在室温下振摇反应5
分钟;然后,向反应液中加入20μl admi衍生化试剂[50μm,溶解于acn/h2o(8:2,v/v)],然后在37℃下反应30分钟。反应结束时,向反应液中加入200μl乙酸乙酯和200μl去离子水,将产物萃取到有机层中;萃取结束后转移上层有机层,然后重复萃取步骤两次;最后合并ea层并用氮气吹干;吹干样品经meoh 复溶上样uplc-q/tof ms分析。
[0134]
小鼠黑色素瘤肺转移组织中2/3-ohfas类化合物的鉴定:我们进一步将建立的两步衍生化法应用于分析小鼠肿瘤组织中2/3-ohfa类化合物。我们共收集了肿瘤组与空白组18只小鼠的肺器官,并按照同样的方法进行相应分析。如前所述,ohfa的含量远低于不含羟基的脂肪酸。利用本发明化合物admi 衍生化进行质谱检测,通过筛选m/z 155.1或171.1特征离子碎片,我们从一系列同分异构体中共识别出了3-ohc12、3-ohc12(c=c)、3-ohc14、 3-ohc14(c=c)、2/3-ohc16、2/3-ohc18(表1)八种含量相对较高的2/3-ohfa;然后,我们进一步通过mcpba氧化反应鉴定不饱和酸3-ohc12(c=c)、 3-ohc14(c=c)的双键位置分别在δ-9、δ-5处。此处的mcpba氧化反应鉴定不饱和酸的双键位置的方法为公知技术,具体方法可以参考y.feng;b.chen; q.yu;l.li.anal chem 2019,91,1791-1795.
[0135]
表1.小鼠肿瘤中鉴定获得的差异羟基脂肪酸
[0136][0137]
鉴定了小鼠组织中的2/3-ohfa类化合物后,我们进一步采用t检验的方法来分析黑色素瘤转移肺组织样本与普通肺组织样本中2-ohfas和3-ohfas 之间的含量差异。如图6所示,鉴定到的八种2/3-ohfa在肿瘤组肺组织中的含量都低于正常样本。
[0138]
实验证实,本发明的化合物,能够高效的用于复杂样品中,2-ohfa和 3-ohfa存在有无的判断,还可以进一步其他的定性和定量实验。
[0139]
上述披露的各技术特征并不限于已披露的与其它特征的组合,本领域技术人员还可根据发明之目的进行各技术特征之间的其它组合,以实现本发明之目的,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种改进,均应落入本发明权利要求书确定的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1