三(二烷胺基)环戊二烯基金属配合物的合成方法与流程
1.本发明涉及化学合成技术领域,特别是涉及一种三(二烷胺基)环戊二烯基金属配合物的合成方法。
背景技术:
2.随着超大规模集成电路产业的发展,人们对半导体材料的性质及其制备工艺提出了更加严格的要求。根据摩尔定律,集成电路技术伴随着单个电子器件尺寸的缩小在不断的发展,而栅介质层的厚度是影响电子器件尺寸的决定性因素之一。目前集成电路电子行业普遍使用sio2作为电子器件的栅介质材料,但随着器件特征尺寸的减小,传统sio2栅极介质材料厚度已经接近材料的物理厚度,进而导致器件的功耗大幅增加,难以满足微电子行业器件稳定性要求。寻找高介电常数材料(高k材料)替代传统sio2栅极介质层,通过增加介质层的物理厚度而降低隧穿效应,是提高电子器件稳定性的有效技术手段。
3.zro2薄膜具有适中的介电常数(k~25),且具有与传统硅基集成电路工艺相兼容的优良特性,被看作是最有发展前景的新型栅介质材料。通常使用cvd或ald技术获得氧化物薄膜的过程是首先引入气相的前驱体,随后前驱体在晶圆表面发生化学反应。为能成功用于生产中,理想的前驱体必须有足够的反应活性,并有足够的稳定性来保证操作安全,而且要有合适的蒸汽压,同时也要保证前驱体纯净,以保证得到的薄膜不会导致器件问题(电流泄漏、阈值电压漂移等)。
4.三(二甲胺基)环戊二烯基锆在常温下是液体,不仅具有较好的稳定性、较高的蒸汽压,而且表现出了相当高的反应性,因此三(二甲胺基)环戊二烯基锆的物理化学特性可用于cvd或ald技术以制备锆氧化物薄膜。
5.传统的制备三(二甲胺基)环戊二烯基锆的方法是:先用丁基锂与二甲胺反应,制得二甲胺基锂,然后使其与四氯化锆反应制得四(二甲胺基)锆,最后将四(二甲胺基)锆与环戊二烯反应,得到三(二甲胺基)环戊二烯基锆。该方法的收率较低,最高仅为74%。另外,有方法将上述传统方法的原料采用一锅法投料,没有进行中间体的分离,而且反应结束后未经过滤而直接进行脱溶,随后蒸馏产品,由此减少中间产物与空气的接触,且简化了后处理的工艺,提高了生产效率。但是该方法的收率较传统方法大幅下降,最高仅为35%,同时反应结束后容器清理困难,增加了人工成本。另外,还有方法涉及一种五甲基环戊二烯基三(二甲胺基)锆的制备方法,其先用丁基锂与二甲胺反应,制得二甲胺基锂,然后使其与五甲基环戊二烯基三氯化锆反应,制得五甲基环戊二烯基三(二甲胺基)锆。该制备方法的收率为83%。
6.上述方法中均不可避免地需要使用高度易燃的原料丁基锂,或需要经过如二甲胺基锂和四(二甲胺基)锆等易燃的中间体,存在安全隐患,且丁基锂产品一般都是稀释于烷烃溶剂内,使用过程中会产生大量的废液,因此限制了三(二甲胺基)环戊二烯基锆的工业化生产放大。同时,制备方法的收率也有待进一步提升。
技术实现要素:
7.基于此,本发明提供一种收率高、生产安全性高,且绿色环保的三(二烷胺基)环戊二烯基金属配合物的合成方法。
8.具体技术方案如下:
9.一种三(二烷胺基)环戊二烯基金属配合物的合成方法,包括如下步骤:
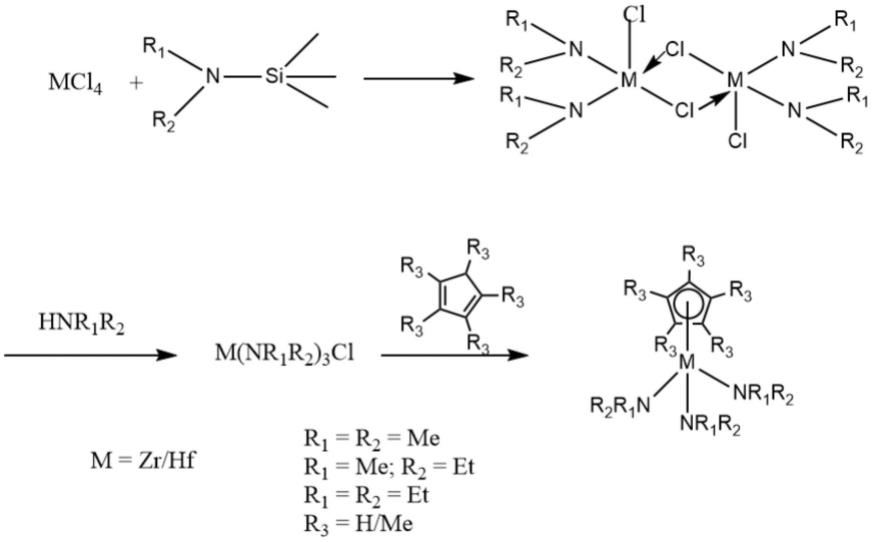
10.(1)将化合物1与mcl4进行反应,制备化合物2;
11.(2)将化合物2与hnr1r2进行反应,制备化合物3;
12.(3)将化合物3与化合物4进行反应,制备所述三(二烷胺基)环戊二烯基金属配合物;
13.其中,化合物1的结构式如下:
[0014][0015]
化合物2的结构式如下:
[0016][0017]
化合物3的结构式如下:
[0018][0019]
化合物4的结构式如下:
[0020][0021]
m为zr或hf;
[0022]
r1、r2分别独立地选自:c1~c5烷基;
[0023]
r3每次出现,分别独立地选自:h或c1~c5烷基;n为0、1、2、3、4或5。
[0024]
在其中一个实施例中,r3为h或甲基。
[0025]
在其中一个实施例中,r1、r2分别独立地选自:甲基或乙基。
[0026]
在其中一个实施例中,mcl4与化合物1的摩尔比为1:(1~4)。
[0027]
在其中一个实施例中,mcl4与hnr1r2的摩尔比为1:(3~6)。
[0028]
在其中一个实施例中,mcl4与化合物4的摩尔比为1:(1~3)。
[0029]
在其中一个实施例中,步骤(1)中,反应的温度为室温、反应的时间为10h~15h。
[0030]
在其中一个实施例中,步骤(2)中,反应的步骤包括:
[0031]
在温度为-10℃~0℃条件下,于化合物2中加入hnr1r2,加入完毕后,在回流条件下反应2h~5h。
[0032]
在其中一个实施例中,步骤(3)中,反应的步骤包括:
[0033]
在温度为-10℃~0℃条件下,于化合物3中加入化合物4,加入完毕后,在室温条件下反应10h~15h。
[0034]
在其中一个实施例中,步骤(1)反应所得反应液无需进行后处理,直接进行步骤(2);及/或,
[0035]
步骤(2)反应所得反应液减压浓缩后,直接进行步骤(3);及/或,
[0036]
步骤(3)反应所得反应液过滤后,进行浓缩和减压蒸馏,制备所述三(二烷胺基)环戊二烯基金属配合物。
[0037]
上述三(二烷胺基)环戊二烯基金属配合物的合成方法,通过合理设置各反应步骤,能够有效提升合成的收率,且原料安全易得,避免使用丁基锂,既大大提高反应的安全系数,又避免在反应中引入杂质金属,便于产物的纯化,能够满足半导体产业对其前驱体源纯度的要求。同时操作简单,无需对中间体进行分离,路线绿色环保,适合工业化放大生产。
具体实施方式
[0038]
以下结合具体实施例对本发明的三(二烷胺基)环戊二烯基金属配合物的合成方法作进一步详细的说明。本发明可以以许多不同的形式来实现,并不限于本文所描述的实施方式。相反地,提供这些实施方式的目的是使对本发明公开内容理解更加透彻全面。
[0039]
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。
[0040]
本文所使用的术语“和/或”、“或/和”、“及/或”的可选范围包括两个或两个以上相关所列项目中任一个项目,也包括相关所列项目的任意的和所有的组合,所述任意的和所有的组合包括任意的两个相关所列项目、任意的更多个相关所列项目、或者全部相关所列项目的组合。
[0041]
本发明中,以开放式描述的技术特征中,包括所列举特征组成的封闭式技术方案,也包括包含所列举特征的开放式技术方案。
[0042]
本发明中,涉及到数值区间,如无特别说明,上述数值区间内视为连续,且包括该范围的最小值及最大值,以及这种最小值与最大值之间的每一个值。进一步地,当范围是指整数时,包括该范围的最小值与最大值之间的每一个整数。此外,当提供多个范围描述特征或特性时,可以合并该范围。换言之,除非另有指明,否则本文中所公开之所有范围应理解为包括其中所归入的任何及所有的子范围。
[0043]
本发明中涉及的百分比含量,如无特别说明,对于固液混合和固相-固相混合均指质量百分比,对于液相-液相混合指体积百分比。
[0044]
本发明中涉及的百分比浓度,如无特别说明,均指终浓度。所述终浓度,指添加成分在添加该成分后的体系中的占比。
[0045]
本发明中的温度参数,如无特别限定,既允许为恒温处理,也允许在一定温度区间
内进行处理。所述的恒温处理允许温度在仪器控制的精度范围内进行波动。
[0046]
本发明中的室温一般指4℃~30℃,较佳地指20
±
5℃。
[0047]
本发明提供一种三(二烷胺基)环戊二烯基金属配合物的合成方法,包括如下步骤:
[0048]
(1)将化合物1与mcl4进行反应,制备化合物2;
[0049]
(2)将化合物2与hnr1r2进行反应,制备化合物3;
[0050]
(3)将化合物3与化合物4进行反应,制备所述三(二烷胺基)环戊二烯基金属配合物;
[0051]
其中,化合物1的结构式如下:
[0052][0053]
化合物2的结构式如下:
[0054][0055]
化合物3的结构式如下:
[0056][0057]
化合物4的结构式如下:
[0058][0059]
m为zr或hf;
[0060]
r1、r2分别独立地选自:c1~c5烷基;
[0061]
r3每次出现,分别独立地选自:h或c1~c5烷基;n为0、1、2、3、4或5。
[0062]
在其中一个具体的示例中,r3为h或c1~c2烷基。进一步地,r3为h或甲基。
[0063]
在其中一个具体的示例中,r1、r2分别独立地选自:c1~c3烷基。进一步地,r1、r2分别独立地选自:甲基或乙基。
[0064]
在其中一个具体的示例中,hnr1r2选自:二甲胺、二乙胺或甲乙胺。
[0065]
在其中一个具体的示例中,化合物1选自:三甲基硅基二甲胺、三甲基硅基二乙胺或三甲基硅基甲乙胺。
[0066]
在其中一个具体的示例中,mcl4与化合物1的摩尔比为1:(1~4)。具体地,mcl4与化
合物1的摩尔比包括但不限于:1:1、1:1.5、1:2、1:2.5、1:3、1:3.5、1:4。
[0067]
在其中一个具体的示例中,mcl4与hnr1r2的摩尔比为1:(3~6)。具体地,mcl4与hnr1r2的摩尔比包括但不限于:1:3、1:3.5、1:4、1:4.5、1:5、1:5.5、1:6。
[0068]
在其中一个具体的示例中,mcl4与化合物4的摩尔比为1:(1~3)。具体地,mcl4与化合物4的摩尔比包括但不限于:1:1、1:1.5、1:2、1:2.5、1:3。
[0069]
在其中一个具体的示例中,步骤(1)中,反应的温度为室温、反应的时间为10h~15h。具体地,步骤(1)中,反应的时间包括但不限于:10h、11h、12h、13h、14h、15h。
[0070]
在其中一个具体的示例中,步骤(1)中,反应采用的溶剂为烷烃类溶剂。进一步地,步骤(1)中,反应采用的溶剂为正己烷。更进一步地,步骤(1)中,反应采用的溶剂为无水正己烷。
[0071]
在其中一个具体的示例中,步骤(2)中,反应的步骤包括:
[0072]
在温度为-10℃~0℃条件下,于化合物2中加入hnr1r2,加入完毕后,在回流条件下反应2h~5h。
[0073]
具体地,步骤(2)中,反应的温度包括但不限于:-10℃、-9℃、-8℃、-7℃、-6℃、-5℃、-4℃、-3℃、-2℃、-1℃。
[0074]
具体地,步骤(2)中,反应的时间包括但不限于:2h、3h、4h、5h。
[0075]
在其中一个具体的示例中,步骤(3)中,反应的步骤包括:
[0076]
在温度为-10℃~0℃条件下,于化合物3中加入化合物4,加入完毕后,在室温条件下反应10h~15h。
[0077]
具体地,步骤(3)中,反应的温度包括但不限于:-10℃、-9℃、-8℃、-7℃、-6℃、-5℃、-4℃、-3℃、-2℃、-1℃。
[0078]
具体地,步骤(3)中,反应的时间包括但不限于:10h、11h、12h、13h、14h、15h。
[0079]
在其中一个具体的示例中,步骤(1)、步骤(2)和步骤(3)在惰性气体环境中进行,使用的惰性气体为高纯氮气或高纯氩气。
[0080]
在其中一个具体的示例中,步骤(1)反应所得反应液无需进行后处理,直接进行步骤(2)。
[0081]
在其中一个具体的示例中,步骤(2)反应所得反应液减压浓缩后,直接进行步骤(3)。
[0082]
在其中一个具体的示例中,步骤(3)反应所得反应液过滤后,进行浓缩和减压蒸馏,制备所述三(二烷胺基)环戊二烯基金属配合物。进一步地,过滤是指采用砂芯进行无水无氧过滤。
[0083]
在其中一个具体的示例中,减压蒸馏之后还包括精馏步骤。
[0084]
以下为具体的实施例,如无特别说明,实施例中采用的原料均为市售产品。
[0085]
实施例中三(二烷胺基)环戊二烯基金属配合物合成方法的路线如下所示:
[0086][0087]
实施例中采用的环戊二烯为刚解聚的环戊二烯,解聚的方法为氮气保护下对二聚环戊二烯进行常压蒸馏解聚,收集41℃~42℃的环戊二烯单体。
[0088]
实施例1:
[0089]
本实施例一种三(二烷胺基)环戊二烯基金属配合物的合成方法,步骤如下:
[0090]
在氮气气氛下,向1000毫升schlenk瓶中加入27.9克四氯化锆,28.2克n,n-二甲基三甲基硅胺和400毫升正己烷,室温搅拌12小时后,冷却至-10℃,加入81克质量浓度20%的二甲胺正己烷溶液,慢慢恢复室温并回流3小时,减压浓缩掉一半体积的溶剂后,将体系降温至-10℃,在氮气保护下慢慢加入刚解聚的环戊二烯7.9克,恢复至室温,并继续搅拌12小时,经过砂芯进行无水无氧过滤,滤液浓缩抽干溶剂,残余物减压蒸馏,得到三(二甲胺基)环戊二烯基锆30.1克,收率87%。
[0091]1hnmr(c6d6):6.06(s,5h),2.92(s,18h)。
[0092]
对终产物进行元素分析检测,理论值:c:45.79,h:8.03,n:14.56。测量值:c:45.62,h:7.97,n:14.02。
[0093]
实施例2:
[0094]
本实施例一种三(二烷胺基)环戊二烯基金属配合物的合成方法,步骤同实施例1,区别在于:将实施例1中二甲胺正己烷溶液的量改为108克。
[0095]
在氮气气氛下,向1000毫升schlenk瓶中加入27.9克四氯化锆,28.2克n,n-二甲基三甲基硅胺和400毫升正己烷,室温搅拌12小时后,冷却至-10℃,加入108克质量浓度20%的二甲胺正己烷溶液,慢慢恢复室温并回流3小时,减压浓缩掉一半体积的溶剂后,将体系降温至-10℃,氮气保护下慢慢加入刚解聚的环戊二烯7.9克,恢复至室温,并继续搅拌12小时,经过砂芯进行无水无氧过滤,滤液浓缩抽干溶剂,残余物减压蒸馏,得到三(二甲胺基)环戊二烯基锆31.5克,收率91%。
[0096]
实施例3:
[0097]
本实施例一种三(二烷胺基)环戊二烯基金属配合物的合成方法,步骤同实施例2,
区别在于:将实施例2中环戊二烯的量改为11.9克。
[0098]
在氮气气氛下,向1000毫升schlenk瓶中加入27.9克四氯化锆,28.2克n,n-二甲基三甲基硅胺和400毫升正己烷,室温搅拌12小时后,冷却至-10℃,加入108克质量浓度20%的二甲胺正己烷溶液,慢慢恢复室温并回流3小时,减压浓缩掉一半体积的溶剂后,将体系降温至-10℃,氮气保护下慢慢加入刚解聚的环戊二烯11.9克,恢复至室温,并继续搅拌12小时,经过砂芯进行无水无氧过滤,滤液浓缩抽干溶剂,残余物减压蒸馏,得到三(二甲胺基)环戊二烯基锆33.6克,收率97%。
[0099]
在千级洁净间使用25厘米高的刺型柱对终产物进行精馏,得到的纯品使用电感耦合等离子体质谱仪(icp-ms)分析其纯度,结果显示产品金属纯度为5n。
[0100]
实施例4:
[0101]
本实施例一种三(二烷胺基)环戊二烯基金属配合物的合成方法,步骤同实施例3,区别在于:将实施例3中环戊二烯改为等当量的五甲基环戊二烯。
[0102]
在氮气气氛下,向1000毫升schlenk瓶中加入27.9克四氯化锆,28.2克n,n-二甲基三甲基硅胺和400毫升正己烷,室温搅拌12小时后,冷却至-10℃,加入108克质量浓度20%的二甲胺正己烷溶液,慢慢恢复室温并回流3小时,减压浓缩掉一半体积的溶剂后,将体系降温至-10℃,氮气保护下慢慢加入五甲基环戊二烯24.5克,恢复至室温,并继续搅拌12小时,经过砂芯进行无水无氧过滤,滤液浓缩抽干溶剂,残余物减压蒸馏,得到五甲基环戊二烯基三(二甲胺基)锆40克,收率93%。
[0103]1hnmr(c6d6):2.93(s,18h),1.98(s,15h)。
[0104]
对终产物进行元素分析检测,理论值:c:53.58,h:9.27,n:11.72。测量值:c:53.44,h:9.21,n:12.03。
[0105]
实施例5:
[0106]
本实施例一种三(二烷胺基)环戊二烯基金属配合物的合成方法,步骤同实施例3,区别在于:将实施例3中四氯化锆改为等当量的四氯化铪。
[0107]
在氮气气氛下,向1000毫升schlenk瓶中加入38.4克四氯化铪,28.2克n,n-二甲基三甲基硅胺和400毫升正己烷,室温搅拌12小时后,冷却至-10℃,加入108克质量浓度20%的二甲胺正己烷溶液,慢慢恢复室温并回流3小时,减压浓缩掉一半体积的溶剂后,将体系降温至-10℃,氮气保护下慢慢加入刚解聚的环戊二烯11.9克,恢复至室温,并继续搅拌12小时,经过砂芯进行无水无氧过滤,滤液浓缩抽干溶剂,残余物减压蒸馏,得到三(二甲胺基)环戊二烯基铪41.5克,收率92%。
[0108]1hnmr(c6d6):6.04(s,5h),2.96(s,18h)。
[0109]
对终产物进行元素分析检测,理论值:c:35.16,h:6.17,n:11.18。测量值:c:35.02,h:5.99,n:11.33。
[0110]
实施例6:
[0111]
本实施例一种三(二烷胺基)环戊二烯基金属配合物的合成方法,步骤同实施例3,区别在于:将实施例3中n,n-二甲基三甲基硅胺改为等当量的n,n-二乙基三甲基硅胺,同时将20%的二甲胺正己烷溶液改为等当量的二乙胺正己烷溶液。
[0112]
在氮气气氛下,向1000毫升schlenk瓶中加入27.9克四氯化锆,34.9克n,n-二乙基三甲基硅胺和400毫升正己烷,室温搅拌12小时后,冷却至-10℃,加入175.5克质量浓度
20%的二乙胺正己烷溶液,慢慢恢复室温并回流3小时,减压浓缩掉一半体积的溶剂后,将体系降温至-10℃,氮气保护下慢慢加入刚解聚的环戊二烯11.9克,恢复至室温,并继续搅拌12小时,经过砂芯进行无水无氧过滤,滤液浓缩抽干溶剂,残余物减压蒸馏,得到三(二乙胺基)环戊二烯基锆41.6克,收率93%。
[0113]1hnmr(c6d6):6.10(s,5h),3.29(q,12h),1.04(t,18h)。
[0114]
对终产物进行元素分析检测,理论值:c:54.78,h:9.47,n:11.27。测量值:c:54.52,h:9.11,n:11.43。
[0115]
对比例1:
[0116]
本对比例一种三(二烷胺基)环戊二烯基金属配合物的合成方法,步骤同实施例3,区别在于:将实施例3中环戊二烯改为等当量的环戊二烯基锂。
[0117]
在氮气气氛下,向1000毫升schlenk瓶中加入27.9克四氯化锆,28.2克n,n-二甲基三甲基硅胺和400毫升正己烷,室温搅拌12小时后,冷却至-10℃,加入108克质量浓度20%的二甲胺正己烷溶液,慢慢恢复室温并回流3小时,减压浓缩掉一半体积的溶剂后,将体系降温至-10℃,氮气保护下慢慢加入环戊二烯基锂13.0克,恢复至室温,并继续搅拌12小时,经过砂芯进行无水无氧过滤,滤液浓缩抽干溶剂,残余物减压蒸馏,得到三(二甲胺基)环戊二烯基锆31.2克,收率90%。
[0118]
在千级洁净间使用25厘米高的刺型柱对终产物进行精馏,得到的纯品使用电感耦合等离子体质谱仪(icp-ms)分析其纯度,结果显示产品中含锂52ppm,金属纯度为4n。
[0119]
测试例:
[0120]
将非掺杂的硅片衬底依次用丙酮、无水乙醇、去离子水、rca溶液、去离子水和无水乙醇依次超声清洗,并用氮气吹干,烘干后放入原子层沉积腔室中;调节原子层沉积腔室温至260℃,并抽真空;加热实施例3的三(二甲胺基)环戊二烯基锆液体源至150℃,利用高纯氩气将蒸发的三(二甲胺基)环戊二烯基锆分子送入原子层沉积腔室内,待三(二甲胺基)环戊二烯基锆分子通入结束后,继续通入氩气10s,以便清洗残余zr源与反应副产物;利用氩气将h2o脉冲进入原子层沉积腔室,待脉冲h2o分子通入结束后,继续通入氩气10s,以便清洗残余h2o分子与反应副产物;重复上述过程,经过200个循环得到二氧化锆薄膜,通过椭偏仪检测薄膜的厚度。部分测试数据统计如下表1:
[0121]
表1
[0122][0123]
由表1可知,采用本发明实施例制备的三(二甲胺基)环戊二烯基锆在不同的脉冲时间、吹扫时间、h2o脉冲时间和吹扫时间条件下,均能够实现具有良好均匀性的二氧化锆薄膜的制备,满足半导体产业对其前驱体源纯度的要求。
[0124]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0125]
以上所述实施例仅表达了本发明的几种实施方式,便于具体和详细地理解本发明的技术方案,但并不能因此而理解为对发明专利保护范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。应当理解,本领域技术人员在本发明提供的技术方案的基础上,通过合乎逻辑的分析、推理或者有限的试验得到的技术方案,均在本发明所附权利要求的保护范围内。因此,本发明专利的保护范围应以所附权利要求的内容为准,说明书可以用于解释权利要求的内容。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1