一种黄精多糖及其制备方法和用途
1.本发明属于生物医药领域,尤其涉及一种黄精多糖及其制备方法和用途。
背景技术:
2.阿尔兹海默症(alzheimer’s disease,ad)是一种起病隐匿进行性发展的中枢神经系统退行性疾病,在65岁以上人群中患病率随年龄逐渐增加。 ad患者主要临床表现为渐进性认知功能退化,包括记忆力功能减退、思维缓慢、行为异常和社交障碍等,最终表现为日常生活完全不能自理并引发一系列并发症,危害十分严重。ad的主要病理特征为大脑海马区和皮质区神经细胞外存在大量由β-淀粉样蛋白(aβ)异常聚集形成的老年斑(senile plaques, sps)。目前,ad一线治疗药物主要为乙酰胆碱酯酶抑制剂(多奈哌齐、加兰他敏、卡巴拉汀)和n-甲基-d-天冬氨酸(nmda)受体阻断剂(美金刚)。从长期的临床使用结果来看,这些药物只能减轻患者症状,并不能延缓或阻止ad的进展。
3.传统中药黄精(polygonatumpolysaccharide)具有补肾益精、益智延年、强精固髓等功效,《神仙之草经》记载“黄精宽中益气,服之可使五脏调良,多年不老、发白更黑之功效。黄精目前是湖南“湘九味”重点培育的大宗药材,在湖南邵阳和新化等地区大面积培育种植。黄精中富含的多糖(polygonatum sibiricum polysaccharides,psp)成分是黄精的主要活性物质。中南大学湘雅二医院药学部联合老年病科长期关注psp的神经保护及抗衰老作用。对ad的研究表明,psp可显著抑制aβ
1-42
注射导致的大鼠海马组织细胞凋亡,改善大鼠的学习记忆能力;在app转基因小鼠实验中, psp可显著减少aβ斑块沉积,增加小鼠海马ca1区突触数量,减小突触面积和减轻突触变性程度,同时促进突触小泡分泌,保护小鼠海马ca1 区突触结构和功能。以上研究证实了黄精多糖具有改善学习记忆功能以达到缓解ad症状的功效。鉴于现有技术已经有报道称黄精多糖能够改善学习记忆,对缓解阿尔兹海默病症状有良好效果,但是并未开发效果良好的单体药物用于治疗该疾病。同时,本领域需要更多可用于治疗阿尔兹海默病的药物。
技术实现要素:
4.本发明的主要目的在于克服现有技术的缺点与不足,提供一种黄精多糖及其制备方法和用途,具体采用以下技术方案:
5.本发明第一方面提供了一种黄精多糖,如式(i)所示:
淀粉样蛋白(aβ)沉积;
25.进一步的,所述预防和/或治疗阿尔兹海默病的药物具有恢复肠道菌群平衡的作用;更进一步的,所述恢复肠道菌群平衡包括抑制肠道促炎菌 helicobacter typhlonius和helicobacter mastomyrinus生长以及促进益生菌嗜黏蛋白阿克曼菌(akkermansiamuciniphila)生长的作用;
26.本发明的第四方面提供一种如式(i)所述化合物的制备方法,包括以下步骤:
27.s1水提醇沉:取干燥黄精药材粉末加水加热提取,减压浓缩干燥粉碎后获得干燥粉末,脱脂后加水分散,然后加70~95%v/v乙醇至样品溶液中乙醇浓度达到60~80%,静置过夜,过滤,重复上述醇沉以及过滤操作1~2次获得黄精多糖浸膏;
28.s2脱蛋白:取黄精多糖浸膏,按照黄精多糖浸膏:去离子水质量比为1:10~1:50加去离子水混悬形成混悬液,然后加入所述混悬液质量 1/4~1/2的sevage试剂,剧烈振摇20~30min,静置分层,缓慢弃去下层有机溶剂及中间层变性蛋白质,保留上清液,重复以上操作6~10次,合并上清液后浓缩得黄精多糖浓缩液,透析12h以上,透析液冷冻干燥,获得黄精粗多糖;
29.s3纤维素柱分离:取黄精粗多糖溶于其质量1/200~1/100的去离子水中,待完全溶解后离心,取上清液过滤,采用纤维素柱分离,依次采用浓度为0.05、0.1、0.2、0.3、0.5mol/l nacl水溶液分段洗脱,并采用苯酚
‑ꢀ
硫酸法跟踪检测洗脱液的吸光值,合并收集主峰,分别透析除盐、减压浓缩、冷冻干燥,获得到7个流分fr.1~fr.7;
30.s4凝胶色谱柱分离:取多糖含量最多的洗脱组分fr.1,用其质量 1/100~1/50的去离子水溶解,待完全溶解后离心,取上清液过滤,采用凝胶柱分离,0.05~0.2mol/l nacl水溶液为洗脱剂,苯酚-硫酸法跟踪洗脱液的吸光度值,合并收集主峰,浓缩干燥后,得到所述黄精多糖。
31.有益效果
32.本发明对黄精的有效部位多糖进一步进行分离纯化,获得均一化黄精多糖,并对其进行了结构鉴定(式(i))。对该多糖进行药理活性测试,证明其具有显著改善学习记忆功能,减少脑内β-淀粉样蛋白沉积,并能恢复宿主肠道菌群稳态,尤其是抑制肠道促炎菌helicobacter typhlonius和 helicobacter mastomyrinus生长,以及促进益生菌akkermansia muciniphila 生长。能够用于治疗阿尔兹海默病,且活性好于多糖混合物,能够作为有效的先导化合物开发临床药物。
附图说明
33.图1:式(i)的分离和表征谱。图1a:deae-52纤维素柱分离图;图1b:凝胶柱分离图;图1c:式(i)的红外光谱图;图1d:式(i)的紫外光谱图。
34.图2:式(i)分子量测定。图2a:式(i)的hpgpc色谱图;图2b:式(i)的分子量结果测定图。
35.图3:式(i)1hnmr图谱
36.图4:式(i)的
13
c nmr图谱
37.图5:式(i)的hsqc图谱
38.图6:式(i)的cosy图谱
39.图7:式(i)的hmbc图谱
40.图8:式(i)的noesy图谱
41.图9:式(i)改善5xfad小鼠的空间学习和记忆功能的药理实验图
42.图10:式(i)减少5xfad脑内aβ沉积
43.图11:给式(i)前后5xfad小鼠肠道菌群16s rrna检测图
44.图12:式(i)的活性与黄精粗多糖相比较
具体实施方案
45.下面结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
46.实施例1多糖的提取、分离以及纯化
47.1.1多糖提取
48.取干燥黄精药材粉末加10倍水量,沸水提取2次,每次2h。抽滤合并减压浓缩,获得干燥粉末。依次用丙酮、甲醇索式提取12h,脱脂。倒出残留粉末在50℃下干燥24h,得脱脂后黄精干燥粉末。取脱脂黄精干燥粉末,加入4倍量的95%乙醇至样品溶液中乙醇浓度达到80%,充分搅拌后,4℃冷藏放置过夜,过滤,按照同样方法再沉淀2次,合并沉淀物并减压浓缩,获得黄精多糖浸膏。
49.1.2脱蛋白
50.取20g含量黄精多糖浸膏,加入400ml去离子水溶解,移入分液漏斗中,加入其体积1/3的sevage试剂(氯仿:正丁醇=4:1),剧烈振摇 20~30min,振摇大量絮状物,静置分层,缓慢弃去下层有机溶剂及中间层变性蛋白质,保留上清液,加入sevage试剂反复萃取7次,合并上清液后减压浓缩得黄精多糖浓缩液。将黄精多糖浓缩液于透析袋(截留分子量为3500da)在去离子水中透析24h,透析液冷冻干燥,获得黄精粗多糖 (psp)。
51.1.3多糖的分离纯化
52.1.3.1 deae-52纤维素预处理
53.取80g deae-52纤维素干粉在去离子水中充分膨胀,洗涤除去杂质。先用0.5m naoh浸泡半小时,在布氏漏斗中除去碱液,去离子水洗至中性,再用0.5m hcl浸泡半小时,在布氏漏斗中除去酸液,去离子水洗至中性;重复两次后,再用去离子水洗至中性。
54.1.3.2装柱
55.装柱前先用去离子水将玻璃柱中底部空气排空,将预处理好的 deae-52纤维素轻轻搅拌混匀,使deae-52纤维素:去离子水(v/v)= 3:1,利用玻璃棒引流尽可能一次性将倾入色谱柱中,混悬液沿色谱柱内壁留下,防止气泡产生。装柱后检查柱床是否规整、是否有气泡或界面存在。
56.1.3.3柱平衡及洗脱
57.为了保证纤维素材料的功能团与起始的洗脱液达到平衡稳定,用起始洗脱液0.05m nacl洗脱5个柱体积。取600mg psp溶于4ml去离子水中溶解,1000rpm离心5min,上清液过滤膜(0.75μm)进一步除去杂质后全部均匀上样。洗脱液先后依次浓度为0.05、0.1、
0.2、0.3、0.5m nacl 分段洗脱。苯酚-硫酸法跟踪检测洗脱液在490nm的吸光值(图1a)。合并收集主峰,得到7个组分fr.1~fr.7。透析除盐,浓缩,冷冻干燥。
58.1.3.4 sephacryl s-200凝胶预处理
59.sephacryl s-200凝胶用去离子水浸泡半小时,待填料充分溶胀后,用去离子水反复浸洗,用布氏漏斗真空抽滤除去醇味,去离子水悬浮凝胶颗粒。
60.1.3.5 sephacryl s-200凝胶装柱
61.除去sephacryl s-200悬浮液中气泡,装柱前先用去离子水将色谱柱底部空气的空气排出,使sephacryl s-200:去离子水(v/v)=3:1,轻轻搅拌混匀,使用玻璃棒引流尽可能一次性将介质倾入色谱柱中。
62.1.3.6 sephacryl s-200的平衡及洗脱
63.用5个柱体积0.1m nacl替换装柱所用的去离子水,取多糖含量最多的洗脱组分fr.1,溶解于用其质量1/50的去离子水中,待完全溶解后离心,取上清液过滤后均匀上洋,以0.1m nacl为洗脱剂,苯酚-硫酸法跟踪洗脱液在490nm的吸光值(图1b)。合并收集主峰,减压浓缩后真空干燥后,得到多糖式(i),称重。
64.实施例2多糖式(i)的结构鉴定
65.2.1测定多糖式(i)的uv、ir以及分子量
66.2.2.1式(i)的uv分析
67.取1mg多糖式(i)溶于2ml水中,稀释到浓度为0.5mg/ml;以蒸馏水为空白对照,于紫外可见光谱200~800nm波长区域扫描。结果见图1c,式(i)在260nm处没有吸收峰,说明没有核酸成分;在280nm处曲线基本平滑,表明式(i)不含蛋白质。uv总体说明式(i)为纯化的多糖。
68.2.2.2式(i)的ir分析
69.将kbr置于玛瑙研钵中充分研磨并压片,作为空白对照。取5mg式 (i)与kbr粉末以1:100比例放入玛瑙研钵中均匀研磨,压片。用傅里叶红外光谱仪扫描分析,扫描范围为4000~400-1
,仪器分辨率2cm-1
,扫描次数16次,得到红外光谱图。如图1d所示,在3371.73cm-1
处有一个较宽的特征峰是分子间o-h的伸缩振动峰;在2927.39cm-1
间有吸收峰是 c-h的伸缩振动;1374.32cm-1
为c-h弯曲振动峰;1703.20cm-1
、 1647.61cm-1
处的吸收峰应为羧基(-coo-)伸缩振动;1200cm-1
~1000cm-1
处的特征峰为吡喃环的c-o-c和c-o-h的变角振动;861.81cm-1
为糖类β-糖苷键的特征峰。ir总体说明式(i)为多糖结构。
70.2.2式(i)的分子量分析
71.2.2.1试剂与仪器
72.2.2.1.1试剂
73.使用的主要试剂反映在下表1中。
74.表1
[0075][0076]
2.2.2主要仪器
[0077]
使用的主要仪器反映在下表2中。
[0078]
表2
[0079][0080]
2.2.3样品信息
[0081]
对样品进行编号,如下表3所示。
[0082]
表3
[0083]
[0084]
2.2.4实验方法
[0085]
2.2.4.1分子量校正曲线建立
[0086]
称量不同分子量的右旋糖酐标准品(分子量1000、5000、12000、 25000、50000、80000、150000、270000、410000、670000系列分析标准品),分别加入0.05m nacl溶液配制成5mg/ml右旋糖酐的标准溶液,使用0.22μm的微孔滤膜过滤备用,采用hpgpc法,使用高效凝胶渗透色谱串联柱来进行检测,采用waters empower软件对结果进行分析,以标准品相对分子质量mp的对数值为纵坐标,以相应色谱峰的保留时间为横坐标进行线性回归,得校正曲线。
[0087]
2.2.4.2供试样品溶液的制备
[0088]
称取纯化多糖样品,向样品中加入0.05m nacl溶液,配制成5mg/ml 供试样品溶液,取上清液用0.22μm的微孔滤膜过滤,然后将样品转置于 2ml进样瓶中备用。
[0089]
2.2.4.3色谱方法
[0090]
采用hpgpc法,使用高效液相色谱仪配示差检测器,聚合物基质水溶性sec(gfc)色谱柱ohpak sb-803hq、ohpak sb-804hq、ohpaksb-805hq(8
×
300mm)串联柱检测,流动相0.05m nacl溶液,流速 0.6ml/min,柱温40℃,进样量30μl。
[0091]
2.2.4.4实验结果
[0092]
(1)分子量校正曲线结果
[0093]
采用waters empower软件,以标准品相对分子质量mp的对数值为纵坐标,以相应色谱峰的保留时间(t)为横坐标进行三阶线性回归,得到校正曲线,如下所示:
[0094]
log mp-t校正曲线方程为:log mp=-0.0004t3+0.0500t
2-2.1475 t+36.9321,r2=0.9997。
[0095]
(2)式(i)分子量测试
[0096]
根据标准品校正曲线,得出计算公式进而计算出式(i)的分子量范围。具体结果见图2,图2b中psp-1-2即为式(i)化合物的表征。
[0097]
2.3式(i)的nmr测定
[0098]
2.3.1试剂与仪器
[0099]
(1)试剂:氘代水(d2o),光谱级,上海阿拉丁生化科技股份有限公司;3-(三甲基甲硅烷基)丙酸-d4钠盐(tmsp),98atom%d, sigma公司。
[0100]
(2)仪器:600mhz bruker核磁共振光谱仪(brukeravance hd iii 600mhz spectromete),德国bruker公司。
[0101]
2.3.2核磁共振测试
[0102]
冻干后的样品溶于0.5ml d2o,加入tmsp作为内标。使用600mhzbruker核磁共振光谱仪,测定一维核磁1h-nmr、
13
c-nmr和二维核磁1h-1
h cosy、tocsy、hsqc、hmbc、noesy。定标:hdo氢δh=δ4.70ppm,tmsp的甲基碳δc=δ-1.80ppm。。
[0103]
为了进一步得到样品的结构特征信息,对其进行一维核磁1h-nmr、
13
c-nmr和二维核磁1h-1
h cosy、hsqc、hmbc、noesy测定,其谱图如图2~7所示,获取各个主要糖残基的全部h和c化学位移信息,推断各个糖残基之间的连接顺序。
[0104]
采用一维核磁共振氢谱(1h-nmr,参见图3)进一步鉴定麦芽糊精样品的糖苷键构型,多糖的氢谱信号大多数在δ3.0~5.5ppm,通常δ 4.5~5.5ppm之间为异头质子(h-1)共
振区。麦芽糊精样品的1h-nmr谱图上大量质子共振信号集中在δ3.0-5.5ppm区域,信号重叠严重,在异头区域发现3个主要异头质子偶合信号,异头质子信号分别为δ5.31ppm、δ 5.25ppm、δ4.87ppm,显示可能含有3种单糖残基,根据它们的化学位移信号强度,将这对应的3种糖残基分别标记为a、b、c,其它的氢信号均集中在δ4.5~3.0ppm区域,信号重叠比较严重,难以归属。
[0105]
表4多糖样品中各糖残基的1h和
13
c的化学位移归属
[0106][0107]
“‑‑”
表示未发现信号
[0108]
通过
13
c nmr谱图(图4)和hsqc谱图(图5)异头区域的交叉峰,确定残基a、b、c的异头碳信号分别为δ98.59ppm、δ99.98ppm、δ 98.05ppm。在异头信号归属确定之后,通过cosy(图6)、hmbc(图7)和noesy(图8),结合1h-nmr、
13
c-nmr谱图对照相关文献的化学位移数据,将样品中主要类型糖残基的1h和
13
c化学位移信号进行归属,结果见表1。各糖残基结构分析具体如下:
[0109]
以糖残基a为例,说明各个糖残基1d和2d nmr解析过程:异头信号δ5.31ppm(h-1)和δ98.59ppm(c-1)表明糖残基a为α构型。根据1h nmr确定的糖残基a的h-1化学位移δ5.31ppm,通过cosy图谱交叉峰明确了h-2、h-3、h-4、h-5信号,糖残基a的h-2、h-3、h-4和 h-5化学位移分别归属为δ3.53ppm、δ3.86ppm、δ3.56ppm和δ3.74ppm,其h-6可借助hsqc相关谱进行归属,为δ3.74ppm。在归属完糖环上氢的化学位移后,可通过hsqc相关谱归属该糖环上各碳的化学位移,分别为δ98.59ppm、δ71.33ppm、δ73.27ppm、δ76.62ppm、δ71.19ppm和δ 60.36ppm,见表1。其中c-1和c-4的化学位移向低场偏移,表明该残基在糖环c-1和c-4位置发生了取代,结合文献报道,推断糖残基a为
→
4)-α-d-glcp-(1
→
。
[0110]
按照类似方法,再推导得到其它主要残基的氢和碳信号,多糖样品中主要单糖残基h和c化学位移归属,如表1所示,结合文献报道,推断 b为
→
4,6)-α-d-glcp-(1
→
,推断c为α-d-glcp-(1
→
。
[0111]
通过hmbc远程相关谱上异头氢与各个糖残基上碳的偶合信号,或是异头碳与各个糖残基上氢的偶合信号,可以进一步地推断出各个糖残基之间的相互连接顺序。样品的hmbc相关谱如图7所示,从图中可找到如下偶合信号,残基a的h-1(δ5.31ppm)与残基a的c-4(δ76.62ppm) 有耦合信号(ah-1/ac-4),残基a的h-1(δ5.31ppm)与残基b的c
‑ꢀ
4(δ76.58ppm)有耦合信号(ah-1/b c-4),残基a的h-4(δ3.56ppm) 与残基a的c-1(δ98.59ppm)有偶合信号(a h-4/a c-1)。多糖样品的残基连接顺序进一步通过noesy谱(图7)来验证,noesy谱中残基a 的h-1(δ5.31ppm)与残基a的h-4(δ3.56ppm)存在交叉峰(a h
‑ꢀ
1/a h-4),残基b的h-1(δ5.25ppm)与残基a的h-4(δ3.56ppm) 存在交叉峰(b h-1/ah-4),残基c的h-1
(δ4.87ppm)与残基b的h
‑ꢀ
6(δ3.84ppm)存在交叉峰(c h-1/b h-6)。由此可表明样品是由残基a 通过1
→
4糖苷键与残基a和b相连构成糖主链,支链上残基b通过6位与残基c相连。
[0112]
综上所述一维和二维核磁信息分析,推断出的样品初步结构是一 种以
→
4)-α-d-glcp-(1
→
为主链的葡聚糖,α-d-glcp-(1
→
连接6位上, 核磁h谱积分显示,
→
4,6)-α-d-glcp-(1
→
与
→
4)-α-d-glcp-(1
→
的比 例约为1:6,再结合其分子量信息(图2b),糖单元连接过程中脱-h2o 基,推测n的范围为13~20。确定(i)结构如下所示:
[0113][0114]
其中,式(i)中的n是选自13~20的一个或多个整数。
[0115]
实施例3式(i)(psp-1)的药理活性测试
[0116]
3.1改善5xfad小鼠的空间学习和记忆功能实验
[0117]
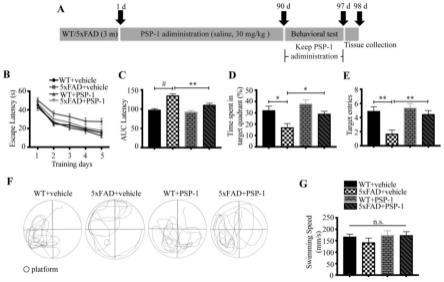
3月龄spf饲养级野生型(wt)小鼠和ad转基因模型小鼠5xfad,各分为2组,每组9只,称重记录。实验组每天灌胃给予30mg/kg式 (i),对照组每天灌胃给予等体积的生理盐水,持续给药3个月后,进行水迷宫行为学实验。行为学测试期间保持给药剂量和频率(图9a)。图9b和9c显示经过5天训练后,给药组的5xfad小鼠明显比生理盐水组的5xfad小鼠到达逃生平台的时间缩短;图9d和9e显示撤掉逃生平台后,给药组的5xfad小鼠明显比生理盐水组的5xfad小鼠在平台象限停留的时间以及穿越原平台位置的次数明显增多;图9f为小鼠水迷宫运动轨迹图;图9g显示给组小鼠的游泳速度没有发生改变。数据为平均值
±
标准偏差(n=9);*,**,#分别表示p<0.05,p<0.01和p<0.001, n.s.表示无统计学意义。以上结果说明,灌胃给予式(i)能明显改善ad 小鼠的空间学习记忆功能。
[0118]
3.2 5xfad脑组织切片aβ斑块染色检测
[0119]
取wt小鼠,5xfad小鼠生理盐水组和5xfad小鼠psp-1组小鼠脑组织,进行冰冻切片,脑切片厚度为30μm。然后使用thioflavin s染色和免疫组化染色(使用特异性aβ抗体)方法,分别对脑海马区和皮层区进行染色,并进行统计学差异分析。结果显示,图10a和10b表明psp-1能明显减少aβ斑块在脑海马区的沉积;图10c和10d表明psp-1能明显减少 aβ斑块在脑皮层区的沉积。代表性图片标尺为100μm。数据为平均值
±
标准偏差(n=5);**表示p<0.01。以上结果表明psp-1能减少ad小鼠脑组织内aβ的沉积。
[0120]
3.3 5xfad小鼠肠道菌群16s rrna检测
[0121]
收集5xfad小鼠给药前(3月龄),给药后5xfad小鼠生理盐水组(6 月龄),5xfad小鼠psp-1组(6月龄)粪便,通过16s rrna检测肠道菌群。图11a和11b说明肠道菌群5xfad小鼠溶酶组(6月龄)较5xfad小鼠给药前(3月龄)有明显区别,说明6月龄5xfad小鼠会出现肠道菌群紊乱,然而给药后的5xfad小鼠肠道菌群得到恢复;图11c显示肠道菌群的
‑ꢀ
多样性没
有发生改变,说明psp-1不改变菌群的种类,但图11d显示肠道菌群的β-多样性在给药后明显区别与生理盐水组,说明各菌群丰度出现改变,psp-1重塑了宿主肠道菌群。图11e、11f和11g分别说明psp-1对肠道促炎菌helicobacter typhlonius和helicobacter mastomyrinus有抑制作用,而对于益生菌akkermansia muciniphila有促生长作用,psp-1有益生元的功能。其中图11a中,(a):5xfad小鼠给药前(3月龄),(b): 5xfad小鼠生理盐水组(6月龄),(c):5xfad小鼠psp-1组(6月龄)。数据为平均值
±
标准偏差(n=6);*和**p<0.05和p<0.01和p<0.001, n.s.表示无统计学意义。以上结果表明口服psp-1可以重塑宿主肠道菌群,是肠道菌群回归稳态。
[0122]
3.4式(i)的活性与黄精粗多糖相比较
[0123]
3月龄5xfad小鼠,分成两组,每组9只。分别灌胃给与黄精粗多糖和psp-1,浓度均为30mg/kg。灌胃3个月后进行水迷宫行为学实验。行为学实验期间,保持口服灌胃。图12显示给予psp-1的5xfad小鼠在训练的第4天和第5天小鼠找到逃生平台的时间均比明显黄精粗多糖短,说明psp-1改善ad小鼠的空间学习记忆功能的活性比黄精粗多糖强。
[0124]
以上所述仅为本发明的优选实施例,并非因此限制本发明的专利范围,凡是在本发明的构思下,利用本发明说明书及附图内容所作的等效结构变换,或直接/间接运用在其他相关的技术领域均包括在本发明的专利保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1