一种固体超强酸的制备方法与流程
本发明涉及超强酸制备技术领域,尤其涉及一种固体超强酸的制备方法。
背景技术:
硬脂酸正丁酯是化学纤维、塑料和橡胶的增塑剂,多种树脂成型加工的脱模剂和润滑剂,金属抽丝润滑剂,混凝土和织物的防水剂,鞋油光泽剂和化妆品的重要成分。硬脂酸正丁酯因无毒可用于食品包装材料、化妆品助剂和唇膏保湿剂。工业上硬脂酸正丁酯通过硬脂酸和正丁醇在硫酸作用下酯化,经高温减压脱醇、水洗、压滤而制得。但该方法以浓硫酸为催化剂存在腐蚀设备、后续处理困难、污染环境等问题,因此寻找一种清洁型催化剂具有重大意义。
自1979年日本学者Hin.M.等人报道了用SO42-作促进剂合成SO42-/MxOy型超强酸催化剂后,人们主要集中于研究这类无卤素单组分固体超强酸催化剂的制备与应用,合成了各种含SO42-促进剂的各种催化剂,极大地丰富了催化反应与应用领域,主要有锆系(SO42-/ZrO2)、钛系(SO42-/TiO2)、铁系(SO42-/Fe2O3),并用于多种催化反应体系。固体超强酸螯合双配位IR指纹区:1240~1230cm-1,1125~1090cm-1,1035~995cm-1和960~940cm-1,可分别归属为结构中的S=O双键与S-O单键;桥式配位IR指纹区:1195~1160cm-1,1110~1105cm-1,1035~1030cm-1和990~960cm-1。除了各指纹区不同外,螯合双配位比桥式配位在最高频区可区别于硫酸盐。此外,金属氧化物与SO42-作用有多种形式,利用原位IR吡啶还可定性测定超强酸催化剂表面酸的种类,B酸位在1540cm-1、L酸在1450cm-1有特征吸收指纹。
现有技术中,超强酸及其制备方法得到了广泛的报道,例如,中国专利CN104945248A公开了一种钛基固体超强酸对尼龙酸二异丁酯的酯化反应表现出了较高的酯化率。生成物经过过滤和蒸馏得到含量≥99.00%,酸值≤0.3mgKOH/g,水份≤0.10%,色泽(铂钴比色)10~20号的成品,经过滤得到的催化剂还能进行多次有效重复使用。中国专利CN102008968A报道了一种固体超强酸介孔材料及其制备方法,属于无机孔材料和催化剂制备领域,具体而言,是一种以碳纳米管为模板剂制得的具有双孔结构的SO42-/ZrO2-SiO2固体超强酸介孔材料,直接在超强酸介孔材料的制备过程中引入了孔径均匀的碳纳米管作为硬模板剂,与有机模板剂共同合成了具有双孔结构的锆硅超强酸介孔材料,在提高材料的比表面积、提高大分子扩散性能的同时,增加了超强酸的酸中心和酸位可及性,从而有望在大分子催化裂解、酯化、酰基化中得到广泛的应用。
中国专利CN101229999A报道了一种利用固体超强酸作为催化剂制备油酸醇酯的方法,所用固体超强酸的制备过程中,将ZrO2载体加入到硫酸促进剂中进行超声波浸渍,浸渍20~40分钟后再过滤烘干,所述固体超强酸作为催化剂的用量为油酸质量的0.05%~0.5%。但是,现有技术中报道的超强酸作为催化剂的催化活性不稳定,不适用于催化制备硬脂酸正丁酯。
技术实现要素:
本发明要解决的技术问题在于提供一种固体超强酸的制备方法,在硬脂酸正丁酯合成中表现出较高活性,绿色环保。
有鉴于此,本发明提供了一种固体超强酸的制备方法,包括以下步骤:提供硝酸铁溶液和硝酸铝的混合溶液;向所述混合溶液中加入分散剂、稀土助剂和沉淀剂,反应,然后陈化,第一次干燥,研磨,得到混合粉末;将所述混合粉末加入促进剂中过量浸渍,抽滤干燥,得到前驱体粉末;向所述前驱体粉末中加入粘结剂,增强剂,润滑剂和去离子水,捏合得到膏状物;将所述膏状物挤出、切粒、第二次干燥,焙烧,得到固体超强酸。
优选的,所述混合溶液中铁铝摩尔比为1:0.25-4。
优选的,所述分散剂为聚乙二醇。
优选的,所述稀土助剂为镧源化合物和/或铈源化合物。
优选的,所述沉淀剂选自碳酸氢钠、氢氧化钠、氢氧化钾、碳酸氢铵和氨水中的一种或几种。
优选的,所述促进剂为氯酸、硝酸、硫酸或过硫酸铵。
优选的,所述粘结剂选自拟薄水铝石、田菁粉、氧化铝和氧化硅中的一种或几种。
优选的,所述增强剂为玻璃纤维。
优选的,所述第二次干燥的温度为60-120℃。
优选的,所述焙烧的温度为200-700℃,焙烧的时间为3-5小时。
本发明提供了一种固体超强酸的制备方法,包括以下步骤:提供硝酸铁溶液和硝酸铝的混合溶液;向所述混合溶液中加入分散剂、稀土助剂和沉淀剂,反应,然后陈化,第一次干燥,研磨,得到混合粉末;将所述混合粉末加入促进剂中过量浸渍,抽滤干燥,得到前驱体粉末;向所述前驱体粉末中加入粘结剂,增强剂,润滑剂和去离子水,捏合得到膏状物;将所述膏状物挤出、切粒、第二次干燥,焙烧,得到固体超强酸。以硬脂酸和正丁醇为原料,在本发明制备的固体超强酸的催化作用下经酯化反应可得到硬脂酸正丁酯。本发明制备的固体超强酸酸度强,活性稳定,在硬脂酸正丁酯合成中表现出较高活性,活性组分不容易脱落流失,与反应物容易分离,重复性能良好。此外,本发明催化剂具有绿色环保,能耗低,反应转化率高,产品质量稳定等特点。
附图说明
图1为硬脂酸丁酯的合成反应装置;
图2为本发明实施例4制备的固体超强酸的红外谱图。
具体实施方式
为了进一步理解本发明,下面结合实施例对本发明优选实施方案进行描述,但是应当理解,这些描述只是为进一步说明本发明的特征和优点,而不是对本发明权利要求的限制。
本发明实施例公开了一种固体超强酸的制备方法,包括:提供硝酸铁溶液和硝酸铝的混合溶液;向所述混合溶液中加入分散剂、稀土助剂和沉淀剂,反应,然后陈化,第一次干燥,研磨,得到混合粉末;将所述混合粉末加入促进剂中过量浸渍,抽滤干燥,得到前驱体粉末;向所述前驱体粉末中加入粘结剂,增强剂,润滑剂和去离子水,捏合得到膏状物;将所述膏状物挤出、切粒、第二次干燥,焙烧,得到固体超强酸。
作为优选方案,所述硝酸铁和硝酸铝的混合溶液优选按照如下方法制备:将硝酸铁溶液和硝酸铝溶液混合,得到硝酸铁和硝酸铝的混合溶液。所述混合溶液中铁铝摩尔比优选为1:0.25-4,更优选为1:0.5。
本发明采用的分散剂优选为非离子表面活性剂,优选为聚乙二醇,更优选为聚乙二醇200-10000,最优选为聚乙二醇2000。所述分散剂的添加量优选为0.5-2wt%,更优选为0.5%。所述稀土助剂优选为镧源化合物或铈源化合物,更优选为硝酸镧或硝酸铈。稀土助剂的添加量优选为0.1-2wt%,更优选0.5wt%。
所述沉淀剂选自碳酸氢钠、氢氧化钠、氢氧化钾、碳酸氢铵和氨水中的一种或几种,所述沉淀剂优选以水溶液的形式加入。其中,所述氨水的碱液浓度优选为0.5-20%,更优选14%。本发明采用的陈化温度优选为0℃。
本发明采用的促进剂优选为氯酸、硝酸、硫酸、过硫酸铵中的一种,更优选为过硫酸铵,促进剂的摩尔浓度优选为0.25mol/L-1mol/L,更优选为0.5mol/L。
铁/铝体系在H2SO4进行浸渍时,当硫酸浓度高于0.25mol/L时,样品会被溶解成为硫酸盐,所以当用H2SO4浸渍催化剂时,浓度不能高于0.25mol/L,本发明优选采用(NH4)2S2O8为促进剂,因为S2O82-是强的氧化剂,同摩尔浓度的浸渍液中,过硫酸铵能提供更多的硫原子和总酸量,这时得到的催化剂酸性更强,环境更友好。
固体酸强度是表示把“中性”即不带电荷的碱转化为相应共轭酸的能力,用Hammett函数H0表示
H0=pKa+log[B]/[BH+]
当指示剂由碱型色变为酸型色,表示固体酸的H0等于或小于该指示剂的pKa值,反之,其H0大于指示剂的PKa值。
所述粘结剂优选选自拟薄水铝石、田菁粉、氧化铝和氧化硅中的一种或几种,更优选为田菁粉;粘结剂的添加量优选为2-8wt%。所述增强剂优选为玻璃纤维,添加量优选为5-10wt%。所述润滑剂优选为甘油,添加量优选为3-5wt%。所述前驱体粉末、粘结剂,润滑剂粉末的粒度优选为150-200目,增强剂的粒度优选为50-80目。
本发明将混合物捏合成膏状物,经过挤条机挤出,切粒,成球后,与60℃-120℃下进行干燥,优选110℃。
本发明中,将干燥后的球经过一定温度的焙烧活化,焙烧温度优选为200℃-700℃,更优选为450℃-600℃,焙烧的时间优选为为3-5小时。
本发明制备的固体超强酸球,比表面积30-200m2/g,平均孔径5nm-15nm,孔分布呈高度集中,H0均小于-12.7。
以硬脂酸和正丁醇为原料,在该球状固体超强酸的催化作用下发生酯化反应得到硬脂酸正丁酯。
催化剂的性能评价
以硬脂酸丁酯的酯化合成反应测试固体超强酸催化剂的活性大小。硬脂酸丁酯的合成反应装置如图1所示,其中,1-铁架台;2-恒温加热磁力搅拌器;3-冷凝管;4-传感器;5-三口烧瓶;6-采样口;7-磁子。
实验步骤:称取5.7g硬脂酸与5.5ml正丁醇(酸醇比为1:3),加入催化剂,于85℃恒温反应2小时。反应过程如下:
产物分析及计算方法
1酸值滴定:
a)0.1M氢氧化钾 b)0.02M氢氧化钾
c)混合试剂(乙醚:乙醇=2:1体积比)用前需用a溶液滴定至中性
d)1%酚酞乙醇溶液
2实验步骤:
准确称取定量样品0.1g于锥形瓶中,加入混合试剂10ml摇匀,加入3d酚酞,用b滴至微红,30s不消失,计消耗b的溶液体积。
酸价计算公式:
CKOH:氢氧化钾浓度(mol·L-1)
VKOH:消耗氢氧化钾体积(ml)
W样品:称取样品的重量(g)
Xi:i时刻体系的酸值
X0:反应起始体系的酸值
为了进一步理解本发明,下面结合实施例对本发明提供的技术方案进行详细说明,本发明的保护范围不受以下实施例的限制。
本发明实施例采用的原料和化学试剂均为市购。
实施例1
将0.5mol/L硝酸铁,0.5mol/L硝酸铝按摩尔比2/1混合搅拌,添加硝酸镧为助剂,添加量为0.05%(wt),聚乙二醇2000为表面活性剂,添加量为0.05%(wt),在强烈搅拌下逐滴滴加浓度为14%氨水,进行共沉淀,生成沉淀后放入冰水浴中进行陈化,洗涤抽滤于110℃下干燥后研细,用浓度为0.25mol/L的(NH4)2S2O8进行过量浸渍,然后经干燥,研磨,得到成型前粉末。
成型方法:催化剂粉末添加粘结剂田菁粉(2%wt),增强剂玻璃纤维(5%wt),润滑剂甘油(3%wt),去离子水捏合可成型的膏状物。将膏状物料通过挤条机挤出圆柱条,圆柱条放入普通制丸机中进行切粒,搓圆成d=2mm的球。
将球状催化剂550℃焙烧4小时,得到固体球状催化剂。
实施例2
0.5mol/L硝酸铁,0.5mol/L硝酸铝按摩尔比1/1混合搅拌,添加硝酸镧为助剂,添加量为0.05%(wt),聚乙二醇2000为表面活性剂,添加量为0.05%(wt),在强烈搅拌下逐滴滴加浓度为14%氨水,进行共沉淀,生成沉淀后放入冰水浴中进行陈化,洗涤抽滤于110℃下干燥后研细,用浓度为0.25mol/L的(NH4)2S2O8进行过量浸渍,然后经干燥,研磨,得到成型前粉末。
成型方法:催化剂粉末添加粘结剂田菁粉(2%wt),增强剂玻璃纤维(5%wt),润滑剂甘油(3%wt),去离子水捏合可成型的膏状物。将膏状物料通过挤条机挤出圆柱条,圆柱条放入普通制丸机中进行切粒,搓圆成d=2mm的球。
将球状催化剂550℃焙烧4小时,得到固体球状催化剂。
实施例3
0.5mol/L硝酸铁,0.5mol/L硝酸铝按摩尔比4/1混合搅拌,添加硝酸镧为助剂,添加量为0.05%(wt),聚乙二醇2000为表面活性剂,添加量为0.05%(wt),在强烈搅拌下逐滴滴加浓度为14%氨水,进行共沉淀,生成沉淀后放入冰水浴中进行陈化,洗涤抽滤于110℃下干燥后研细,用浓度为0.25mol/L的(NH4)2S2O8进行过量浸渍,然后经干燥,研磨,得到成型前粉末。
成型方法:催化剂粉末添加粘结剂田菁粉(2%wt),增强剂玻璃纤维(5%wt),润滑剂甘油(3%wt),去离子水捏合可成型的膏状物。将膏状物料通过挤条机挤出圆柱条,圆柱条放入普通制丸机中进行切粒,搓圆成d=2mm的球。
将球状催化剂550℃焙烧4小时,得到固体球状催化剂。
实施例4
0.5mol/L硝酸铁,0.5mol/L硝酸铝按摩尔比2/1混合搅拌,添加硝酸镧为助剂,添加量为0.05%(wt),聚乙二醇2000为表面活性剂,添加量为0.05%(wt),在强烈搅拌下逐滴滴加浓度为14%氨水,进行共沉淀,生成沉淀后放入冰水浴中进行陈化,洗涤抽滤于110℃下干燥后研细,用浓度为0.5mol/L的(NH4)2S2O8进行过量浸渍,然后经干燥,研磨,得到成型前粉末。
成型方法:催化剂粉末添加粘结剂田菁粉(2%wt),增强剂玻璃纤维(5%wt),润滑剂甘油(3%wt),去离子水捏合可成型的膏状物。将膏状物料通过挤条机挤出圆柱条,圆柱条放入普通制丸机中进行切粒,搓圆成d=2mm的球。
将球状催化剂550℃焙烧4小时,得到固体球状催化剂。
实施例5
0.5mol/L硝酸铁,0.5mol/L硝酸铝按摩尔比2/1混合搅拌,添加硝酸镧为助剂,添加量为0.05%(wt),聚乙二醇2000为表面活性剂,添加量为0.05%(wt),在强烈搅拌下逐滴滴加浓度为14%氨水,进行共沉淀,生成沉淀后放入冰水浴中进行陈化,洗涤抽滤于110℃下干燥后研细,用浓度为0.75mol/L的(NH4)2S2O8进行过量浸渍,然后经干燥,研磨,得到成型前粉末。
成型方法:催化剂粉末添加粘结剂田菁粉(2%wt),增强剂玻璃纤维(5%wt),润滑剂甘油(3%wt),去离子水捏合可成型的膏状物。将膏状物料通过挤条机挤出圆柱条,圆柱条放入普通制丸机中进行切粒,搓圆成d=2mm的球。
将球状催化剂550℃焙烧4小时,得到固体球状催化剂。
实施例6
0.5mol/L硝酸铁,0.5mol/L硝酸铝按摩尔比2/1混合搅拌,添加硝酸镧为助剂,添加量为0.05%(wt),聚乙二醇2000为表面活性剂,添加量为0.05%(wt),在强烈搅拌下逐滴滴加浓度为14%氨水,进行共沉淀,生成沉淀后放入冰水浴中进行陈化,洗涤抽滤于110℃下干燥后研细,用浓度为1mol/L的(NH4)2S2O8进行过量浸渍,然后经干燥,研磨,得到成型前粉末。
成型方法:催化剂粉末添加粘结剂田菁粉(2%wt),增强剂玻璃纤维(5%wt),润滑剂甘油(3%wt),去离子水捏合可成型的膏状物。将膏状物料通过挤条机挤出圆柱条,圆柱条放入普通制丸机中进行切粒,搓圆成d=2mm的球。
将球状催化剂550℃焙烧4小时,得到固体球状催化剂。
实施例7
0.5mol/L硝酸铁,0.5mol/L硝酸铝按摩尔比2/1混合搅拌,添加硝酸镧为助剂,添加量为0.05%(wt),聚乙二醇2000为表面活性剂,添加量为0.05%(wt),在强烈搅拌下逐滴滴加浓度为14%氨水,进行共沉淀,生成沉淀后放入冰水浴中进行陈化,洗涤抽滤于110℃下干燥后研细,用浓度为0.5mol/L的(NH4)2S2O8进行过量浸渍,然后经干燥,研磨,得到成型前粉末。
成型方法:催化剂粉末添加粘结剂田菁粉(2%wt),增强剂玻璃纤维(5%wt),润滑剂甘油(3%wt),去离子水捏合可成型的膏状物。将膏状物料通过挤条机挤出圆柱条,圆柱条放入普通制丸机中进行切粒,搓圆成d=2mm的球。
将球状催化剂450℃焙烧4小时,得到固体球状催化剂。
实施例8
0.5mol/L硝酸铁,0.5mol/L硝酸铝按摩尔比2/1混合搅拌,添加硝酸镧为助剂,添加量为0.05%(wt),聚乙二醇2000为表面活性剂,添加量为0.05%(wt),在强烈搅拌下逐滴滴加浓度为14%氨水,进行共沉淀,生成沉淀后放入冰水浴中进行陈化,洗涤抽滤于110℃下干燥后研细,用浓度为0.5mol/L的(NH4)2S2O8进行过量浸渍,然后经干燥,研磨,得到成型前粉末。
成型方法:催化剂粉末添加粘结剂田菁粉(2%wt),增强剂玻璃纤维(5%wt),润滑剂甘油(3%wt),去离子水捏合可成型的膏状物。将膏状物料通过挤条机挤出圆柱条,圆柱条放入普通制丸机中进行切粒,搓圆成d=2mm的球。
将球状催化剂500℃焙烧4小时,得到固体球状催化剂。
实施例9
0.5mol/L硝酸铁,0.5mol/L硝酸铝按摩尔比2/1混合搅拌,添加硝酸镧为助剂,添加量为0.05%(wt),聚乙二醇2000为表面活性剂,添加量为0.05%(wt),在强烈搅拌下逐滴滴加浓度为14%氨水,进行共沉淀,生成沉淀后放入冰水浴中进行陈化,洗涤抽滤于110℃下干燥后研细,用浓度为0.5mol/L的(NH4)2S2O8进行过量浸渍,然后经干燥,研磨,得到成型前粉末。
成型方法:催化剂粉末添加粘结剂田菁粉(2%wt),增强剂玻璃纤维(5%wt),润滑剂甘油(3%wt),去离子水捏合可成型的膏状物。将膏状物料通过挤条机挤出圆柱条,圆柱条放入普通制丸机中进行切粒,搓圆成d=2mm的球。
将球状催化剂600℃焙烧4小时,得到固体球状催化剂。
实施例10
0.5mol/L硝酸铁,0.5mol/L硝酸铝按摩尔比2/1混合搅拌,添加硝酸镧为助剂,添加量为0.05%(wt),聚乙二醇2000为表面活性剂,添加量为0.05%(wt),在强烈搅拌下逐滴滴加浓度为14%氨水,进行共沉淀,生成沉淀后放入冰水浴中进行陈化,洗涤抽滤于110℃下干燥后研细,用浓度为0.5mol/L的(NH4)2S2O8进行过量浸渍,然后经干燥,研磨,得到成型前粉末。
成型方法:催化剂粉末添加粘结剂田菁粉(2%wt),增强剂玻璃纤维(5%wt),润滑剂甘油(3%wt),去离子水捏合可成型的膏状物。将膏状物料通过挤条机挤出圆柱条,圆柱条放入普通制丸机中进行切粒,搓圆成d=2mm的球。
将球状催化剂700℃焙烧4小时,得到固体球状催化剂。
对比例
0.5mol/L硝酸铁,0.5mol/L硝酸铝按摩尔比2/1混合搅拌,添加硝酸镧为助剂,添加量为0.05%(wt),聚乙二醇2000为表面活性剂,添加量为0.05%(wt),在强烈搅拌下逐滴滴加浓度为14%氨水,进行共沉淀,生成沉淀后放入冰水浴中进行陈化,洗涤抽滤于110℃下干燥后研细,用浓度为0.25mol/L的H2SO4进行过量浸渍,然后经干燥,研磨,得到成型前粉末。
成型方法:催化剂粉末添加粘结剂田菁粉(2%wt),增强剂玻璃纤维(5%wt),润滑剂甘油(3%wt),去离子水捏合可成型的膏状物。将膏状物料通过挤条机挤出圆柱条,圆柱条放入普通制丸机中进行切粒,搓圆成d=2mm的球。
将球状催化剂550℃焙烧4小时,得到固体球状催化剂。
催化剂比表面积采用氮气物理吸附BET方法测定。
酸强度具体是以苯做溶剂,将指示剂配置成1mg/mL的指示剂溶液,每次取0.2g固体超强酸粉未放入小试管内,加入4-5滴二氯亚砜,然后滴加3-4滴指示剂溶液,观察固体表面颜色的变化。
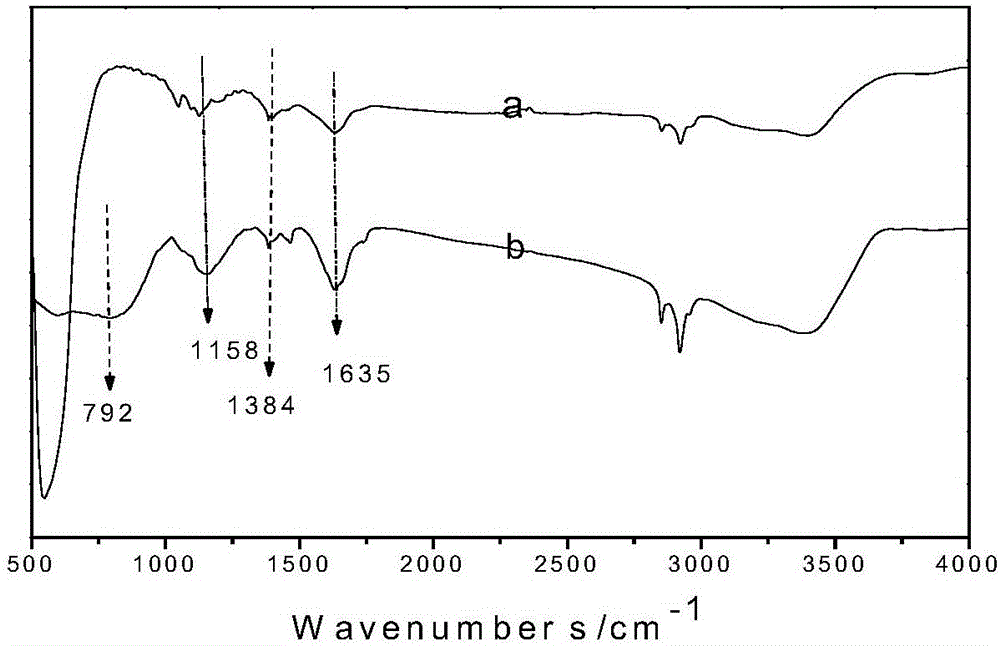
分别对实施例4和对比例制备的固体超强酸进行红外谱图分析,如图2所示,其中,a曲线为对比例制备的固体超强酸的红外谱图,b曲线为实施例4制备的固体超强酸的红外谱图。
对本发明实施例制备的固体超强酸的性能进行检测,结果如表1所述。
表1 本发明制备的固体超强酸球的性能
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!