DDUP作为肿瘤耐药检测、治疗及预后分子靶点的应用
ddup作为肿瘤耐药检测、治疗及预后分子靶点的应用
技术领域
1.本发明属于生物技术和医学领域,具体涉及ddup作为肿瘤耐药检测、治疗及预后分子靶点的应用。
背景技术:
2.恶性肿瘤严重威胁人类的生命健康,1943年,科学家首次将氮芥应用于淋巴瘤的治疗,揭开了现代肿瘤化疗的序幕。随后新的抗肿瘤药物不断出现,化疗成为肿瘤主要的治疗方法之一,但是如同细菌易对抗生素产生耐药性一样,肿瘤细胞也常对化疗药物产生耐药性,这是导致化学治疗失败的主要原因之一。因此肿瘤耐药是恶性肿瘤治疗急需解决的关键问题之一。以三大女性生殖系统恶性肿瘤之一的卵巢癌为例,因其恶性程度高、复发率高、治疗难度大,已严重威胁世界女性健康。由于早期缺乏特异性临床症状,且尚无有效的早期筛查、诊断方法,导致卵巢癌难以筛查,70%以上患者诊断时已处于卵巢癌晚期。目前,卵巢癌细胞减灭术后的标准治疗方案是以铂类药物为基础的化疗方案,卵巢癌在初始治疗后大部分患者都能够获得临床缓解,但70%患者在初始治疗后会出现复发,5年生存率仅39%。其中一个根本的原因是铂耐药,导致治疗过程中癌细胞对铂的反应性逐渐降低,最终导致治疗失败。因此,研究卵巢癌铂耐药的具体机制,寻找克服铂耐药的关键因素,对于提高卵巢癌治疗效果显得尤为重要。
3.肿瘤耐药的形成是多基因、多因素共同相互参与调节的生物学过程,其形成机制复杂,目前仍未研究透彻。铂类药物作用的主要靶点为dna,通过与dna形成单加合物或链内交联形成铂-dna加合物,从而抑制dna复制及转录,导致dna断裂和错误编码,引起的这种dna损伤可以通过激活多种信号传导途径而激活细胞凋亡途径,最终导致细胞死亡。当信号传导途径被异常激活或抑制,铂的流出增加或吸收减少导致细胞内铂积累减少,dna损伤修复通路异常激活等,导致细胞对铂类药物的抵抗、凋亡抑制,促使肿瘤铂化疗耐药的产生。
4.为了保护dna免受内源性或外源性的有毒物质诱导的损伤,哺乳动物细胞进化出一个复杂的信号通路和修复网络以移除或耐受dna损伤,包括dna损伤反应(dna damage response,简称ddr)通路和dna修复蛋白。铂类化疗药物通过共价结合dna的n7位点鸟嘌呤,与dna形成加合物继发诱导dna双链断裂。铂类药物治疗后,癌细胞进行dna损伤修复或凋亡决定了其是存活还是死亡。ddr在维持基因组完整性方面以及耐药性的产生起着关键作用。研究发现基因组不稳定性是癌症的标志,并且ddr缺陷导致肿瘤的风险增加。dna修复途径主要包括:核苷酸切除修复(ner)、错配修复(mmr)、碱基切除修复(ber)、非同源重组修复(nhej)、同源重组修复(hr)、复制后修复(prr)等。不同的修复途径之间相互作用及协调导致dna损伤修复和细胞存活,这些途径的改变(异常激活或沉默)有助于形成对铂类药物的敏感性或者抗性。由于不能正确地检测、修复、或对dna损伤做出正确的反应,导致了化疗耐药表型的产生。因此,进一步深入探究肿瘤细胞化疗耐药机制,为肿瘤的治疗提供更有效的作用靶点成为亟待解决的问题。
技术实现要素:
5.本发明第一方面的目的,在于提供一种ddup。
6.本发明第二方面的目的,在于提供与本发明第一方面的ddup相关的生物材料。
7.本发明第三方面的目的,在于提供本发明第一方面的ddup和/或第二方面的生物材料的应用。
8.本发明第四方面的目的,在于提供检测ddup的物质在制备肿瘤耐药检测产品、肿瘤预后检测产品或预测肿瘤复发检测中的应用。
9.本发明第五方面的目的,在于提供atr抑制剂在制备ddup抑制剂中的应用。
10.本发明第六方面的目的,在于提供ddup抑制剂在制备提高肿瘤对铂类药物敏感性的产品中的应用。
11.本发明第七方面的目的,在于提供一种药物。
12.本发明第八方面的目的,在于提供一种试剂盒。
13.为了实现上述目的,本发明所采取的技术方案是:
14.本发明的第一个方面,提供一种ddup,所述ddup的氨基酸序列包含:
15.a)mwlvectgrdltglscllsmdrqprrrqhvagcrdvppplpqgswgqtsprhsilcsksgcdllgggeyngetsgeeflapawtcraqqaatwlsvqqtshkalgpaggaamssklspeeqflsrihflrtfmcsvagaelpgipqatengegcrpardpasspsslsmasvytqcssaqlvsals(seq id no.2);或
16.b)与seq id no.2所示的氨基酸序列具有95%、96%、97%、98%或99%的序列同一性的序列。
17.本发明的第二个方面,提供与本发明第一方面的ddup相关的生物材料,所述生物材料包含a1)~a4)中的任一种:
18.a1)编码本发明第一方面的ddup的核酸分子;
19.a2)含有a1)所述核酸分子的表达盒;
20.a3)含有a1)所述核酸分子的载体;
21.a4)含有a1)所述核酸分子的细胞系。
22.优选地,所述核酸分子的核苷酸序列如seq id no.1所示。
23.优选地,所述细胞系不包含繁殖材料。
24.本发明的第三个方面,提供第一方面的ddup和/或第二方面的生物材料在调控dna损伤和/或dna损伤修复中的应用。
25.优选地,所述应用为体外非治疗目的。
26.本发明的第四个方面,提供检测ddup的物质在制备肿瘤耐药检测产品、肿瘤预后检测产品或预测肿瘤复发检测中的应用。
27.优选地,所述检测ddup的物质包含定量检测ddup的物质。
28.优选地,所述检测ddup的物质包含在基因水平和/或蛋白质水平上检测ddup的物质。
29.优选地,所述物质包含用于选自下组的一种或多种检测技术或方法中的物质:免疫组织化学法(如免疫荧光分析、反向酶联免疫吸附、免疫胶体金法)、western印迹法、northern印迹法、pcr、生物芯片法。
30.优选地,所述免疫组织化学法选自:免疫荧光分析、反向酶联免疫吸附和免疫胶体
金法。
31.优选地,所述检测ddup的物质选自:对ddup具有特异性的物质,例如其抗体(优选单克隆抗体);ddup特异性的探针、基因芯片、pcr引物等。
32.优选地,所述检测ddup的物质选自:ddup抗体、ddup特异性的探针、基因芯片、pcr引物。
33.优选地,所述ddup的氨基酸序列包含:
34.a)mwlvectgrdltglscllsmdrqprrrqhvagcrdvppplpqgswgqtsprhsilcsksgcdllgggeyngetsgeeflapawtcraqqaatwlsvqqtshkalgpaggaamssklspeeqflsrihflrtfmcsvagaelpgipqatengegcrpardpasspsslsmasvytqcssaqlvsals(seq id no.2);或
35.b)与seq id no.2所示的氨基酸序列具有95%、96%、97%、98%或99%的序列同一性的序列。
36.优选地,所述ddup的核苷酸序列如seq id no.1所示。
37.优选地,所述产品为试剂盒。
38.优选地,所述肿瘤包括卵巢癌、乳腺癌、肝癌、肺癌、胃癌中的至少一种。
39.优选地,所述肿瘤耐药中的药包含铂类药物;进一步优选地,所述肿瘤耐药中的药包括卡铂、顺铂中的至少一种。
40.本发明的第五个方面,提供atr抑制剂在制备ddup抑制剂中的应用。
41.优选地,所述atr抑制剂包含berzosertib。
42.优选地,所述ddup抑制剂包含抑制ddup活性的物质;进一步优选地,所述ddup抑制剂为抑制ddup磷酸化的物质。
43.优选地,所述抑制ddup磷酸化的物质抑制ddup t174位点磷酸化。
44.优选地,所述ddup的氨基酸序列包含:
45.a)mwlvectgrdltglscllsmdrqprrrqhvagcrdvppplpqgswgqtsprhsilcsksgcdllgggeyngetsgeeflapawtcraqqaatwlsvqqtshkalgpaggaamssklspeeqflsrihflrtfmcsvagaelpgipqatengegcrpardpasspsslsmasvytqcssaqlvsals(seq id no.2);或
46.b)与seq id no.2所示的氨基酸序列具有95%、96%、97%、98%或99%的序列同一性的序列。
47.本发明的第六个方面,提供ddup抑制剂在制备提高肿瘤对铂类药物敏感性的产品中的应用。
48.优选地,所述ddup抑制剂包含抑制ddup活性的物质、降解ddup的物质、降低ddup表达水平的物质中的至少一种;进一步包含抑制ddup活性的物质、降低ddup表达水平的物质中的至少一种。
49.优选地,所述抑制ddup活性的物质包含抑制ddup磷酸化的物质;进一步优选地,所述抑制ddup活性的物质包含atr抑制剂;更进一步优选地,所述抑制ddup活性的物质包含berzosertib。
50.优选地,所述降低ddup表达水平的物质包含b1)~b3)中至少一种:
51.b1)靶向ddup的sirna、dsrna、mirna、核酶、grna或shrna;
52.b2)编码b1)的核酸分子;
53.b3)包含b2)的表达盒、载体或转基因细胞系。
54.优选地,所述靶向ddup的shrna的序列如seq id no.3或seq id no.4所示。
55.优选地,所述靶向ddup的grna的序列如seq id no.5或seq id no.6所示。
56.优选地,所述靶向ddup的sirna的序列如seq id no.21或seq id no.22所示。
57.优选地,所述ddup抑制剂包含c1)~c10)中至少一种:
58.c1)berzosertib;
59.c2)靶向ddup的shrna;
60.c3)编码c2)的核酸分子;
61.c4)包含c3)的表达盒、载体或转基因细胞系;
62.c5)靶向ddup的shrna;
63.c6)编码c5)的核酸分子;
64.c7)包含c6)的表达盒、载体或转基因细胞系;
65.c8)靶向ddup的sirna;
66.c9)编码c8)的核酸分子;
67.c10)包含c9)的表达盒、载体或转基因细胞系。
68.优选地,所述ddup的氨基酸序列包含:
69.a)mwlvectgrdltglscllsmdrqprrrqhvagcrdvppplpqgswgqtsprhsilcsksgcdllgggeyngetsgeeflapawtcraqqaatwlsvqqtshkalgpaggaamssklspeeqflsrihflrtfmcsvagaelpgipqatengegcrpardpasspsslsmasvytqcssaqlvsals(seq id no.2);或
70.b)与seq id no.2所示的氨基酸序列具有95%、96%、97%、98%或99%的序列同一性的序列。
71.优选地,所述ddup的核苷酸序列如seq id no.1所示。
72.优选地,所述肿瘤包括卵巢癌、乳腺癌、肝癌、肺癌、胃癌中的至少一种。
73.优选地,所述铂类药物包括卡铂、顺铂中的至少一种。
74.优选地,所述产品为药物。
75.本发明的第七个方面,提供一种药物,包含:ddup抑制剂。
76.优选地,所述包含抑制ddup活性的物质、降解ddup的物质、降低ddup表达水平的物质中的至少一种。
77.优选地,所述ddup抑制剂包含降解ddup的物质、降低ddup表达水平的物质中的至少一种;或降解ddup的物质、降低ddup表达水平的物质中的至少一种以及抑制ddup活性的物质。
78.优选地,所述抑制ddup活性的物质包含抑制ddup磷酸化的物质;进一步优选地,所述抑制ddup活性的物质包含atr抑制剂;更进一步优选地,所述抑制ddup活性的物质包含berzosertib。
79.优选地,所述降低ddup表达水平的物质包含b1)~b3)中至少一种:
80.b1)靶向ddup的sirna、dsrna、mirna、核酶、grna或shrna;
81.b2)编码b1)的核酸分子;
82.b3)包含b2)的表达盒、载体或转基因细胞系。
83.优选地,所述靶向ddup的shrna的序列如seq id no.3或seq id no.4所示。
84.优选地,所述靶向ddup的grna的序列如seq id no.5或seq id no.6所示。
85.优选地,所述靶向ddup的sirna的序列如seq id no.21或seq id no.22所示。
86.优选地,所述ddup抑制剂包含:
87.c2)~c10)中至少一种;或
88.c2)~c10)中至少一种和c1;
89.c1)berzosertib;
90.c2)靶向ddup的shrna;
91.c3)编码c2)的核酸分子;
92.c4)包含c3)的表达盒、载体或转基因细胞系;
93.c5)靶向ddup的shrna;
94.c6)编码c5)的核酸分子;
95.c7)包含c6)的表达盒、载体或转基因细胞系;
96.c8)靶向ddup的sirna;
97.c9)编码c8)的核酸分子;
98.c10)包含c9)的表达盒、载体或转基因细胞系。
99.优选地,所述ddup的氨基酸序列包含:
100.a)mwlvectgrdltglscllsmdrqprrrqhvagcrdvppplpqgswgqtsprhsilcsksgcdllgggeyngetsgeeflapawtcraqqaatwlsvqqtshkalgpaggaamssklspeeqflsrihflrtfmcsvagaelpgipqatengegcrpardpasspsslsmasvytqcssaqlvsals(seq id no.2);或
101.b)与seq id no.2所示的氨基酸序列具有95%、96%、97%、98%或99%的序列同一性的序列。
102.优选地,所述ddup的核苷酸序列如seq id no.1所示。
103.优选地,所述药物用于提高肿瘤对铂类药物敏感性。
104.优选地,所述肿瘤包括卵巢癌、乳腺癌、肝癌、肺癌、胃癌中的至少一种。
105.优选地,所述铂类药物包括卡铂、顺铂中的至少一种。
106.优选地,所述药物还包含铂类药物。
107.优选地,所述药物还包含顺铂、卡铂中的至少一种。
108.优选地,所述药物用于治疗肿瘤。
109.优选地,所述肿瘤包括卵巢癌、乳腺癌、肝癌、肺癌、胃癌中的至少一种。
110.本发明的第八个方面,提供一种试剂盒,包含:检测ddup的物质。
111.优选地,所述检测ddup的物质包含定量检测ddup的物质。
112.优选地,所述检测ddup的物质包含在基因水平和/或蛋白质水平上检测ddup的物质。
113.优选地,所述物质包含用于选自下组的一种或多种检测技术或方法中的物质:免疫组织化学法(如免疫荧光分析、反向酶联免疫吸附、免疫胶体金法)、western印迹法、northern印迹法、pcr、生物芯片法。
114.优选地,所述免疫组织化学法选自:免疫荧光分析、反向酶联免疫吸附和免疫胶体金法。
115.优选地,所述检测ddup的物质选自:对ddup具有特异性的物质,例如其抗体(优选单克隆抗体);ddup特异性的探针、基因芯片、pcr引物等。
116.优选地,所述检测ddup的物质选自:ddup抗体、ddup特异性的探针、基因芯片、pcr引物。
117.优选地,所述ddup的氨基酸序列包含:
118.a)mwlvectgrdltglscllsmdrqprrrqhvagcrdvppplpqgswgqtsprhsilcsksgcdllgggeyngetsgeeflapawtcraqqaatwlsvqqtshkalgpaggaamssklspeeqflsrihflrtfmcsvagaelpgipqatengegcrpardpasspsslsmasvytqcssaqlvsals(seq id no.2);或
119.b)与seq id no.2所示的氨基酸序列具有95%、96%、97%、98%或99%的序列同一性的序列。
120.优选地,所述ddup的核苷酸序列如seq id no.1所示。
121.优选地,所述试剂盒用于d1)~d3)中任一种:
122.d1)肿瘤耐药检测;
123.d2)肿瘤预后检测;
124.d3)预测肿瘤复发检测。
125.优选地,所述肿瘤包括卵巢癌、乳腺癌、肝癌、肺癌、胃癌中的至少一种。
126.优选地,所述肿瘤耐药中的药包含铂类药物;进一步优选地,所述肿瘤耐药中的药包括卡铂、顺铂中的至少一种。
127.本发明的有益效果是:
128.本发明首次公开了一种ddup;该ddup可以调控dna损伤和/或dna损伤修复,与肿瘤患者的铂类药物抵抗性、复发、总体生存时间、无病生存时间显著相关,可作为肿瘤耐药、肿瘤预后、肿瘤复发检测的标志物。
129.本发明首次公开了ddup抑制剂在提高肿瘤对铂类药物敏感性中的应用,通过ddup抑制剂和铂类药物的联用,可以使肿瘤细胞的γ-h2ax显著增加,细胞增殖指标ki67显著降低,细胞凋亡比例显著增加,显著抑制肿瘤的重量、体积、延长患者的生存期,提高肿瘤治疗效果。
附图说明
130.图1是lncrna ctbp1-dt编码的微蛋白-ddup对dna损伤修复的影响图:其中,a是定量蛋白质组学联合核糖体图谱分析鉴定参与dna损伤修复的新因子的结果图;b是lncrna ctbp1-dt编码的微蛋白-ddup的质谱图;c是基因组上ctbp1-dt的位置、假定的ddup orf位于第二个外显子以及全长的ddup用于合成抗-ddup多抗的示意图;d是实施例1中彗星实验结果图;e是实施例1中免疫荧光分析结果图***表示:与vector相比,p<0.001。
131.图2是lncrna ctbp1-dt对dna损伤修复的影响图:其中,a是ddup突变体载体构建示意图及其在cpt处理下的表达结果图;b是ddup突变体载体在cpt处理下的表达的免疫荧光分析结果图;c是实施例2中彗星实验结果图;d是ddup突变体载体在cpt处理下检测γ-h2ax焦点的免疫荧光分析结果图;e是实施例2中免疫印迹实验结果图;***表示:与vector/ctrl相比,p<0.001。
132.图3是ddup的磷酸化对于ddup调控的dna损伤修复的影响图:其中,a是定量蛋白质组学实验鉴定与ddup相互作用的蛋白的结果图;b是co-ip分析ddup与atr、atm、rad18、γ-h2ax和rad51c的相互作用的结果图;c是atr和ddup的分子对接示意图;d是co-ip分析全长
或截短的ddup与atr的相互作用的结果图;e是co-ip分析ddup及其突变体与ptq/sq的相互作用的结果图;f是不同突变体转染的细胞中的γ-h2ax焦点的免疫荧光分析结果图;***表示:与vector相比,p<0.001。
133.图4是ddup对rad18在dna损伤位点的停留的影响图:其中,a是ddup焦点与rad18、γ-h2ax和rad51c焦点共定位的结果图;b是co-ip分析ddup与rad18、γ-h2ax和rad51c相互作用的结果图;c是模拟的ddup/wt、ddup/t174d三维结构图;d是rad18及rad51c焦点随着时间的变化情况结果图;e是实施例4中同源重组修复检测实验结果图;f是免疫共沉淀及免疫印迹实验分析ddup失调对pcna单泛素化的调控的结果图;***表示:与vector/ctrl相比,p<0.001。
134.图5是ddup对体外卵巢癌细胞对顺铂的抗性的影响图:其中,a是ctbp1-dt与肿瘤患者的无进展生存期、进展后生存期或无复发生存期的关系图;b是ddup对铂类药物抵抗性(platinum resistance)和复发(relapse)的影响的结果图;c是ddup对总体生存时间及无病生存时间的影响的结果图;d是患者来源的卵巢癌细胞(pdovc)中染色体结合的和总ddup表达的免疫印迹分析的结果图;e是患者来源的卵巢癌细胞(pdovc)的同源重组修复检测实验的结果图;f是免疫印迹实验分析患者来源的卵巢癌细胞(pdovc)中染色体结合和总单泛素化pcna和ddup的表达的结果图;***表示:与不添加berzosertib的pdovcs#3/pdovcs#4相比,p<0.001。
135.图6是ddup对体内卵巢癌细胞对顺铂的抗性的影响图:其中,a是病人来源卵巢癌异种移植物模型构建的示意图;b是不同处理后异种移植肿瘤的代表性图;c是不同处理对异种移植肿瘤的重量的影响图;d是不同处理对异种移植肿瘤的体积的影响图;e是不同各处理的小鼠的kaplan-meier生存曲线图;f是不同处理的小鼠的肿瘤的免疫组化染色图和tunel图;***表示:与vehicle相比,p<0.001。
具体实施方式
136.以下通过具体的实施例对本发明的内容作进一步详细的说明。
137.应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。
138.下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。本实施例中所使用的材料、试剂等,如无特别说明,为从商业途径得到的试剂和材料。
139.术语解释:
140.质谱数据,是指利用质谱仪得到的肽段信息,二级谱图等数据。
141.核糖体图谱高通量测序数据:是指通过分离核糖体-mrna复合体进行高通量测序得到的数据。
142.喜树碱:一种诱导dna损伤的化疗药物。
143.pdx模型:是将患者手术切除的肿瘤组织接种于免疫缺陷小鼠体内而建立的新一代肿瘤模型。保存了病人肿瘤组织的基因型和表型的多样性,可以比较真实的反映原始肿瘤的特性。
144.焦点:英文为foci,focus的复数形式,其原本含义是焦点。用在dna损伤修复中是指各种损伤修复蛋白被募集到dna损伤位点。当dna发生损伤时,损伤位点会招募修复蛋白
形成焦点,这时用相应蛋白的抗体做细胞免疫荧光染色,在荧光显微镜下可观察到细胞核中出现多个由dna损伤相关蛋白形成的小点。这种方法可以用来检测损伤修复相关蛋白再dna损伤位点的募集。
145.berzosertib:atr激酶的抑制剂。
146.实施例1 lncrna ctbp1-dt编码的微蛋白-ddup促进dna损伤修复
147.(1)质谱鉴定联合核糖体图谱高通量测序鉴定dna损伤修复新蛋白ddup
148.质谱鉴定联合核糖体分析方法:使用喜树碱(camptothecin,cpt)处理293t细胞1小时,诱导293t细胞发生dna损伤。lc-ms/ms质谱鉴定:分别提取对照组(dmso)和cpt处理组的全蛋白,进行酶解后分成10个组分进行质谱鉴定,每组具有两个生物学重复。使用核糖体图谱高通量测序:分别提取对照组和cpt处理组的核糖体rna建库后进行高通量测序。联合质谱分析和核糖体图谱高通量测序结果,鉴定到lncrna ctbp1-dt翻译的蛋白ddup,与dna损伤修复相关。具体步骤如下:
149.1)核糖体图谱高通量测序:
150.①
细胞铺板及药物处理:当细胞的生长密度达到80~90%时,将细胞以3.5
×
106/ml的密度接种在10-cm培养皿上。第二天,弃去培养上清,加入10ml配制相应药物的培养基(chx完全培养基(即含放线菌酮(cycloheximide,#5087390001,sigma-aldrich,st.louis,missouri,100μg/ml)、5%fbs的dmem培养基)。1小时后收集细胞样品。
151.②
收集细胞样品:用预热的pbs润洗细胞,换成chx完全培养基(即含放线菌酮(cycloheximide,#5087390001,sigma-aldrich,st.louis,missouri,100μg/ml)、5%fbs的dmem培养基,于37℃细胞培养箱中培养3~4分钟;弃去培养基,用5ml预冷的pbs洗一遍;每个培养皿加入300μl预冷的lysis buffer(裂解液),lysis buffer:140mm nacl、5mm mgcl2、10mm tris
–
hcl ph 8.0、1%(v/v)triton x-100、0.5%(w/v)sodium deoxycholate(脱氧胆酸钠)、0.4u/μl rnase inhibitor、20mm dtt、0.1mg/ml cycloheximide(环己酰亚胺)、10mm rvc(氧钒核糖核苷复合物)、0.1%(v/v)cocktail),并用细胞刮轻轻将细胞混合液刮下,收集至1.5ml ep管中。用枪反复吹打细胞悬液,孵育15min,期间数次颠倒ep管;16,000
×
g,4℃离心10min。取上清,待用;以上操作在冰上进行。
152.③
制备蔗糖密度梯度:提前配制10%(w/v)和50%(w/v)的蔗糖溶液;在超速离心管中加入5.5ml 10%的蔗糖溶液,用长针头在离心管底部小心加入5.5ml 50%的蔗糖溶液,避免溶液混合;使用密度梯度泵制作蔗糖密度梯。
153.④
样品超速离心及分离:将处理好的样品轻轻地加入至蔗糖密度梯度超速离心管中,sw40ti超速离心转子170,000
×
g,4℃离心2h;用梯度组分分离仪器将超速离心后的样品组分依次分离收取,收集80s之后各组分的样品,同时可获得各组分依次流出的峰图。
154.⑤
组分样品rna的提取和鉴定:将收集的蔗糖梯度样品分别加入等体积的trizol溶液,按照trizol法提取rna。
155.⑥
文库构建与质检:采用ribo-zero
tm kit从总rna样品中去除rrna(部分lncrna具有与mrna相同的polya尾结构,用去除rrna的方法能够最大程度地保留含有polya尾的lncrna)。向富集得到的rna中加入fragmentation buffer将rna打断成小片段。然后以片段化的rna为模板,加入6bp随机引物(random hexamers)反转录合成cdna第一条链,再加入缓冲液、dntps(dntp中的dttp用dutp取代)、dna polymerase i和rnase h合成cdna第二条链。
合成的双链cdna经纯化、末端修复、加a、连接测序接头处理后,用user酶降解含有u的cdna第二链并进行pcr富集,最后用ampure xp beads纯化pcr产物,得到最终的链特异性文库。文库构建完成后,先使用qubit 2.0进行初步定量,稀释文库,随后使用agilent 2100对文库的插入片段大小进行检测,插入片段符合预期后,使用q-pcr方法对文库的有效浓度进行准确定量,以保证文库质量。
156.⑦
上机测序:使用illumina高通量测序平台novaseq hiseq x ten进行测序。
157.2)质谱鉴定:
158.①
细胞铺板及药物处理:当细胞的生长密度达到80~90%时,将细胞以3.5
×
106/ml的密度接种在10-cm培养皿上。第二天,弃去培养上清,加入10ml配制相应药物的培养基(cpt,10μm)。30分钟后收集细胞样品。
159.②
收集细胞样品:弃去培养基,并使用1
×
pbs缓冲液润洗细胞2次。使用细胞刮收取细胞。
160.③
将样品转移至对应的10kd的超滤管中,12,000g离心20min。
161.④
每个超滤管各加200μl buffer1(8m urea/100mm tris-hcl,ph 8.5),充分溶解变性蛋白。
162.⑤
加入20μl dtt(二硫苏糖醇,100mm)溶液,37℃反应2h还原二硫键。
163.⑥
加入20μl iaa(吲哚-3-乙酸,500mm)溶液,室温避光反应15min。
164.⑦
将还原烷基化后的蛋白溶液12,000g离心20min,弃掉收集管底部溶液。加入200ul buffer1离心,重复2次。
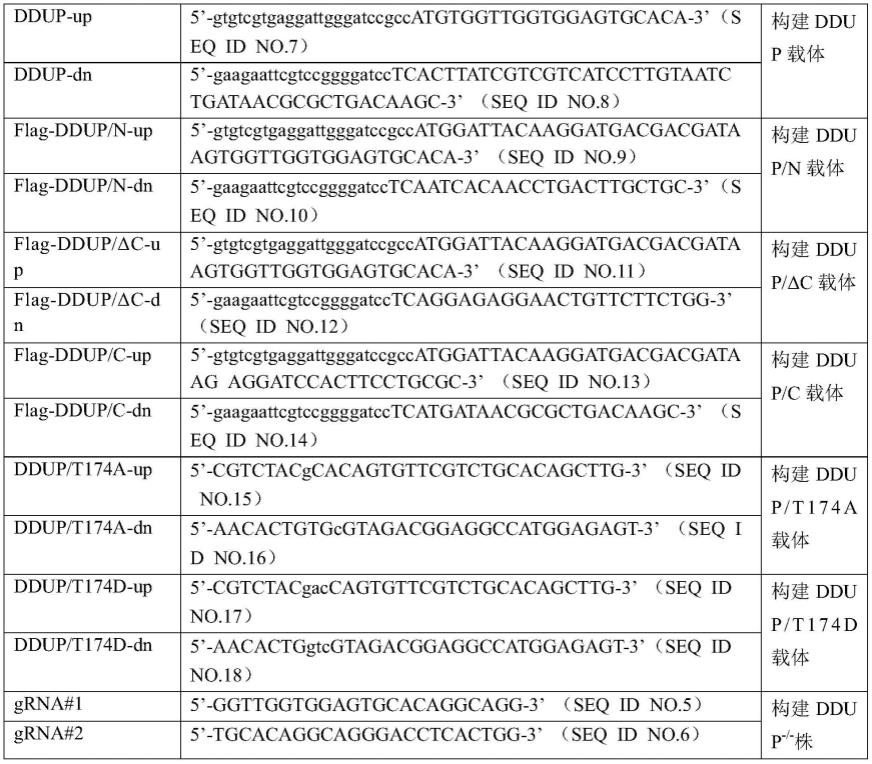
165.⑧
加200μl buffer2(8m urea/100mm tris-hcl,ph 8.0),12,000g离心20min,弃掉收集管底部溶液,重复1次。
166.⑨
加200μl碳酸氢铵溶液(25mm),12,000g离心20min,弃掉收集管底部溶液,重复2次。
167.⑩
更换新收集管,加入100μl胰酶溶液(0.01ug/ul),37℃反应14h。
168.取出超滤管,12,000g离心20min,收集酶解后肽段。
169.超滤管中再加入100μl碳酸氢铵溶液(25mm),12,000g离心10min,收集管底溶液并与的溶液合并;离心、冷冻干燥。
170.冷冻干燥后的样品用100μl流动相a(10mm甲酸铵,5%(v/v)乙腈水溶液,ph 10.0)溶液溶解;肽段分离在agilent 1100hplc上进行,检测波长:紫外210nm,流速:0.25ml/min;分离梯度为60分钟内流动相b(10mm甲酸铵,90%(v/v)乙腈水溶液,ph 10.0)从5~38%(v/v),线性梯度。在梯度范围内每隔1分钟收集1管,共收集15管洗脱溶液,离心干燥待做lc-ms分析。
171.lc-ms/ms分析:使用thermo scientific q exactive质谱仪进行鉴定。
172.结果如图1中a、b所示:通过质谱数据和核糖体图谱测序数据整合分析,鉴定出dna损伤中上调最高的长链非编码rna-ctbp1-dt(序列为:atgtggttggtggagtgcacaggcagggacctcactggactttcctgtctgctcagcatggacaggcagcccaggagaaggcagcacgtggccgggtgcagggacgtaccacccccacttccccaggggagctggggtcagacgagtcccaggcactccatcctctgcagcaagtcaggttgtgatttactagggggtggtgaatataatggagagacttctggggaggaattcctggctcccgcgtggacttgcaga
gctcaacaggcagccacgtggctgagtgtccagcaaacatcacataaggctttgggtcctgcaggtggggctgccatgagcagcaagctcagtccagaagaacagttcctctccaggatccacttcctgcgcacttttatgtgcagtgtagctggagcagagctccccggaattccacaggcaactgagaacggagagggatgcaggccagccagggatccagcgtcttccccatcgtcactctccatggcctccgtctacacacagtgttcgtctgcacagcttgtcagcgcgttatcatga,seq id no.1);cpt(10μm)处理后,产生γ-h2ax,表明cpt处理后细胞发生损伤;lc-ms/ms的分析结果表明,与对照组相比,cpt处理组鉴定到lncrna ctbp1-dt翻译的新蛋白,说明响应dna损伤的压力,lncrna ctbp1-dt可翻译出新蛋白以适应压力,将这个新蛋白命名为dna damage upregulated protein,简称为ddup(氨基酸序列如seq id no.2所示)。
173.(2)制备ddup多抗(如图1中c所示):首先,合成ddup1~83及94~179氨基酸序列作为抗原。利用制备好的ddup抗原免疫两只实验级日本大耳兔子,每只兔子免疫0.25mg抗原。在接下来的120天内,分四次注射抗原,即在第14天,第28天,第42天,第95天,分别免疫每只兔子0.26mg,0.27mg,0.28mg,0.29mg抗原。在最后一次抗原注射后的第14天,分别对两只兔子进行采血。通过elisa实验检测抗体的效价。随后通过将ddup连接到sulfolink柱子进行亲和纯化。
174.(3)彗星实验:
175.①
提前一天,提前分别将293t和hela细胞以3~4
×
105/ml均匀铺到六孔板中。次日进行脂质体转染:
176.a液为:ddup-psin-ef2质粒(购于山东维真生物科技有限公司)1μg、p3000试剂2μl、opti-mem 125μl;或shrna5nm(终浓度)、opti-mem 125μl;
177.b液为:125μl opti-mem溶液加入5μl lipofectamine
tm 3000,混匀。
178.将b液缓慢加入a液中,(切勿颠倒顺序),移液器缓慢混匀后,室温放置5min,将混合物均匀滴加到待转染细胞培养基中。48小时后,弃去培养基,加入含有终浓度为10μm的cpt或加入终浓度为10μm的vp-16完全培养液,孵育30分钟。其中vector是指psin-ef2空载,sh-v为空载体对照。
179.其中沉默ddup的shrna序列分别为:
180.shrna#1:5
’‑
acctcggaatgatgcagactcctatctcaagaggataggagtctgcatcattcctt-3’(seq id no.3);
181.shrna#2:5
’‑
acctcgttcgtctgcacagcttgtcatcaagagtgacaagctgtgcagacgaactt-3’(seq id no.4)。
182.②
按照彗星试验试剂盒(trevigen,md,usa)说明书进行实验。简述如下:弃去培养基,用胰酶消化细胞。用冷的pbs重悬细胞,使细胞浓度为1х105/ml,然后再与融化的lmagarose琼脂糖胶以1:10的比例(v/v)混匀,均匀地铺在预热(37℃)的玻片上。待玻片冷却后,将玻片浸泡在预冷的裂解液中1小时后,再浸泡在50ml中性电泳中30分钟,随后电泳1小时(21v、4℃)。随后去掉电泳液,将样品沉浸在dna沉淀液中30分钟,70%(v/v)乙醇再浸泡30分钟,随后用sybr
tm
gold(1:10000)染色。此实验平行重复3次。
183.③
使用opencomet插件对彗星图像进行测量,每例样本随机选取其中100个细胞计算尾距(tail moment)。
184.实验结果:彗星实验是一种在单细胞水平上检测dna损伤的技术。因为γ-h2ax是dna损伤的标志物,因此可通过γ-h2ax的表达来评价dna损伤。如果dna受损,断裂的碎片进
入到凝胶中,在电场的作用下,dna碎片离开原位向阳极迁移,形成拖尾。彗星实验结果如图1中d所示:在dna损伤条件下,与对照相比,高表达ddup,dna的拖尾明显减少;而敲低ddup,dna的拖尾显著增加;表明高表达ddup抑制dna损伤,敲低ddup,促进dna损伤。
185.(4)免疫荧光分析:首先,提前将细胞以2
×
104/ml铺于覆有cover slip的24孔板中。分别对293t和hela细胞进行ddup过表达或敲低(方法同(3)彗星实验),加入含有终浓度为10μm的cpt完全培养液,孵育30分钟。将经cpt处理的细胞按以下步骤进行处理:
186.①
用pbs洗2次。
187.②
加入4%多聚甲醛,室温固定30分钟。
188.③
吸走固定液,pbs洗3次。
189.④
使用0.5%triton x-100溶液对细胞进行打孔,室温下孵育细胞10分钟。
190.⑤
用1
×
pbst在摇床上清洗,重复3次,每次5分钟。
191.⑥
加入1%bsa,室温封闭45分钟。
192.⑦
使用γ-h2ax抗体配制的一抗(1:800)过夜孵育细胞。
193.⑧
回收一抗孵育液,用1
×
pbst在摇床上清洗,每次5min,重复3次。
194.⑨
孵育二抗:将一抗对应抗性的兔二抗按照1:500配制,并加入孔中与细胞常温避光孵育1h。
195.⑩
用1
×
pbst在摇床上清洗,每次5min,重复3次。
196.用dapi溶液染细胞核,常温5min。用1
×
pbst在摇床上清洗,每次5min,重复3次。
197.晾干后,用抗淬灭封片剂封片,使用共聚焦显微镜zeiss lsm880成像,使用imagej软件对每个细胞核形成的焦点数量进行计数并统计。
198.实验结果:因为γ-h2ax是dna损伤的标志物,因此可通过γ-h2ax的表达来评价dna损伤。免疫荧光实验结果如图1中e所示:在dna损伤条件下,与对照相比,高表达ddup,γ-h2ax形成的焦点明显减少,而敲低ddup,γ-h2ax形成的焦点显著增加;表明高表达ddup抑制dna损伤,敲低ddup,促进dna损伤。
199.实施例2 lncrna ctbp1-dt并不影响dna损伤修复
200.(1)构建ddup突变体载体(图2中a):使用ddup、5’utr-ddup-3’utr、5’utr-atg1(m)-3’utr、5’utr-atg2(m)-3’utr、5’utr-atg1/2(m)-3’utr序列(atg1(m)、atg2(m)分别是指seq id no.1中第一、二个atg的突变为att),通过bamh1和ecor1限制性酶切位点构建于psin-ef1a-puro-oligo载体(addgene#85132),每个载体的ddup orf框都带上一段flag标签。
201.上述载体的构建与鉴定方法具体如下:
202.①
引物设计:本研究所涉及的质粒构建引物见表1。
203.②
目的序列的pcr扩增
204.a.使用trizol(life technologies)提取293t细胞的rna后逆转录为cdna后,以cdna为模板,于0.2ml无菌ep管中配置pcr扩增反应体系(#f1066k,toyobo):293t cdna 100μg,dntps(2.5mm)5μl,10
×
buffer 5μl,dna聚合酶(kod plus neo)1μl,primer-up(10μm)1.5μl,primer-dn(10μm)1.5μl,超纯水35μl。
205.b.充分混匀上述反应液后短暂瞬时离心,放置于pcr仪器中进行pcr扩增,反应条件如下:94℃ for 2min;98℃ 10s,58℃ 30s,68℃ 1min,30个循环;68℃ 7min;
206.c.配置1%琼脂糖凝胶(加入终浓度为0.5μg/ml的溴化乙锭),将pcr扩增产物上样后电泳,在紫外灯照射下根据目的条带大小,将琼脂糖凝胶的相应条带切下来,胶回收并纯化提取扩增产物。
207.③
目的序列的pcr扩增
208.通过qiaquick gel extraction kit试剂盒进行胶回收纯化,按照试剂盒中的说明书进行以下操作:
209.a.将上述切下来的琼脂糖凝胶转移到无菌1.5ml ep管,切碎,称重胶块的重量;
210.b.按照1μg/1μl加入3倍凝胶体积的buffer qg,充分混合后50℃水浴加热10min,直至胶块完全融化,期间每5min摇晃并混匀胶溶液;
211.c.将融化后的凝胶混合液转移至安置于收集管上的spin column,做好标记;
212.d.将收集管放入离心机中,12000rpm,1min,弃去滤液;
213.e.spin column中加入750μl bufferpe,重新离心,12000rpm,30s,弃滤液;
214.f.将收集管放到离心机中空离,12000rpm,2min;
215.g.将spin column装置到新的1.5ml无菌ep管上,做好标记。使用65℃预热elution buffer后,在spin column膜的中央处垂直加入50μl elution buffer,室温静置1min;
216.h.离心机中12000rpm,离心1min,洗脱下来的液体即是收集到的dna溶液。
217.④
.载体质粒与pcr产物的双酶切
218.a.质粒载体与pcr产物的酶切反应体系如下((new england biolabs,ma,usa)):psin-ef1a-puro 1μl,bamh1(#r0136s)1μl,ecor1(#r0101s)1μl,100
×
bsa 0.5μl,10
×
buffer 5μl,无酶水41.5μl。pcr产物42.5μl,bamh1(#r0136s)1μl,ecor1(#r0101s)1μl,100
×
bsa 0.5μl,10
×
buffer 5μl。
219.b.充分混匀后瞬时离心,37℃ 2-3h,产物用1-2%琼脂糖凝胶(含终浓度为0.5μg/ml的溴化乙锭)电泳,在紫外灯下根据目的条带显示位置将琼脂糖凝胶切下来进行胶回收纯化,回收步骤参见
③
。
220.⑤
目的序列与载体质粒的连接
221.a.连接反应的配置体系如下(new england biolabs,ma,usa):目的序列3μl,psin-ef1a-puro 1μl,10
×
ligase buffer 1μl,t4 dna ligase(#m0202s)0.5μl,超纯水4.5μl.
222.b.轻轻吹打使混合液充分混匀后,连接2-3h,室温。
223.⑥
产物转化
224.a.从-80℃中取出感受态细菌(#cb101,tiangen)100μl,放置于冰上融化后,轻轻吹打充分混匀;
225.b.取10μl上述的连接产物缓慢加入感受态细菌溶液中,轻柔混匀,冰上放置30min;
226.c.将上述b的混合液放于42℃水浴锅中,90s进行热休克反应后,立即取出置于冰上,2~3min;
227.d.在c的混合液中加入900μl lb培养液(分别称量10g蛋白胨(tryptone)、5g酵母提取物(yeast extract)及10g nacl粉末,溶解并定容至1l ddh2o(蒸馏水)中,高温高压灭菌备用),放到摇床摇菌1h,230rpm,37℃;
228.e.取400μl转化菌液混合溶液滴在含固态培养基的培养皿(称量15g agarose琼脂糖粉末加至1l lb培养基中,溶解并后高温高压灭菌,并倒入100mm培养皿中冷却成固态培养基)上,倾斜培养皿使菌液分布均匀,室温静置10min;
229.f.随后把培养皿倒置于37℃恒温培养箱中进行培养,待12~16h并长出肉眼可见的独立菌落后进行挑菌鉴定。
230.⑦
重组质粒dna的提取
231.在培养皿中选取独立生长、大小适中的菌落,并转移到2ml lb培养液(含氨苄青霉素)中,放于摇床上,230rpm摇菌过夜(14~16h),37℃。通过qiaprep spin miniprepkit进行小量质粒抽提,按说明书操作:
232.a吸取2ml的浑浊菌液到2ml无菌ep管,放于离心机中,以12000rpm离心1min,弃上清;
233.b.在ep管中加入250μl buffer p1(含rnase a1),吹打使沉淀菌体充分重选充分悬浮;
234.c.接着加入250μl buffer p2,轻柔上下翻转ep管,混合4~6次,待菌液变成透明的粘稠溶液,即菌液已裂解完全;
235.d.在ep管中加入400μl buffer n3,轻柔上下翻转ep管,混合4~6次,待混合液中出现白色絮状沉淀,即裂解已被中止,将ep管放到离心机离心,12000rpm,10min,室温;
236.e.小心吸取上清并转移至qiaprep spin column管中,12000rpm,离心1min后弃滤液;
237.f.加入500μlbuffer pb至spin column中,12000rpm,离心30s后弃滤液;
238.g.加入750μl buffer pe至spin column中,12000rpm,离心30s后弃滤液;
239.h.将弃完滤液的column管放于离心机中空离2min,12000rpm;
240.i.将spin column放于新的无菌1.5ml ep管上,elution buffer在65℃水浴锅中提前预热,然后往spin column膜的中央处小心加入60μl elution buffer,室温静置1min后离心,12000rpm,1min,洗脱下来的溶液即dna溶液。
241.表1质粒构建引物序列
[0242][0243]
按照实施例1中脂质体转染的方法,将对应质粒转染进293t细胞,48小时后,弃去培养基,加入含有终浓度为10μm cpt的完全培养液孵育三十分钟,随后提蛋白进行免疫印迹实验,具体步骤如下:
[0244]
①
提取蛋白质:使用1
×
pbs缓冲液润洗细胞2次,最后一次彻底吸除润洗液。加入适量细胞裂解液(#89901,thermofisher scientific),室温裂解5~10min。随后用超声破碎仪充分破碎裂解细胞,至液体澄清且流动顺畅。根据bca蛋白定量试剂盒(#23225,thermofisher scientific)说明书的操作,对蛋白样品进行定量。
[0245]
②
配胶:配制浓度为15.5%的聚丙烯凝胶。
[0246]
③
上样:按照实验分组依次上样,若相邻两种组样品体积相差超过2倍,应使用1
×
loading buffer(#p0015l,碧云天)补齐。
[0247]
④
电泳:80v恒压浓缩,120v恒压分离,待样品中的溴酚蓝迁移至分离胶底部即可停止。
[0248]
⑤
转膜:将转膜夹浸泡在1
×
transfer buffer(10
×
transfer buffer:称取145g glycine粉末及29g trisbase粉末溶解于ddh2o,充分搅拌后用无菌水定容至1000ml,室温保存。使用前取100ml 10
×
transfer buffer,加900ml无菌水稀释成1
×
transfer buffer)中,充分湿润,随后将黑面朝下,依次放入滤纸、凝胶和pvdf膜(#1620177,bio-rad),注意上下对齐、避免产生气泡,最后用滤纸覆盖并卡紧转膜夹。将转膜夹按照正确的方向放入转膜槽,迅速加入1
×
transfer buffer至水平线,将转膜装置转移至冷库,260ma恒流转膜2h。
[0249]
⑥
封闭:转膜结束后迅速将pvdf膜取出,立即浸泡到5%脱脂牛奶中,室温封闭30min。
[0250]
⑦
一抗:利用5%脱脂牛奶配制flag一抗(#14793,cst)稀释液(1:1000),将pvdf膜和一抗同时放入抗体孵育袋中,挤压排尽气泡后封口,置于水平摇床4℃孵育过夜。
[0251]
⑧
清洗:回收一抗稀释液,将pvdf膜浸泡到1
×
tbst中,置于垂直摇床清洗多余抗体,洗3次,每次5min。
[0252]
⑨
二抗:同样利用5%脱脂牛奶配制鼠二抗(#pr30012,proteintech)稀释液(1:2000),将pvdf膜和二抗同时放入抗体孵育袋中,排尽气泡后封口,置于水平摇床常温孵育45min。
[0253]
⑩
清洗:回收二抗稀释液,将pvdf膜浸泡到1
×
tbst中,置于垂直摇床清洗多余抗体,洗3次,每次5min。
[0254]
显影:提前将清洗后的pvdf膜放入暗盒,立即覆盖上透明塑料薄膜,避免干膜。在暗室中,取等体积的化学发光液a液和b液(#wp2005,thermofisher scientific),充分混匀后立即滴加在pvdf膜表面,轻柔旋转暗盒使发光液充分覆盖在整张膜上。静置30s后再次盖上塑料薄膜,并用吸水纸吸除多余的发光液,迅速覆盖3~4张x光胶片进行曝光,根据条带亮度调整曝光时间,随后显影冲洗。
[0255]
实验结果如图2中a所示:与5’utr-atg1(m)-3’utr、5’utr-atg1/2(m)-3’utr组相比,ddup、5’utr-ddup-3’utr、5’utr-atg2(m)-3’utr实验组在dna损伤诱导剂的作用下,可翻译出ddup蛋白,由于ddup orf表达框融合了标签蛋白flag,因此可使用flag抗体进行检测。说明ddup的翻译依赖于第一个atg,而第二个atg不起作用。
[0256]
(2)ddup突变体载体在cpt处理下的表达的免疫荧光分析:实验步骤与实施例1中(4)免疫荧光分析相同,区别仅在于转染了不同的质粒(本实施例(1)中的ddup突变体载体)及使用的一抗不同(本实施例的一抗是抗-flag抗体(#14793,cst)),实验结果如图2中b所示:与5’utr-atg1(m)-3’utr、5’utr-atg1/2(m)-3’utr组相比,ddup、5’utr-ddup-3’utr、5’utr-atg2(m)-3’utr实验组在dna损伤诱导剂的作用下,可翻译出ddup蛋白,由于ddup orf表达框融合了标签蛋白flag,因此可使用flag抗体进行检测;说明ddup的翻译依赖于第一个atg,而第二个atg不起作用,更重要的是,ddup在dna损伤诱导下可以形成焦点。
[0257]
(3)彗星实验:实验步骤与实施例1中(3)彗星实验相同,区别仅在于转染了不同的质粒(本实施例(1)中的ddup突变体载体),实验结果如图2中c所示:ddup、5’utr-ddup-3’utr、5’utr-atg2(m)-3’utr实验组,在dna损伤诱导剂的作用下,由于ddup的大量表达,dna几乎没有拖尾,dna损伤被有效地修复,dna损伤更少;相反,在5’utr-atg1(m)-3’utr、5’utr-atg1/2(m)-3’utr组,由于无法表达ddup蛋白,dna拖尾很长,dna损伤得不到修复。
[0258]
(4)ddup突变体载体在cpt处理下检测γ-h2ax焦点的免疫荧光分析:实验步骤与实施例1中(4)免疫荧光分析相同,区别仅在于转染了不同的质粒(本实施例(1)中的ddup突变体载体),实验结果如图2中d所示:ddup、5’utr-ddup-3’utr、5’utr-atg2(m)-3’utr实验组,在dna损伤诱导剂的作用下,由于ddup的大量表达,几乎见不到γ-h2ax形成的焦点,说明dna损伤被有效地修复;相反,在5’utr-atg1(m)-3’utr,5’utr-atg1/2(m)-3’ut组,由于无法表达ddup蛋白,可以大量见到γ-h2ax形成的焦点,dna明显受到损伤。
[0259]
(5)免疫印迹实验:实验步骤与本实施例中(1)相同,区别仅在于采用细胞系不同
以及使用ddup的重组蛋白作为指示,具体如下:在hela细胞利用crispr-cas9系统构建了ddup敲除细胞系:广州genechem公司负责设计ddup的特异性sgrna并构建gv392质粒后提供相应的病毒液,hela细胞按照15moi病毒浓度感染cas9病毒液,感染24小时后,被感染的细胞用0.5μg/ml puromycin筛选7天,筛选稳定的hela/cas9细胞再次感染gv392-gfp-trim37 grna的病毒液(15moi)。通过gfp的感染效率保证》95%的细胞感染成功后,利用流式细胞仪将感染成功的细胞分离成单个细胞克隆。ddup grna#1和grna#2分别表示单克隆1和单克隆2。hela/ddup grna#1和grna#2的细胞dna通过qiagen基因dna试剂盒提取后,grna的靶基因序列被pcr扩增,并通过t7内源性核酸酶1(t7e1)将扩增产物消化酶切后,通过peasy-t1克隆试剂盒检测靶基因的敲除效率。所使用的grna序列分别为:grna#1:5
’‑
ggttggtggagtgcacaggcagg-3’(seq id no.5);grna#2:5
’‑
tgcacaggcagggacctcactgg-3’(seq id no.6)。实验结果如图2中e所示:利用crispr/cas9基因敲除系统对hela中的ddup进行完全敲除后,免疫印迹实验表明cpt诱导的ddup蛋白表达明显受到限制;更重要的是,免疫荧光实验cpt诱导的dna损伤更加明显,表现为γ-h2ax焦点显著增加(对照组为野生型的hela细胞,r-ddup是ddup的重组蛋白(ddup的全长蛋白用于重组蛋白制备。体外合成ddup表达框序列,构建到pete-28a载体,插入位点为(nco1/xho1)。表达菌株为:e.coli rosetta),定制于武汉爱博泰克生物科技有限公司)。
[0260]
实施例3 ddup与ddr蛋白的相互作用
[0261]
(1)定量蛋白质组学实验鉴定与ddup相互作用的蛋白(图3中a):
[0262]
①
按照实施例1中(3)彗星实验的实验方法进行脂质体转染(ddup-psin质粒)和加药处理。
[0263]
②
将经cpt处理的细胞放在冰面上。用1
×
pbs清洗细胞两遍后,彻底移除pbs。
[0264]
③
按每100mm培养皿加入1ml含蛋白酶抑制剂cocktail(#p8340,sigma-aldrich)的细胞裂解液,用细胞刮铲刮取收集细胞裂解液,收集到1.5ml离心管中,插在冰上,每10min涡旋振荡30s,总裂解时间30min。
[0265]
④
裂解液12000rpm,4℃离心15min,收集上清,且留取30μl作为input对照,-40℃保持待用。
[0266]
⑤
提前将ddup抗体偶联到珠子(#20423,thermofisher scientific)上,取40μl实施例1中(2)得到的抗体至新的1.5ml离心管中,取1
×
pbs 500μl/管,重悬冲洗珠子2~3次。
[0267]
⑥
取预处理后的珠子加入至
④
中裂解液中,4℃旋转摇床孵育过夜。
[0268]
⑦
次日,将珠子-蛋白质混合物用洗涤液轻柔上下颠倒清洗,重复5~6次。
[0269]
⑧
如果是后续进行质谱分析,则将珠子-蛋白质混合物按照实施例1中质谱鉴定的方法进行酶解并使用质谱仪进行检测。
[0270]
⑨
如果是后续进行免疫印迹实验,则将input样本取出,加入30μl细胞裂解液(#89901,thermofisher scientific),ip样本中也加入30μl细胞裂解液(#89901,thermofisher scientific)。随后均按1:5加入5
×
loading buffer(#p0015l,碧云天),100℃金属浴加热变性10min。样品后续进行免疫印迹进行实验。结果如图3中a所示:在dna损伤后,一共鉴定到12个蛋白质,其中有4个已知的与dna损伤相应相关的蛋白质,包括atr、γ-h2ax、rad18、rad51c。
[0271]
(2)co-ip分析ddup与atr、atm、rad18、γ-h2ax和rad51c的相互作用:
[0272]
实验步骤与本实施例中(1)相同,区别仅在于:使用或不使用atr的抑制剂-berzosertib(终浓度为80nm);用cpt或cddp(终浓度为10μm)诱导dna损伤。通过免疫共沉淀将ddup免疫共沉淀下来,对ddup结合的蛋白做进一步的免疫印迹实验。结果如图3中b所示:在dna损伤时(无论是cpt还是cddp处理),ddup都与atr,γ-h2ax,rad18,rad51c结合,而不与atm结合,说明dna损伤的情况下,ddup与atr,γ-h2ax,rad18,rad51c相互作用。这个结果与图3中a的质谱分析结果一致。
[0273]
(3)分子对接:
[0274]
利用i-tasser(https://zhanglab.ccmb.med.umich.edu/i-tasser/)服务器,得到模拟的ddup三维结构;atr的三维结构(pdb id:5yz0)则从rcsb蛋白数据库下载;利用cluspro(https://cluspro.org)服务器进行分子对接,结果如图3中c所示:ddup的n端,即21~53aa区域与atr结合,而atr的heat repeats区域与ddup结合,进一步从结构模拟上证明ddup与atr结合。
[0275]
(4)co-ip分析全长或截短的ddup与atr的相互作用:
[0276]
实验步骤与本实施例中(1)相同,区别仅在于:脂质体转染含ddup及其截短体的载体(ddup截短体包含flag-ddup/n(包含ddup的第1~62位氨基酸)、flag-ddup/δc(包含ddup的第1~124位氨基酸)、flag-ddup/c(包含ddup的第125~186位氨基酸),载体构建方法同实施例2中(1));使用atr抗体偶联珠子(#20423,thermofisher scientific)进行免疫共沉淀。结果如图3中d所示:atr都与flag-ddup/n、flag-ddup/δc结合,而不与flag-ddup/c结合,说明atr与ddup的n端结合,这也与前面的分析结果一致。
[0277]
(5)co-ip分析ddup及其突变体与ptq/sq的相互作用
[0278]
实验步骤与本实施例中(1)相同,区别仅在于:脂质体转染含ddup/wt(实施例2中的ddup-psin载体)或ddup的突变体ddup/t174a的载体(实施例2中的ddup/wt载体通过点突变技术将174位点的t突变为a),载体构建方法同实施例2中(1);使用或不使用atr的抑制剂-berzosertib(终浓度为80nm);使用或不使用γ-ppase蛋白磷酸酶(2u/μg)。结果如图3中e所示:通过分析ddup的序列,找到atr特异的磷酸化位点t174,并发现在突变t174位点后,atr不能在dna损伤的情况下磷酸化ddup;同样,在atr的抑制剂berzosertib存在的情况下,atr也不能磷酸化ddup。以上结果说明atr特异性磷酸化ddup t174位点。
[0279]
(6)不同突变体转染的细胞中的γ-h2ax焦点:
[0280]
实验步骤与本实施例中(1)相同,区别仅在于:在ddup敲除的hela细胞中转染不同的ddup突变体(ddup/wt和ddup/t174a、ddup/t174d)或空载体(vector),使用或不使用atr的抑制剂-berzosertib(终浓度为80nm)。结果如图3中f及图4中c所示:在hela/ddup-/-细胞中,在dna损伤时,突变t174a位点可观察的到γ-h2ax焦点的大量形成;相反,突变t174d位点几乎观察不到γ-h2ax焦点;ddup/t174d可以模拟ddup被atr磷酸化的状态,整个结构呈打开的状态,更有利于ddup与其他蛋白的结合;在ddup/wt的稳转细胞株中,dna损伤时,berzosertib可显著诱导γ-h2ax焦点;可见,ddup促进dna损伤修复,减少γ-h2ax焦点是依赖于atr的磷酸化。
[0281]
实施例4 ddup增强rad18在dna损伤位点的停留
[0282]
(1)ddup焦点与rad18、γ-h2ax和rad51c焦点共定位:
[0283]
实验步骤与实施例1中(4)免疫荧光分析相同,区别仅在于:每个实验组使用了两
种不同的一抗(ddup/rad18、ddup/γ-h2ax、ddup/rad51c抗体组合)。结果如图4中a所示:在dna损伤时,ddup形成的焦点可以分别与rad18、γ-h2ax、rad51c形成的焦点共定位。
[0284]
(2)co-ip分析ddup与rad18、γ-h2ax和rad51c相互作用:
[0285]
实验步骤与实施例3中(1)相同,区别仅在于:脂质体转染了ddup、rad18、h2a.x的干扰rna,序列如表2所示。结果如图4中b所示(scr为对照):当敲低了ddup,在dna损伤时,使用γ-h2ax免疫共沉淀下来的蛋白进行免疫印迹实验,结果表明γ-h2ax可以与rad18、rad51c结合,说明γ-h2ax与rad18、rad51c结合不依赖于ddup;而敲低了rad18,在dna损伤时,对ddup免疫共沉淀下来的蛋白进行免疫印迹实验,结果表明ddup只可以与γ-h2ax结合,说明ddup不直接与rad51c结合;而敲低了rad18,在dna损伤时,对rad18免疫共沉淀下来的蛋白进行免疫印迹实验,结果表明rad18可以与ddup、rad51c结合,说明rad18与ddup、rad51c的结合不依赖于γ-h2ax。综合以上结果表明,ddup直接与γ-h2ax、rad18结合形成复合物。
[0286]
表2干扰rna的序列
[0287]
si rad18#15
’‑
gaccaaagagacacgttctgt-3’(seq id no.19)si rad18#25
’‑
gctgtttatcacgcgaagaga-3’(seq id no.20)si ddup#15
’‑
ggaaugaugcagacuccuauc-3’(seq id no.21)si ddup#25
’‑
gttcgtctgcacagcttgtca-3’(seq id no.22)si h2ax#15
’‑
tggactaattttattaaaggatt-3’(seq id no.23)si h2ax#25
’‑
gactaattttattaaaggattgt-3’(seq id no.24)
[0288]
(3)模拟ddup磷酸化:
[0289]
利用i-tasser(https://zhanglab.ccmb.med.umich.edu/i-tasser/)服务器,得到模拟的ddup/wt三维结构。将ddup的t174位点突变为d174,利用i-tasser(https://zhanglab.ccmb.med.umich.edu/i-tasser/)服务器,得到模拟的ddup/t174d三维结构。结果如图4中c所示:ddup/t174d可以模拟ddup被atr磷酸化的状态,整个结构更松散,更有利于ddup与其他蛋白的结合。
[0290]
(4)免疫荧光实验分析rad18及rad51c焦点随着时间的变化情况:
[0291]
实验步骤与实施例1中(4)相同,区别仅在于:在ddup-/-敲除细胞中,诱导dna损伤,分别在0、0.5、6、12小时,这四个时间点收细胞进行免疫荧光实验。每组随机计数100个细胞,每个细胞核大于10个焦点的细胞为阳性细胞。结果如图4中d所示:在对照细胞里,未发生dna损伤时(即0小时),免疫荧光的结果表明rad18和rad51均匀的分布在细胞核中,很少形成焦点;而发生dna损伤10分钟后,rad18和rad51焦点迅速增加,到30分钟时,达到平台期;随后随着时间的推移,rad18和rad51焦点慢慢减少,到第24小时,回复到背景值;相反,在ddup-/-细胞里,由于ddup的完全敲除,在dna损伤达到平台期后,rad18和rad51焦点迅速减少,到第12小时,回复到背景值;以上结果说明ddup可以长时间在损伤的染色体位点上维持rad18和rad51焦点,以促进dna修复。
[0292]
(5)同源重组修复检测实验:
[0293]
①
将hela、hela/ddup-/-‑
#1、hela/ddup-/-‑
#2细胞分别以3
×
105/ml的密度均匀地铺到六孔板中。
[0294]
②
第二天,待细胞培养到约70%的密度,按照上述脂质体转染的方法,将空载和
ddup质粒(施例1的(3)中ddup-psin质粒)转染到hela细胞中;将ddup/wt、ddup/t174a质粒(实施例3的(5)中ddup/t174a载体)分别转染到ddup-/-‑
#1、ddup-/-‑
#2细胞中。
[0295]
③
24小时后更换培养基,将dr-gfp质粒(addgene#26475)和pcbascei(addgene#26477)质粒分别共转染进hela、hela/ddup-/-‑
#1、hela/ddup-/-‑
#2细胞中。
[0296]
④
48小时后,用胰蛋白酶消化并收集细胞;1
×
pbs洗涤细胞,1000rpm离心,5min,重复两遍。
[0297]
⑤
用200μl 1
×
pbs重悬细胞,用流式细胞仪检测gfp阳性的细胞。
[0298]
通过绿色荧光蛋白gfp指示同源重组修复的效率。结果如图4中e所示:通过流式细胞仪的细胞分选,在hela细胞里,过表达ddup可显著增加gfp阳性细胞,表明同源重组修复功能增强;而在ddup-/-‑
#1、ddup-/-‑
#2细胞中,过表达ddup/wt同样可以增加gfp阳性细胞;然而,过表达ddup/t174a,gfp阳性细胞与对照组相比没有变化;说明ddup可以在dna损伤时促进同源重组修复。
[0299]
(6)免疫共沉淀及免疫印迹实验分析ddup失调对pcna(增殖细胞核抗原)单泛素化的调控:
[0300]
①
将hela、hela/ddup-/-‑
#1、hela/ddup-/-‑
#2细胞分别以3
×
105/ml的密度均匀地铺到六孔板中。
[0301]
②
第二天,待细胞培养到约70%的密度,按照上述脂质体转染的方法,将空载和ddup质粒(实施例1的(3)中ddup-psin质粒)转染到hela细胞中;将ddup/wt、ddup/t174a质粒(实施例3的(5)中ddup/t174a载体)分别转染到ddup-/-‑
#1、ddup-/-‑
#2细胞中;48小时后,分别用cddp(终浓度为5μm)、uv(60j/m2)诱导dna损伤。
[0302]
③
用细胞刮收集细胞至1.5ml ep管中,用1
×
pbs清洗2遍;弃去上清,用200μl缓冲液a((10mm kcl,15mm mgcl2,10mm hepes,ph 7.9,0.34mm sucrose(蔗糖),1mm dithiothreitol(二硫苏糖醇),10%glycerol(甘油),0.1%triton x-100和蛋白酶抑制剂混合物cocktail((#p8340,sigma-aldrich))重悬细胞,冰上孵育10分钟。
[0303]
④
离心,1300g,5分钟,弃上清,收集细胞沉淀。
[0304]
⑤
用缓冲液a清洗2遍。
[0305]
⑥
加入200μl裂解液b(0.2mm egta,1mm dithiothreitol,and protease inhibitor mixture),涡旋震荡30秒,冰上裂解20分钟,每隔五分钟重复震荡一次。
[0306]
⑦
心收集染色质组分,1700g,5分钟,4℃。
[0307]
⑧
用裂解液b润洗一遍,用sample buffer重悬后进行免疫印迹实验。
[0308]
结果如图4中f所示:在hela细胞里,过表达ddup可显著增加染色质上的单泛素化的pcna,同时,免疫共沉淀实验也表示pcna可以与泛素结合,说明在dna损伤时,ddup可以促进更多的pcna到染色质位点,并且促进pcna单泛素化;而在ddup-/-‑
#1、ddup-/-‑
#2细胞中,与对照相比,在dna损伤时,染色质上的单泛素化pcna明显减少;对细胞核的pcna焦点进行统计也发现,未发生dna损伤时(即0小时),pcna很少形成焦点;而发生dna损伤10分钟后,pcna焦点迅速增加,到30分钟时达到平台期;随后随着时间的推移,pcna焦点慢慢减少,到第24小时,回复到背景值;相反,在ddup-/-细胞里,由于ddup的完全敲除,在dna损伤打到平台期后,pcna焦点迅速减少,到第12小时,回复到背景值;以上结果说明ddup可以长时间在损伤的染色体位点上维持pcna焦点,以促进dna修复。
[0309]
实施例5 ddup的表达在体外可作为卵巢铂类药物耐药的诊断标记
[0310]
(1)ctbp1-dt与肿瘤患者的无进展生存期、进展后生存期或无复发生存期的关系:
[0311]
使用kaplan-meier plotter(https://kmplot.com/analysis/)在线工具分析对ctbp1-dt进行无疾病进展分析(progression free survival,pfs)、进展生存率分析(post progression survival,pps)、无复发生存率分析(relapse free survival,rfs),结果如图5中a所示:无论在卵巢癌、肺癌、胃癌,还是乳腺癌中,高表达的ctbp1-dt都与病人更差的生存相关;可见,ctbp1-dt是一个广泛性的癌基因。
[0312]
(2)免疫组化分析:
[0313]
采用367例经石蜡包埋的卵巢癌铂耐药组织和4例新鲜卵巢癌组织以及患者的临床资料。上述所有样品均由中山大学肿瘤防治中心妇科进行手术取样,并送至本院病理科进行组织包埋、切片、诊断。本研究使用的石蜡组织切片由本院病理科提供,且该样品的使用已经征得患者的知情同意和中山大学伦理委员会的批准。在367例基于铂类药物治疗的卵巢癌临床样本中,通过免疫组化实验检测ddup蛋白水平的表达值,再通过367例临床样本分析结果对其相关的临床信息进行分析分类。根据免疫组化的结果将样品分为ddup高表达组和ddup低表达组两组,并分析两组样品与铂类药物抵抗性(platinum resistance)和复发(relapse)的相关性以及两组样品的总体生存率及复发生存率。具体实验方法如下:
[0314]
①
染色步骤:
[0315]
a.脱腊:将组织切片置于65℃恒温培养箱烤片2h,立刻浸泡于新鲜二甲苯溶液,10~15min,更换新鲜二甲苯后继续浸泡10~15min;
[0316]
b.水化:将组织切片依次浸泡于无水乙醇、95%乙醇、75%乙醇、蒸馏水,每种溶液处理3~5min;
[0317]
c.去酶:将组织切片浸泡于3%h2o2溶液中灭活内源性过氧化物酶,室温处理15min;
[0318]
d.抗原修复:将组织切片浸泡于柠檬酸钠缓冲液中进行微波修复,高火5min,解冻2min,中高火20min,结束后取出置于通风橱冷却;
[0319]
e.封闭:冷却的组织切片经1
×
pbs涮洗2~3次,甩干,用免疫组化笔勾勒组织边缘,在组织上滴加100μl的封闭工作液,室温处理20min;
[0320]
f.一抗:甩去封闭工作液,滴加100μl新鲜配制的目的抗体(ddup抗体)溶液,轻轻晃动以充分覆盖组织,4℃孵育过夜;
[0321]
g.洗涤:使用1
×
pbs洗涤3次以去除未结合的抗体,每次5min;
[0322]
h.二抗:将组织切片甩干后滴加100μl生物素标记山羊抗小鼠/兔igg溶液,室温孵育20min;
[0323]
i.洗涤:再次以1
×
pbs洗涤3次,每次3~5min;
[0324]
j.连接:甩去二抗溶液,滴加100μl辣根酶标记链霉卵白素工作液,室温反应20min;
[0325]
k.洗涤:使用1
×
pbs洗去多余的连接液,重复3次,每次3~5min;
[0326]
l.显色:甩干切片,在组织上滴加30min内配制的dab显色液,在显微镜下观察,待组织出现阳性染色立即用蒸馏水终止显色反应;
[0327]
m.洗涤:所有切片显色结束后,经自来水涮洗3~5次;
[0328]
n.复染:将组织切片浸泡于苏木精溶液复染2~3min,经自来水涮洗5~8次;
[0329]
o.分化:将组织切片浸泡于盐酸酒精溶液中5~10s,肉眼可见组织出现玫红色即可立即置于自来水中终止反应;
[0330]
p.返蓝:利用自来水轻柔冲洗切片,约30min,经显微镜观察细胞核呈现天蓝色即可终止;
[0331]
q.脱水:将组织切片依次浸泡于75%乙醇、95%乙醇、无水乙醇进行梯度脱水,各3~5min;
[0332]
r.封片:切片充分干燥后滴加适量中性树脂、盖上盖玻片,轻压玻片去除气泡。
[0333]
②
评分:
[0334]
本研究所有免疫组化的染色结果均由两位专业的病理科医师在双盲条件下进行独立评分,以两者打分的平均值作为最终得分。免疫组化评分采用半定量法,结合染色强度和染色范围两项指标评判表达结果。其中,染色强度计分0~3分(0:染色为阴性、1:染色为弱阳性、2:染色为阳性、3:染色为强阳性);染色范围按阳性细胞所占的百分比计为0~4分(0:没有阳性细胞、1:阳性细胞《10%、2:阳性细胞达到10%~35%、3:阳性细胞达到36~75%;4:阳性细胞》75%。最终将染色强度和染色范围得分相乘,所得的分数(即si)为最终的评分。si≥8为高表达,si《8为低表达。
[0335]
实验结果如图5中b、c所示:ddup在铂类耐药的组织中染色程度明显加深,表明ddup与卵巢癌病人耐药相关;卡方检验显示,ddup表达水平与患者铂类耐药、复发相关;kaplan-meier生存分析显示,ddup表达水平较高的铂类药物治疗患者,其总体生存时间及无病生存时间均低于ddup表达水平低的铂类药物治疗患者;cox生存分析显示,ddup的高表达水平,预示更高的死亡、铂类耐药及复发,因此可作为判断肿瘤患者耐药、预后、肿瘤复发的独立因素。
[0336]
(3)患者来源的卵巢癌细胞(pdovc)中染色体结合的和总ddup表达的免疫印迹分析:
[0337]
实验步骤与实施例4中(6)相同,区别仅在于:细胞是临床病人来源的新鲜组织分离出来的细胞;使用或不使用atr的抑制剂-berzosertib(终浓度为80nm);使用或不使用cddp(终浓度为5μm)。结果如图5中d所示:相对于pdovcs#1、pdovcs#2,在dna损伤时,pdovcs#3和pdovcs#4染色质上展现出更高水平的ddup,表明pdovcs#3和pdovcs#4对铂类药物耐药,而pdovcs#1、pdovcs#2对铂类药物相对敏感;而当cddp与berzosertib联合使用,可以观察到铂类药物耐药的#3、#4样本染色质上的ddup消失,而全细胞裂解液中的ddup不变,说明berzosertib可以抑制染色质上的ddup。
[0338]
(4)患者来源的卵巢癌细胞(pdovc)的同源重组修复检测实验:
[0339]
实验步骤与实施例4中(6)相同,区别仅在于:细胞是临床病人来源的新鲜组织分离出来的细胞;使用cddp(终浓度为5μm);使用或不使用atr的抑制剂-berzosertib(终浓度为80nm)。结果如图5中e所示:相对于pdovcs#1、pdovcs#2,在cddp处理时,pdovcs#3和pdovcs#4同源重组修复效率更高,因此对铂类药物更加耐受;相反,pdovcs#1、pdovcs#2同源重组修复效率很低;而当cddp与berzosertib联合使用,可以观察到铂类药物耐药的#3、#4样本同源重组修复被抑制,说明berzosertib可通过抑制ddup进而抑制同源重组修复,这与图5中d的实验结果一致。
[0340]
(5)免疫印迹实验分析患者来源的卵巢癌细胞(pdovc)中染色体结合和总单泛素化pcna和ddup的表达:
[0341]
实验步骤与实施例4中(6)相同,区别仅在于:细胞是临床病人来源的新鲜组织分离出来的细胞;使用cddp(终浓度为5μm)或uv(60j/m2)。结果如图5中f所示:相对于pdovcs#1、pdovcs#2,在dna损伤时,pdovcs#3和pdovcs#4染色质上展现出更高水平的单泛素化pcna;而当cddp与berzosertib联合使用,可以观察到铂类药物耐药的#3、#4样本染色质上的单泛素化pcna消失,说明berzosertib可以抑制染色质上的单泛素化pcna。
[0342]
实施例6在体内ddup可作为卵巢癌耐药的治疗靶点
[0343]
(1)建立病人来源卵巢癌异种移植物模型:
[0344]
取回新鲜的临床病人组织后(pdoc#3和pdoc#4),在超净台中,用pbs清洗两次后,将组织剪成20~0mm的小块,使用穿刺针将组织块接种于nod-scid il-2rγ-/-(nsg)小鼠(江苏集萃药康生物科技有限公司)的卵巢上,每只接种一个部位(如图6中a)。两周后,将接种了病人组织的小鼠分为四组(每组6只),分别为对照组,卡铂组,berzosertib组及卡铂,berzosertib联合处理组,分别给予对应的药物治疗:
[0345]
②
对照(vehicle)组:dmso溶液腹腔注射,100μl/次,3次/周;
[0346]
③
卡铂(carboplatin)组:腹腔注射,50mg/kg,100μl/次,3次/周;
[0347]
④
berzosertib组:腹腔注射,60mg/kg,100μl/次,3次/周;
[0348]
⑤
卡铂+berzosertib联合组:卡铂,50mg/kg,100μl/次;berzosertib,60mg/kg,100μl/次;3次/周。
[0349]
经过药物处理6周后,对四组小鼠的生存进行统计,并且取出小鼠体内的肿瘤组织,并对组织进行肿瘤体积,重量;将上述中各组的小鼠腹腔内的肿瘤组织取出后,包埋切片,并进行免疫组化分析和tunel免疫荧光染色。结果如图6中b~f所示:在pdx模型中,经过6个周期的药物处理,卡铂+berzosertib联合使用,可以显著抑制肿瘤的重量、体积、延长小鼠的生存期,而且这种差异在药物处理3周的时候已经有明显的区别;免疫组化结果也提示卡铂和atr抑制剂berzosertib联合处理,使得肿瘤细胞的γ-h2ax显著增加,细胞增殖指标ki67显著降低,细胞凋亡比例显著增加。说明卡铂和atr抑制剂berzosertib联合使用能有效地抑制ddup高表达的卵巢癌肿瘤。
[0350]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1