含NO的组合物的制作方法
本发明涉及no释放组合物,特别是nonoate和n-亚硝基组合物的形成,其能够实现no和任选的其它抗微生物剂的可控释放。
发明背景
控制抗微生物剂或其它活性剂从组合物和材料中释放的特性的设备对于其抗感染或污染的有效性是关键的。
活性剂释放的初始速率和半衰期可能在材料或组合物的效力上是重要的因素。此外,通过改变环境因素引发或刺激一种活性剂或其组合或多种药剂的释放的设备也可以通过例如使得实现活性剂的有目标的释放而起着很重要的作用。
一氧化氮(no)是具有抗微生物性质的小分子,其已经吸引了相当大的兴趣,因为其在生物工艺过程中也是重要的。它是增加通过动脉和静脉的血流量的血管扩张剂,并且也是控制血小板粘附和聚集的重要因素。其在免疫系统中也起着关键的作用。关于一氧化氮的作用方式目前很多都是已知的,并且显然其在医药生物技术中在体内和体外应用方面都具有巨大的潜力。
一氧化氮的可控释放可能在治疗中是重要的。例如,一氧化氮可以预防闭塞动脉中的气囊血管成形术和支架插入后的血栓形成和再狭窄(国际专利申请wo95/24908)。将一氧化氮递送至皮肤对于具有病症如关节炎和雷诺综合症中可能发生的周围循环问题的患者也可以具有治疗益处。一氧化氮对伤口愈合和血管再生也有影响,并当例如年老患者可能出现治愈缓慢时,将一氧化氮递送至伤口可以是有益的(m.shabani等,实用局部涂抹的一氧化氮释放聚合物改善伤口修复(enhancementofwoundrepairwithatopicallyappliednitricoxide-releasingpolymer)woundrepairandregeneration,4,353,1996和s.frankh.kampfer,c.wetzler,j.pfeilschifer,一氧化氮驱使皮肤修复:已知的媒介物的新功能(nitricoxidedrivesskinrepair:novelfunctionsofanestablishedmediator)kidneyinternational,61,882,2002)。
然而,一氧化氮递送至需要的区域以及以所需的最佳剂量递送常常是困难的,因为一氧化氮是气体。一氧化氮的递送在体外例如生物技术应用中和在体内例如医药应用中都是困难的。
递送一氧化氮的各种方法已知有例如:
(a)自发释放no的分子;
(b)被代谢而提供no的分子;
(c)通过光活化no释放的分子;
(d)从聚合物和聚合物包衣释放no;
(e)从沸石和金属有机结构(mof)释放no。
(a)类分子包括一氧化氮亲核体配合物(nonoate)(c.m.maragos等,作为用于血管舒张剂效果的一氧化氮的可控生物释放的药剂的no与亲核体的配合物(complexesofnowithnucleophilesasagentsforthecontrolledbiologicalreleaseofnitric-oxide-vasorelaxanteffects)j.med.chem,34,3242,1991)。许多官能团能够与no配位,但是最普遍的是由伯胺和仲胺形成nonoate(例如,如在us专利4954526中描述的),其中no与胺部分结合,优选与仲胺结合。这些nonoate也可以称为二醇二氮烯鎓(diazeniumdiolates)。
目前,nonoate在治疗中的应用时有限的,因为它们遍布于体内,而这可能损害选择性。另一个问题在于,许多nonoate是固有地不稳定的,即使在高ph仍具有短的保存期限,且在生物条件下具有较短的半衰期(大约数分钟)。
依然,一些nonoate已经表现出有希望作为可控释放物质,用于通过温度、ph的变化,暴露于水中或通过光活化而触发一氧化氮的释放。
(alokasrinivasan,naodkebede,josephe.saavedra,alexanderv.nikolaitchik,daniela.brady,emilyyourd,keithm.davies,larryk.keefer和johnp.toscanoj.am.chem.soc.2001,13;123(23):5465-72)描述了光活化的no从nonoate的释放。然而,由备选的分解途径所产生的潜在毒性的(例如致癌物)产物是一个问题。
lehmann等eur.j.med.chem.,19/01/2005报导了释放no的不同二醇二氮烯鎓(nonoate)的用途。其中,环丙沙星-二醇二氮烯鎓杂化化合物被证明利用ph-温度触发因素释放no。据报导仅仅在ph7.0-8.0的缓冲水溶液中释放no,并且归因于触发因素的“爆发效应(bursteffect)”是相对适度的且具有短的半衰期。此外,从溶液中以这种方式可控释放不适于许多用途(例如创伤敷料)。
wo2014/012074描述了释放no的昔布(coxib)化合物(昔布化合物具有作为抗癌剂的应用),其中含有no部分的不同分子通过代谢前体药物机制递送气体(即(b)类)。
(b)类分子也包括甘油三硝酸酯和硝普酸钠(l.j.ignarro,源于内皮的一氧化氮的生物合成和代谢(biosynthesisandmetabolismofendothelium-derivednitric-oxideann.rev.pharmacol.toxicol.30,535,1990)。这些化合物目前广泛地用作血管舒张药,然而长时间的使用可能导致有毒副产物如氰化物。
此外,因为(b)类分子必须代谢而释放no,因此no靶向于具体位置可能是差的,这导致趋向于是系统性的影响。
除了某些二醇二氮烯鎓(nonoate),(c)类包括金属配合物,如c.works,c.j.jocher,g.d.bart,x.bu,p.c.ford,光化学一氧化氮前体(photochemicalnitricoxideprecursors)inorg.chem.,41,3728,2002所描述的钌配合物。然而,总体而言,其中光引发的no释放是可能的化学物质的范围是有限的。此外,在no释放后留下的小分子和配合物典型地不表现出其它作用并且甚至可能与更长期限的毒性问题相关。
(d)类一氧化氮的释放通过将一氧化氮释放化合物负载于固体制品上而将一氧化氮递送至特定的目标位置,减轻了与体系活性相关的问题。这样的no释放化合物可以是可以被涂覆于医疗器械上的聚合材料,所述医疗器械可以被用于被治疗的身体的目标特定区域。聚合物可以包含例如在化学反应后释放no的n2o2基团(国际专利申请wo95/24908和美国专利申请2002094985)。然而,在这种情况下no的释放可能难以控制并且目前所需的材料的制备可能是昂贵的。已经在心血管问题例如,再狭窄中显示了这些聚合物的可能应用。
(e)类通过从称为沸石的晶体金属交换的多孔铝硅酸盐多孔骨架材料释放一氧化氮,减轻了与体系活性相关的问题(如本申请人的早期国际专利申请wo2005/003032中所述的)。所报导的这些材料的性能是可接受的,为约1mmolno/g沸石且所述材料已经显示为具有抗血栓性能(wheatley等,journaloftheamericanchemicalsociety,128,502-509,2006)。
例如,在本申请人的早期国际专利申请wo2008/020218,wo2012/020214和wo2013/186542中,已经报导了no从的金属有机骨架材料(mof)的储存和释放。mof用于no的储存和可控释放的应用也被r.e.morris和p.s.wheatley,angew.chem.int.ed.,2008,47,4966报导,其报导了从cpo-27结构的mof通过暴露于空气和潮湿中随时间吸附和释放no的优越性能。
金属有机骨架(metal-organicframeworks,mof)是一类纳米多孔材料。在这些固体中,金属离子(mn+)与有机单元(ly-)连接在一起以形成三维网络。这些网络中有许多显示出良好的热稳定性并且极度多孔的,其自由容积高达~90%(o.m.yaghi等nature,423,705,2003(b)h.li等nature402,276,1999.(c)wo200288148-a)。
然而,许多容易获得的且潜在有用的mof材料没有表现出这种朝no的储存和释放的性质。此外,尽管通过与水接触而触发的或仅仅通过与水接触而触发的释放对于某些应用是理想的,但是这种性质对于其他的应用可能是不适宜的。另外,对于负载no的mof储存在干燥惰性条件下的要求可能也是限制。
有一些no-释放分子用作mof和相关材料中的连接基的报导。a.lowe,p.chittajallua,q.gongb,j.lib,k.j.balkusjr.micropor.mesopor.mat.2013,181(17-22)和j.l.nguyen,k.k.tananbe,和s.m.cohen,crystengcomm2010,12,2335-2338已经报导了制备成具有含有仲胺的连接基的mof结构。这些氨基可以用作一氧化氮的结合位置(与例如如上面提及的morris等所述的金属位置相反)。当暴露于高水平的潮湿,高温和/或ph变化下,所得的骨架nonoate基团能够随时间释放no,但是迄今为止所报导的材料的总no性能是有限的。
也有在其它类型的多孔材料中的no储存的报导。例如,s.diring,k.kamei和s.furukawanaturecommunications2013,2684(4)描述了能够通过暴露于uv光中而释放no的硼咪唑骨架。然而,因为一氧化氮为连接基的一部分,因此no释放降解了连接基,因此降解了骨架。因此no释放是不可逆的。
b.j.heilman,s.tr.j.oliver和p.k.mascharakj.am.chem.soc.,2012,134(28),第11573-11582页描述了能够光活性释放no的锰亚硝酰基配合物。该配合物整体上可以被吸附到多孔材料(例如al-mcm-41)中,但是这阻塞了孔隙并且防止了任何其它分子的吸附。
因此,仍然需要改进的用于抗微生物剂的可控释放的材料和组合物以解决或减轻上述缺陷中的一个或多个。
发明概述
本发明的一个方面是提供一种通过含氮官能团与no配位从而形成nonoate或n-亚硝基化合物的生物活性分子,其中所述n-亚硝基化合物是通过将no与胺或亚胺部分或两者配位而形成的。
根据本发明的另一个方面,提供一种多孔的骨架材料,如金属有机骨架材料(mof),其包含在该多孔材料或mof的内部孔隙和/或通道内的选自nonoate和/或n-亚硝基化合物的外骨架no配位的化合物。在一些实施方案中,多孔骨架材料不包括mof。
nonoate化合物由具有能够与一氧化氮分子配位的给电子部分的有机前体化合物形成。然后如此形成的反应中间体可以与另外的一氧化氮分子配位。因此,nonoate能够释放对应于前体的每一个给电子部分1当量或2当量以上,通常为2当量的no(典型地导致前体化合物本身的形成或再形成)。
n-亚硝基配合物包括与氮原子结合的单个no部分。典型地,no所结合的n原子属于胺基,然而,本发明人已经发现,no也可以与亚胺部分结合。本发明的一个特别的方面是令人惊奇地发现,在同时含有胺和亚胺部分的化合物如胍和双胍中,no可以优先地与亚胺n结合以形成新的部分,如:
所述nonoate和/或n-亚硝基化合物能够释放no且本发明人已经发现,这些化合物在这方面的性质基本上不受多孔材料/mof骨架中的吸附的影响。例如,no从nonoate和/或n-亚硝基化合物的可控释放(例如响应刺激如光,温度变化,ph等)仍然可以由材料/mof的内部孔隙和/或通道内的外骨架nonoate和/或n-亚硝基化合物实现。此外,由于其高的内部表面积,mof的高的nonoate和/或n-亚硝基化合物负载是可以的。mof的结构和性能也基本上不受nonoate和/或n-亚硝基化合物客体种类的影响。因此,本发明提供与mof材料的已知性能和应用相结合提供的nonoate和/或n-亚硝基化合物的no储存和释放性能。
金属有机骨架(mof)是一类晶体多孔材料,其中金属离子(mn+)或金属离子簇与连接基(ly-)连接在一起以形成三维网络,从而限定扩展的分析尺寸的孔隙和通道。通道网络可以一维、二维或三维地扩展。通道网络可以交叉并且可以限定内部空腔。因此金属有机骨架可以被视为限定大的内部(和外部)表面积的材料。mof可以被视为一类沸石型(zeotype)材料。
应注意,尽管说明书主要涉及mof的应用,但是这不应被解释为限制性的,并且其余的扩展的多孔材料也是已知的,如2d和1d配位聚合物,沸石和沸石型,介孔材料和有机骨架固体。这些材料应被认为在本发明的范围内,并且下文所提及的mof不应被解释为限制性的。
mof可以包含单一类型的金属离子,或多种类型金属离子。mof可以包含一种或多种过渡金属、碱金属、碱土金属的金属离子和/或其它适合的金属阳离子,如例如铝。mof可以包含多种氧化态的金属元素的离子。
mof可以包含一种或多种骨架过渡金属离子,其选自(但是不限于):tin+,vn+,crn+,mnn+,fen+,con+,nin+,cun+,znn+,agn+,ru,rh,其中n为1,2,3或4,这取决于金属和金属的氧化态。
mof可以包含选自以下的一种或多种骨架过渡金属离子:cu+,cu2+,mn2+,mn3+,zn2+,fe2+,fe3+,v3+,v4+,ag+,ru3+,rh3+,ni2+,cr2+,co2+和co3+。例如,一种或多种骨架金属离子可以选自cu+,cu2+,cr2+,zn2+,co2+,co3+,ag+,ni2+,mn2+和mn3+。优选一种或多种骨架金属离子选自cu2+,zn2+,ag+,ni2+,mn2+。
mof可以包含一种或多种骨架碱金属离子,其选自例如:na+和k+。
mof可以包含一种或多种骨架碱土金属离子,其选自(但是不限于)ca2+和mg2+,特别是mg2+。
可以存在的其余骨架金属离子(备选地或除上述之外)可以包括例如al3+。
在一些实施方案(例如对于生物、医疗和/或化妆品用途)中,可以优选存在于mof中的金属离子为被认为是对于这些用途在毒理学上可接受的那些,例如被认为具有可接受/有限的毒性的那些金属。这些考虑因素取决于应用情况并且可以由技术人员适当地决定。例如,mg,ca,fe或mn的离子可以被认为具有低的毒性。在可接受的毒性和抗微生物效力之间可以存在平衡。例如,已知相对于ni,cu和zn的离子(毒性较低但是通常抗微生物活性较低),ag表现出抗微生物性质且具有可接受的毒性。
mof可以包含单一类型的配体,或多种类型的配体。包含多种的配体和/或金属的mof可以被称为“混合组分”mof,或更具体为混合配体或混合金属mof。
每一种配体可以包含一个配位官能团,或多个配位官能团。配位官能团可以包含与给定的金属离子配位的一个原子或多个原子。
例如,每一种配体可以包含2-10个配位位置,例如2-6个配位位置,或2-4个配位位置;例如2或3个配位位置。
mof可以包含多齿配体,例如二齿,三齿或具有另外的齿数的配体。
mof可以包含羧酸酯或聚羧酸酯配体,例如,苯聚羧酸酯配体。聚羧酸酯配体可以是多齿(例如二齿或三齿)连接配体。
苯聚羧酸酯配体可以包含苯环和至少两个羧酸酯基团以及苯环的任选的一个或多个另外的取代基。
mof可以为任何适宜的具有足够大以容纳生物活性的结合no的分子的孔隙尺寸(如最小尺寸大于4埃)的mof。mof可以为任何适宜的具有足够大以容纳生物活性的胍、双胍或多胍类分子的孔隙尺寸的mof。
mof可以包含例如1,4-苯二羧酸酯,1,3,5-苯三羧酸酯(btc),二羟基苯二羧酸酯,特别是2,5-二羟基对苯二甲酸酯(dhtp)等。
mof可以是例如,mof-74(cpo-27),hkust-1,stam-1,mil101和sip-3。
mof可以包含生物活性的配体。例如:mof可以包含烟酸酯或异烟酸酯配体(即烟酸的共轭碱,或具有吡啶环和至少一个悬垂的羧酸酯基团的相关种类)。mof可以包含富马酸酯配体(即富马酸的共轭碱)或另外的不饱和二羧酸酯,例如如imaz等,chem.commun.,2011,467,7287-7302所述的mof。mof可以包含琥珀酸酯(即琥珀酸的共轭碱)或另外的石蜡族二羧酸酯。当从mof释放时(例如当mof随时间分解时),所述配体可以是生物活性剂,因此提供本发明的mof的另外的作用方式。
mof可以包含胺配体,例如,1,4-联吡啶等。
mof可以包含具有多个共配位部分的配体,如5-磺基间苯二甲酸酯配体等。
mof可以包含多种的配体。
mof可以包含或含有除上述那些之外的物质,例如,另外的金属或其它的带正电的离子,或其它的阴离子种类。
另外的阴离子可以包括卤素,例如cl-,f-,br-或i-或其它的阴离子,例如oh-或so4-。
特别是金属有机骨架可以在客体位置如骨架中形成的孔隙或通道包含种类/分子。这些种类可以是例如水,溶剂或例如源于在骨架的制备中使用的组分的其它分子。
一个或多个水分子可以以水合水分子的形式存在且结合至网络结构,例如结合至骨架金属离子或骨架配体。
如技术人员所理解的,一个或多个水分子可以解离,例如,以与另外的水分子一起形成质子化的“h3o+”种类和配位oh-配体。
水的总量可以根据例如归因于环境潮湿、温度、与生物液接触等的mof水合程度变化。
氢氧根配体可以形成骨架结构的一部分且可以与骨架结构内的多个金属离子配位。
本发明不限于特定的mof形貌或结构类型。mof可以具有例如结构类型stam-1,cpo-27或hkust-1,其合成和性能概述于技术人员所涉及的wo2008/020218,wo2012/020214和wo2013/186542中。mof可以具有结构类型sip-3。
外骨架种类,如nonoate和/或n-亚硝基化合物或另外的客体种类可以被描述为已经被吸附到mof中。这样的种类可以被视为以及被吸附到mof的孔隙或通道内的内部表面上。
nonoate和/或n-亚硝基化合物可以被吸附到mof,即被吸附到其内部和外部表面上。
nonoate和/或n-亚硝基化合物可以被物理吸附或化学吸附到mof。
nonoate和/或n-亚硝基化合物可以例如通过静电力和/或范德华力,包括色散力而非共价结合至表面(例如mof内部)。nonoate和/或n-亚硝基化合物可以与表面化学相互作用,包括与一个或多个表面原子形成共价键或与一个或多个表面原子配位。
nonoate和/或n-亚硝基化合物可以与骨架金属离子,骨架配体和/或其它的外骨架种类如水,或外骨架阳离子或阴离子相互作用(包括于其化学或物理结合)。
nonoate和/或n-亚硝基化合物可以可逆地被吸附。
nonoate和/或n-亚硝基化合物可以在一定时间内从mof扩散(defuse)出来。nonoate和/或n-亚硝基化合物可以能够通过将mof暴露于其中而被另外的种类如水或另外的小分子置换。备选地或除此之外,nonoate和/或n-亚硝基化合物可以能够响应刺激,如温度或压力变化,或用光辐照,或与可以置换来自mof的nonoate和/或n-亚硝基的另外的种类接触而从mof被释放。nonoate和/或n-亚硝基化合物从mof释放的速率可以通过刺激而改变(例如加速)。
no可以在一定时间内从mof(或从nonoate和/或n-亚硝基,一旦从mof本身释放的话)自发地被释放。例如,nonoate和/或n-亚硝基化合物可以在一定时间内分解以释放no。
nonoate和/或n-亚硝基化合物可以能够在通过将mof暴露于其中(或通过将游离nonoate和/或n-亚硝基化合物暴露于其中)与另外的种类如水或另外的小分子相互作用之后释放no。nonoate,和/或n-亚硝基(游离的和/或在mof中)化合物可以能够响应刺激,如温度或压力变化,或用光辐照,或与另外的种类接触而释放no。由nonoate和/或n-亚硝基化合物(和/或由mof)释放no的速率可以通过刺激而改变(例如加速)。
nonoate,和/或n-亚硝基可以被不可逆地吸附以无限期地保留在mof中。nonoate和/或n-亚硝基化合物可以在没有具体的外部刺激的情况下被不可逆地不可释放地吸附以无限期地保留在mof中,但是如果施加外部刺激则可以从中被释放。类似地,在从mof释放no之后留下的外骨架化合物可以类似地被可逆地、不可逆地、或不可逆地可释放地吸附。no的不可逆地可释放的吸附具体可以是指no在没有水的情况下无限期地留在mof中,但是当与水分/潮湿接触时其从mof中被释放。
nonoate和/或n-亚硝基化合物可以由具有给电子部分的前体化合物形成,其中给电子部分为胺或亚胺,或由胺和亚胺官能团如胍或双胍/多胍的组合构成的基团。给电子部分可以备选地包含另外的杂原子作为电子给体-如氧,磷或硫。例如,nonoate可以是磺基-nonoate,其由包含亚硫酸酯部分的前体形成。类似地,亚硝基化合物可以采取n-氧化物磺胺或s-亚硝基硫醇,或p-亚硝基化合物的形式。
优选与本发明相关使用的nonoate和/或n-亚硝基化合物在环境温度和压力下和在没有外部刺激(如与水接触,用光辐照,ph变化)的情况下是稳定的,或具有很长的半衰期(约数天,数周或数月)。
nonoate可以包含一个或多个以下通式结构x的官能团:
其中r1和r2可以独立地为碳或杂原子官能团,如亚胺,酰胺,烷基,芳基,烯丙基等,或h。r1和r2可以一起形成脂环基或杂环基。
n-亚硝基化合物可以包含一个或多个以下通式结构a-d的官能团:
其中r1-r12可以独立地包括取代或未取代的c1-c10烷基-,芳基-,醛-,羧酸-,酯-,硫醇-,膦酸酯-,氧膦基-,磺酸酯-,硼-和/或胺-基部分,h和/或卤素。两个以上r基团可以一起形成包含一个或多个取代或未取代的环的杂环结构的一部分。取代基为oh,卤素,nh3,氧代,c1-c6烷基,苯基等。r3-r7中的至少一个可以任选地为no,在一些情况下仅仅结构c和d中的r6和/或r7可以是no。r8-r12各自是任选的,但是当存在时导致其所结合的n原子带正电。所述官能团可以连接至聚合链或大分子或形成其一部分。
在一些实施方案中结构为上述c和d。
在一个实施方案中,r1-r12可以独立地包括取代或未取代的c1-c10烷基-,芳基-,醛-,羧酸-和/或酯-基部分,h和/或卤素。取代基为oh,卤素,c1-c6烷基,或苯基。r3-r7中的至少一个可以任选为no。r8-r12是任选的,且当存在时对它们所结合的氮原子引正电荷。
在另一个实施方案中r1-r5可以独立地包括取代或未取代的c1-c8烷基和/或苯基(例如苯基)部分,或h。r6和/或r7为no。取代基为c1-c6烷基,苯基,卤素。r8-r12不存在。
前体化合物的给电子部分可以是伯胺。因此,结构a中的r1和r2中的一个可以是h,且r8不存在。
前体化合物的给电子部分可以是仲胺。前体化合物可以是例如胍,二胍或双胍化合物。
前体化合物的给电子部分可以是叔胺。前体化合物可以是例如甲硝哒唑或咖啡因。
又一方面,提供一种双胍化合物,其包含与其结合的一个或多个no分子,其中所述双胍可通过以下方法获得:使具有以下通式结构i的前体化合物:
(i)
与no气体或亚硝基化剂反应以生成包含与其结合的一个或多个no分子的双胍化合物;其中
r1-r5可以独立地包括取代的和/或未取代的c1-c10烷基部分,和/或芳基部分(例如苯基)。r1-r6可以是h,条件是当r1为h时r2不是h,且当r3为h时r4不是h。
r6和r7可以独立地为h,c1-c10烷基部分和/或芳基部分(例如苯基),或者一起或独立地可以表示配位的金属离子,如,但是不限于,银,铜,镍,锌,镁和钙。
r8-r12各自是任选的,但是当存在时导致其所结合的n原子带正电,且可以独立地包括h,c1-c10烷基部分和/或芳基部分(例如苯基)。取代基可以包含c1-c10烷基,苯基和/或卤素部分。
r1-r7可以与另外的结构i(如本文所定义的且通常等同于第一结构)结合以形成例如双结构或三结构,如双胍或三胍。
所述结构可以是单独的,或可以是其中所述结构也可以重复一次或多次的聚合链或大分子的一部分。
合宜地,当存在时,r5-r7和r8-r12可以为h。优选r8-r12不存在且r2,r3和r5-7为h。
上述结构可以是n,n’-双取代的,n,n,n’-三取代的,n,n’,n’-三取代的或n,n,n’,n’-四取代的分子,其中每一个取代基为非h的取代基。
适合的亚硝基化剂可以是nocl+ccl4)。
根据上述,前体化合物可以与no以不同的方式形成配合物。可能的形式的两个(非限制性的)实例为nonoate和n-亚硝基配合物。
n-亚硝基配合物包括单个与氮原子结合的no部分。可能的结合位置如下所示:
其中r1/r2和r3/r4中的至少一个为非h取代基,其与上面关于结构i的定义一样。确切的结合位置取决于取代基r-基团的性质。上述结构图示为no基团所连接的氮是季氮。然而,应理解所述氮也可以是叔氮,使得r基团显示为连接至不存在的氮。例如,n-亚硝基化合物可以是
可能的nonoate结合位置如下所示:
其中r2,r4和r5为h。如上所述一个以上季氮可以是叔氮。
根据本发明使用的示例性的化合物包括氯己定,阿来西定,氯胍,氯丙胍(chlorproguanil),聚氨基双胍和聚六甲基双胍。
nonoate和/或n-亚硝基化合物可以以盐的形式(使得mof可以包含外骨架抗衡离子,例如碱金属的外骨架抗衡离子),或以共轭酸或碱的形式存在。
前体化合物可以是生物活性剂,如抗生素,杀虫剂,抗真菌剂,杀孢子剂等。前体化合物可以是适合对人类或动物给药(局部地,静脉内,口服等)的药物化合物。例如,前体化合物可以是喹诺酮如环丙沙星,或双胍如氯己定或其相关化合物、配合物或盐,或磺胺,如呋塞米。
因此,有利地,由活性前体形成的nonoate和/或n-亚硝基化合物可以释放no以重新形成活性剂,所述活性剂形成nonoate和/或n-亚硝基化合物。因此,负载nonoate和/或n-亚硝基的mof可以具有第一作用方式(释放抗微生物的no)和第二作用方式,该作用方式与形成nonoate和/或n-亚硝基化合物的生物活性剂的释放相关。
前体化合物本身可以具有多种的作用方式。例如,前体化合物可以为金属盐的形式,其中金属离子本身具有抗微生物性质(例如银盐,或镍,锌或铜盐,或混合金属盐)。这些盐可以负载于mof的孔隙/通道内使得mof包括外骨架nonoate,和/或n-亚硝基种类(典型地为阴离子的)和外骨架金属离子,其可以能够从mof被释放。
已知的是,mof材料本身可以包含配体和/或具有生物活性的金属离子。因此本发明可以提供负载nonoate和/或n-亚硝基的mof材料,其具有与在mof材料的最终分解期间从nonoate和/或n-亚硝基化合物释放no、形成nonoate和/或n-亚硝基化合物的前体化合物的释放和在一些实施方案中另外的生物活性剂和/或形成mof本身的生物活性剂的释放相关的多种作用方式。
如下面更详细描述的,在根据本发明的mof具有多种作用方式的情况下,每一种作用方式可以在不同的时段内发生和/或响应不同的刺激而被引发或加速。
环丙沙星为具有以下结构的化合物:
氯己定为具有以下结构的化合物:
呋塞米为具有以下结构的化合物:
某些氯己定盐可以备选地被视为或称为氯己定配合物,即,氯己定和酸性或碱性化合物或盐的配合物。
本发明人已经发现,氯己定和相关分子及其盐(包括但不限于)醋酸氯己定,盐酸氯己定和葡萄糖酸氯己定,和金属氯己定盐,如银氯己定)可以被制备以形成稳定的nonoate和/或n-亚硝基化合物。本发明的一个特别的方面是出人意料地发现了在同时含有胺和亚胺部分的化合物如胍和双胍中,no可以优先地与亚胺n结合以形成新的部分如:
相关的氯己定分子可以采取以下形式:
其中a和a1各自独立地表示任选地被含1至4个碳原子的烷基或烷氧基,硝基,或卤素原子取代的苯基;含1至12碳原子的烷基;或含4至12个碳原子的脂环族基;x和x1各自独立地表示含1至3个碳原子的亚烷基;z和z1各自独立地可以为0或1;r和r1各自独立地表示氢,含1至12个碳原子的烷基,或含7至12个碳原子的芳烷基;且n为2至12的整数。
一种示例性的化合物为阿来西定。
nonoate和/或n-亚硝基化合物也可以由其它生物活性前体,如环丙沙星或环丙沙星盐或配合物,呋塞米或呋塞米盐或配合物制备。此外,从所得的nonoate,和/或n-亚硝基释放no的特性可以响应采用光的辐照和/或水分的刺激而改变,从而允许从这些nonoate和/或n-亚硝基化合物可控地释放no。例如,从这些材料释放no可以被触发(例如通过用光辐照)以提供no的初始爆发和随后持续的释放(例如当nonoate和/或n-亚硝基化合物在环境条件下分解时)。因此,本发明可以提供与前体化合物/盐本身相比,显著的抗微生物活性的延长。
因此,另一方面,本发明扩展至nonoate和/或n-亚硝基化合物或盐,其包含具有至少一种仲胺的生物活性前体,所述仲胺与no配位以形成具有结构x或a,b,c,或d的nonoate和/或n-亚硝基部分。生物活性前体种类可以是氯己定,所定义的相关分子,或其盐,氯己定盐或配合物,环丙沙星或环丙沙星盐或配合物,呋塞米或呋塞米盐或配合物。
另外,本发明的另一方面提供一种释放no的方法,所述方法包括:
提供包含生物活性前体种类的nonoate和/或n-亚硝基化合物或盐,所述生物活性前体种类具有至少一种仲胺,所述仲胺与no已经配位以形成具有结构x.a、b、c或d的nonoate和/或n-亚硝基部分;其中所述生物活性前体种类可以是例如氯己定,所定义的相关分子或其盐,氯己定盐或配合物,环丙沙星或环丙沙星盐或配合物,呋塞米或呋塞米盐或配合物;和
用uv光辐照所述nonoate和/或n-亚硝基化合物或盐以由其释放no和/或
使所述nonoate和/或n-亚硝基化合物或盐暴露于水分/潮湿下以由其释放no。
本发明人还已经令人惊奇地发现,使用以前应用于以no负载分子筛材料(如沸石和mof)的方法-例如如由morris和wheatley(angew.chem.int.ed.,2008,47,4966)或lowe等(micropor.mesopor.mat.2013,181(17-22))的方法(其中分子筛首先被活化,然后负载no),可以由这些生物活性前体化合物形成nonoate和/或n-亚硝基化合物。
又一方面,本发明还扩展至由具有一个仲胺部分的生物活性前体(如氯己定,所定义的相关化合物或其盐,氯己定盐或配合物,环丙沙星或环丙沙星盐或配合物,呋塞米或呋塞米盐或配合物)制备nonoate和/或n-亚硝基的方法,所述方法包括:
通过加热前体和/或使所述前体暴露于减压下活化所述生物活性前体;和
将活化的前体暴露于no中。
加热和/或暴露前体于减压下被认为移除分子,否则所述分子会与仲胺结合或稳定仲胺,使得更高比例的仲胺可用于后续与no配位以形成nonoate和/或n-亚硝基化合物。
nonoate和/或n-亚硝基化合物可以由已经吸附到mof的孔隙和/或通道中的生物活性前体形成。
所述方法可以包括将前体暴露于下高真空(例如大约少于10-2,10-3或10-4托)。
所述方法可以包括将活化的前体暴露于一种以上的no气氛中。所述方法可以包括将活化的前体暴露于约两种、三种或四种no气氛中。
所述前体可以被活化任何适宜的时间段,其可以根据例如组成、前体化合物的纯度或形态或活化条件而变化。典型地,例如,活化可以发生约1小时的时间。
类似地,活化的前体可以在no中暴露适宜的时长例如1小时、2小时以上。
技术人员应理解步骤中最佳条件和持续时间可以通过监控反应(例如通过监控压力变化,重量分析,光谱等)的进行来确定。
根据本发明的mof可以包括另外的生物活性剂作为mof的孔隙和/或通道内的客体种类。所述另外的生物活性剂可以被结合且储藏在mof的孔隙/通道内且自发地(例如通过扩散)和/或响应刺激(如暴露于水分或化学药剂,温度升高等)被释放到环境中。
另外的生物活性剂可以被释放的时间类似于或短于/长于no从nonoate和/或n-亚硝基化合物释放的时间。另外的生物活性剂可以以与no相补充的方式或协同的方式起作用。
另外的生物活性剂可以是no。mof可以包含不可逆地可释放地吸附的no。
mof可以合宜地同时以nonoate和/或n-亚硝基化合物形式负载有不可逆地可释放地吸附的no,或者同时由前体原位形成nonoate和/或n-亚硝基化合物。
负载有外骨架nonoate和/或n-亚硝基化合物(如上所述)的mof可以自发地和/或响应刺激释放no。
许多另外的生物活性剂可以被设想到。所述活性剂可以是小分子(如一氧化碳,硫化氢,氮氧化物等),或有机活性剂(包括但不限于,下列类别-青霉素类(例如阿莫西林,青霉素),头孢菌素类(所有代),氨基苷类(例如新霉素,链霉素),糖肽类(万古霉素等),大环内酯类(红霉素等)。抗菌剂,抗病毒剂和/或抗真菌剂可以以类似方式被储藏和吸附。所述活性剂也可以是外骨架金属离子或金属纳米簇(例如银,铜,锌,镍等)。
活性剂可以是抗微生物剂,和/或可以用于使与其接触的微生物对no更敏感。备选地,另外的客体种类可以仅是被设计为排斥微生物的排斥分子,其可以在抗污应用中使用。
活性剂可以包含生理活性的药物分子(由此本文包括中性或离子型种类两者,使得外骨架抗衡离子也可以存在)。药物分子本身可以不具有抗菌活性(例如抗癌药物如多柔比星或另外的药物如咖啡因)。no和药物分子的组合可以帮助战胜具体的病症并且防止治疗过程中的感染或细菌污染。已经提出活性剂的组合的递送延缓耐菌性的发展。
活性剂可以具有抗生物膜的活性,即,它可以导致细菌生物膜分别(具体为d-氨基酸如d-亮氨酸,d-蛋氨酸,d-酪氨酸,d-色氨酸等,或氨基酸的混合物)。
mof可以包含两种以上的另外的生物活性剂。
根据本发明的另一方面,提供一种制备包含外骨架nonoate和/或n-亚硝基化合物的mof,特别是根据其他方面的mof的方法,所述方法包括:
由适合的前体化合物形成nonoate和/或n-亚硝基;和
使mof与nonoate和/或n-亚硝基化合物接触,以将nonoate和/或n-亚硝基化合物吸附到mof的孔隙和/或通道内;
或者所述方法包括:
形成mof,所述mof包含在该mof的孔隙和/或通道内的前体化合物,所述前体化合物能够反应以形成nonoate和/或n-亚硝基化合物;和
通过使所述mof与no接触而原位形成外骨架nonoate和/或n-亚硝基化合物。
所述方法可以包括使mof与能够反应以形成nonoate和/或n-亚硝基的前体化合物接触,以将所述前体化合物吸附到mof的孔隙和/或通道中。
所述方法可以包括:例如通过使用前体化合物(或nonoate和/或n-亚硝基化合物)作为用于mof合成的模板分子,在前体化合物(或nonoate和/或n-亚硝基化合物)周围形成mof。
因此,在本发明的一个方面中,提供一种金属有机骨架材料(mof),其包含在mof的内部孔隙和/或通道内的外骨架前体化合物,所述外骨架前体化合物能够反应以形成nonoate和/或n-亚硝基化合物。
所述方法可以包括活化mof和/或前体化合物。
术语“活化的”是指mof以其中相对于其他情况外骨架或客体种类的吸附更易接受的状态存在。类似地,术语“活化的”是指前体化合物以其中nonoate和/或n-亚硝基配合物的形成更易接受的状态存在。活化可以提高吸附/反应的速率。活化可以增加吸附/反应的程度。
活化可以涉及多余的分子/种类的移除。
例如,mof的活化可以涉及从骨架的孔隙和/或通道移除客体分子/种类(例如在合成mof之后存在的种类,或者其已经从周围如水扩散至mof),从而允许其它种类更容易被吸附到mof中。这样的客体分子/种类可以阻挡经由mof的孔隙/通道的扩散,使得其移除可以有助于用另外的种类(例如no)负载。mof的活化可以导致mof,例如骨架金属离子,变得更少配位饱和的,从而提供大多数和/或更具反应性的配位或吸附位置。
可获得的位置可以能够强烈地(不可逆地,或不可逆地可释放地)结合至随后引入的客体种类。不可逆地或不可逆地可释放地吸附的客体种类的存在可以由在吸附/脱附等温线的吸附和脱附臂之间的强烈的滞后指示。相反地,可逆地被吸附的客体种类可以是更弱地结合的。
类似地,前体化合物的活化可以涉及分子/种类的移除,否则所述分子/种类可能阻碍no例如结合至前体的胺部分。
mof的活化还可以涉及改变骨架的结构以促进客体种类(例如nonoate和/或n-亚硝基化合物)的吸附。此外,前体化合物的活化可以涉及化学变化,如质子化或去质子化,其可能使得前体更易于结合至no。
根据具体情况而定,mof可以在前体化合物或nonoate和/或n-亚硝基化合物的吸附之前被活化。
前体化合物可以在吸附到mof的孔隙/通道中之后被原位活化。
单个的活化步骤或工序可以导致mof和被吸附在其中的前体化合物两者的活化。
在一些实施方案中,mof可以内在地允许客体分子(例如nonoate和/或n-亚硝基化合物)不可逆地被吸附,在此情况下,可能无需活化,或者活化可以用于增加可能被吸附的客体分子的量。
活化可以包括加热mof和/或前体化合物。可以用于活化的典型温度包括至多约450℃,例如,约20℃至约250℃,优选地约50℃至约150℃,最优选约80℃至约120℃,例如约110℃的温度。更低的温度对于前体化合物的活化或者对于其中已经吸附前体化合物的mof的活化可能是适合的。
活化可以包括使mof和/或前体化合物暴露于减压(在环境温度或高温)下,例如通常如上关于某些生物活性前体化合物所述的。
可以用于活化的典型压力包括低于大气压的压力,例如低于lbar,如约1x10-4mbar至约lbar。
活化可以包括用光辐照,例如紫外光。
活化,特别是mof的活化,可以通过化学方法实现。化学活化可以使用化学处理方法如将mof暴露于所需的化学品或化学品的混合物实现。适合的化学品的实例包括溶剂如乙腈(ch3cn),二甲基甲酰胺(dmf),乙醇(etoh)或甲醇(meoh),超临界二氧化碳等。
化学活化可以通过客体分子被所选择的活化化学种类的分子的化学置换,从mof骨架移除多余的分子,例如多余的客体分子。
活化可以包括这些步骤的组合。例如,活化可以以化学方法实现,随后是一个以上的非化学活化步骤,或者反之亦然。
通过仅仅移除所存在的客体种类的一部分或者通过仅仅移除某些类型的客体种类,mof可以被部分地活化,或者通过从骨架移除基本上全部的客体种类,mof可以是“充分”活化的。
所述方法可以包括使mof和/或前体(可以已经被吸附到mof中)与一种以上的no气氛接触。与no接触可以在在两种、三种或四种no气氛中进行。
使mof和/或前体与no在高压力下接触可以促进nonoate和/或n-亚硝基化合物的形成和/或将no吸附到mof骨架本身中。
使mof和/或前体与no在高压力下接触可以导致no的更大负载。
使mof和/或前体与no在高压力下接触可以提供更温和的活化条件。例如,为了实现给定的no负载(作为不可逆地可释放地储藏的no和/或作为nonoate和/或n-亚硝基化合物的被吸附到mof中的no的负载),可以需要更低的活化温度,或者可以仅仅通过减压实现活化。
因此,在一个方面中本发明进一步扩展至制备包含不可逆地可释放地储藏的no的mof,所述方法包括在高的压力使mof暴露于no。所述方法可以包括将mof在使其与no在高的压力下接触之前活化。mof可以通过使mof暴露于减压下而被活化。
所述方法可以包括使mof(包括负载有前体,或nonoate和/或n-亚硝基化合物的mof)与另外的生物活性剂接触。
根据情况而定,mof可以在其与nonoate和/或n-亚硝基化合物或前体化合物接触之前、之后或同时与另外的生物活性剂接触。
另外的生物活性剂可以是no,使得所述方法包括制备包含吸附的外骨架nonoate和/或n-亚硝基化合物和吸附的no的mof,所述no可以是不可逆地可释放地吸附的no。
在所述方法包括接触负载有前体化合物(即被吸附到mof的孔隙和/或通道中)的mof的情况下,使mof与no接触可能导致在mof的孔隙和通道内的外骨架nonoate和/或n-亚硝基化合物的形成。
使mof与no接触可能导致no被吸附到mof的孔隙和/或通道中。将no吸附到mof中和原位形成nonoate和/或n-亚硝基化合物可以同时进行。
所吸附的no可以被不可逆地可释放地吸附。
所述方法可以包括将mof在与no接触之前活化。
所述方法可以包括使用单一的活化步骤。例如,可以活化负载有前体化合物的mof(即,为了至少部分地同时活化前体和mof本身),然后使其与no接触以原位形成外骨架nonoate和/或n-亚硝基化合物且不可逆地可释放地吸附no。
备选地,mof可以被活化,然后同时负载有nonoate和/或n-亚硝基化合物和no(例如通过使活化的mof与用于制备nonoate和/或n-亚硝基化合物的反应混合物接触),或甚至负载有任何其他的生物活性剂。
所述方法可以包括将mof(在上述那些步骤的中间步骤之前、之后或作为中间步骤)离子交换。离子交换可以通过任何适合的方法例如通过在过量的金属离子或离子的溶液中洗涤mof实现。离子交换可以包括在随后用另一种金属离子的溶液洗涤之前,用螯合剂,或一些其他的药剂或能够优先地与存在的外骨架金属离子结合的配体的溶液洗涤mof。
mof可以通过如本领域中已知的任何适宜的方法制备。例如,mof可以合宜地通过如本申请人的共同未决的申请pct/gb2013/051520中所述的低温水溶液合成制备。
本发明的另一方面提供一种释放no的方法,所述方法包括提供在mof的内部孔隙和/或通道内具有外骨架nonoate和/或n-亚硝基化合物的mof,使所述mof与no释放到其中的介质接触,和将no释放到所述介质中。
所述方法可以包括例如通过在环境条件下扩散,将nonoate和/或n-亚硝基化合物从mof的孔隙和/或通道释放一段时间。所述方法可以包括将no从nonoate和/或n-亚硝基化合物释放一段时间。no可以在nonoate和/或n-亚硝基化合物mof内的同时从nonoate和/或n-亚硝基化合物释放,和/或从已经从mof释放的nonoate和/或n-亚硝基化合物释放。
所述方法可以包括对mof施加外部刺激。外部刺激可以包括升温,ph变化,用光辐照,压力变化或这些的组合。
外部刺激可以包括使mof与另一种种类如水接触,以从mof置换nonoate和/或n-亚硝基化合物和/或与nonoate和/或n-亚硝基化合物相互作用以释放no。
no的释放(和/或nonoate和/或n-亚硝基化合物从mof的释放)可以通过外部刺激而引发。no的释放速率可以通过刺激而改变(例如加速)。
因此,本发明提供以可控的方式释放no(可能存在的任何另外的生物活性剂)。可控的释放曲线可以根据具体要求通过选择金属有机骨架材料本身,例如通过通道/孔隙尺寸,或骨架离子或配体的吸附性能进一步被调整。
所述方法可以包括以特定的频率辐照(例如采用uv光)。例如,某些nonoate和/或n-亚硝基化合物对于用uv光辐照是敏感的,其可能导致nonoate和/或n-亚硝基化合物分解以释放no。本发明人已经发现,nonoate和/或n-亚硝基化合物的这种性能不受其结合mof中的不利影响。
所述方法可以包括将另外的生物活性剂从mof孔隙和/或通道释放到所述介质中。
如上所述,nonoate和/或n-亚硝基化合物可以由适合的前体化合物形成,所述前体化合物本身可以是生物活性的(例如氯己定化合物,相关的化合物或环丙沙星化合物)。此外,一旦nonoate和/或n-亚硝基化合物已经释放no,前体化合物就可以保留。因此,另外的生物活性剂可以是所述前体化合物。
所述方法可以包括从mof的孔隙和/或通道释放金属离子。金属离子可以以外骨架金属离子的形式存在于mof中。金属离子可以是生物活性的(例如ag+,其是抗菌活性的)。
所述方法可以包括将mof的组分,即,骨架金属离子和/或骨架配体释放凹所述介质中。mof可以包含作为生物活性剂的骨架金属离子和/或配体。
mof可以能够提供多种的“作用方式”。
例如,mof可以能够释放多种的生物活性剂,其每一种可以表现出不同的生物活性(例如如no和氯己定)。确实地,生物活性剂可以协同地起作用,使得其组合效力超过当单独施用时的任何一种或两种药剂的效果。
mof可以能够在多于一个时间段期间(可以特征在于不同的半衰期)释放生物活性剂。第一生物活性剂(例如no)可以在第一个时间段期间(例如在短的初始爆发中)被释放且第二生物活性剂(例如a前体化合物)可以在第二个时间段期间(典型地更长的时间段)被释放。
确实地,mof可以能够在多于一个时间段期间释放给定的生物活性剂,特别是no。例如,在mof中的nonoate和/或n-亚硝基化合物可以在环境条件下具有长的半衰期(约数小时或数天)且还能够响应外部刺激暴露于如uv辐照下而以更大的速率(更短的半衰期)释放no。
mof可以能够响应于多种的外部刺激而释放生物活性剂。例如,mof可以包括不可逆地可释放地吸附的no和外骨架nonoate和/或n-亚硝基化合物。所述方法可以包括通过使mof与水分接触释放不可逆地可释放地吸附的no,以及通过用uv光辐照而从nonoate和/或n-亚硝基化合物释放no。
因此所述方法可以包括施加多种的刺激。
此外,令人惊奇地发现,某些包含吸附的no的mof也能够通过用uv光辐照而释放no。
对于包含不可逆地可释放地吸附的no的mof也观察到了这种性质,所述mof已经显示出能够通过与水分接触而释放no(这描述于例如技术人员所涉及的下列参考文献3-6和本申请人的国际申请wo2008/020,218,wo2012/020,214和wo2013/186,542)。
所述方法可以应用于显示出潮湿触发的no释放的mof以提供对从这些材料释放no的引发和特征(曲线)的另外的控制。
对于以前被认为基本上不可逆地吸附no的mof如mg-cpo-27或hkust-1(铜基mof)也观察到该性质。在这些情况下,可以将样品在低或无no释放的情况下长时间暴露于空气(和其中的水分)中,且可以通过用uv光辐照而选择性地实现no释放。
因此,另一方面,本发明扩展至一种释放no的方法,所述方法包括:提供在mof的孔隙和/或通道中优选不可逆地或不可逆地可释放地吸附有no的mof;和用uv光辐照mof以从中释放no。
可以使mof与no将释放在其中的介质接触,然后辐照,或者反之亦然。no将释放在其中的介质可以包括本身可以刺激no的释放的种类如水。
no可以与骨架金属离子结合。
mof可以是任何适合的具有足够大以容纳生物活性的no结合分子的孔尺寸的mof。mof可以是任何适合的具有足够大以容纳如本文所述的生物活性的胍类、双胍类或多胍类分子的no结合分子的孔尺寸的mof。
mof可以具有结构类型cpo-27或hkust-1。mof可以具有结构类型mil-101、stam-1或sip-3。
mof可以基于选自上面列举的那些的一种以上骨架金属离子。特别是,mof可以包含mg2+(例如mg-cpo-27),和/或zn2+和/或ni2+(例如zn-cpo-27,ni-cpo-27,mgni-cpo-27,mgnizn-cpo-27,nizn-cpo-27),或cu+(例如cu-hkust-1)。
所述方法可以包括辐照包含以下类型的骨架配体中的一种以上的mof:二或三羧酸酯(例如苯羧酸酯如苯1,3,5-三羧酸酯,或对苯二甲酸酯如2,5-二羟基对苯二甲酸酯或1,4-对苯二甲酸酯);胺配体(如1,4-联吡啶)。
mof可以包括具有多于一个的配位部分的配体(例如5-磺基间苯二甲酸酯配体)。
mof可以包含多种的配体。
no以及在一些实施方案中一种以上另外的生物活性剂的释放可以在动物体内进行,对动物体局部地进行或者在非身体应用中以体外方式如从表面如临床和食品制备场所释放进行。
所述方法可以应用人类或动物的治疗。因此,作为一个另外的方面,本发明还提供一种有需要的个体的治疗或预防方法,所述方法包括提供根据本发明的其他方面的mof,使mof与所述个体接触,且释放no(和任选的一种以上另外的生物活性剂)。
no(和/或一种或多种另外的生物活性剂)可以从mof的孔隙和/或通道被释放。
所述方法还可以包括对mof施加外部刺激。外部刺激作为当与个体接触时mof暴露于生理条件(例如水分,温度)的结果可以而施加。外部刺激可以包括例如,用uv光辐照。因此,所述方法可以包括通过例如在经过一段时间后(例如对于mof足以系统性地分布的或消化的)选择性地施加外部刺激,和/或通过使个体的特定区域暴露于外部刺激(例如uv光)下有目标的释放no(和/或一种或多种另外的生物活性剂)。
根据本发明的另一个方面,提供一种用于手术和/或治疗的金属有机骨架材料(mof),其包含在mof的内部孔隙和/或通道内的外骨架nonoate和/或n-亚硝基化合物(其优选的和任选的特征对应本发明的其它方面的那些)。
另一方面,本发明扩展至一种药物、保健品或化妆品制剂,其包含mof,该mof包含在mof的内部孔隙和/或通道内的外骨架nonoate和/或n-亚硝基化合物,以及用于其的药物/保健品/化妆品载体。
又一方面,本发明提供金属有机骨架材料(mof)用于制备治疗或预防疾病的药物的用途,所述金属有机骨架材料包含在mof的内部孔隙和/或通道内的外骨架nonoate和/或n-亚硝基化合物,以及用于其的药物/保健品/化妆品载体。
可以被治疗的疾病或医学病症包括皮肤感染,包括皮肤真菌,利什曼病,软疣和乳头状瘤病毒,以及分枝杆菌感染。另外的用途包括伤口和/或烧伤康复。对于其他细菌问题的治疗包括降低严重的脚部或身体气味问题和治疗耐甲氧西林的金黄色葡萄球菌感染。
在本发明的又一方面中,提供一种医疗制品,其包含mof,所述mof包含在该mof的内部孔隙和/或通道内的外骨架nonoate和/或n-亚硝基化合物。
适用于本发明的医疗制品包括支架,导管,伤口敷料,绷带,自粘性膏剂和贴剂。例如,光纤导管可以浸渍有或包覆有nonoate和/或n-亚硝基化合物或负载nonoate和/或n-亚硝基的mof。光如uv光可以通过光纤传递通过到导管,并且这可以触发no的释放。
mof可以以医疗制品的涂层的形式例如以油漆或聚合涂层的形式提供。mof可以以制备医疗制品的全部或一部分的材料的组成部分的形式。
mof可以例如结合到塑料制剂中,所述塑料制剂转而可以被模塑或形成为织物。因此,本发明还扩展至一种塑料组合物,其包含mof,该mof包含在该mof的内部孔隙和/或通道内的外骨架nonoate和/或n-亚硝基化合物。
可以在化妆品和个人卫生用途中有利地利用no的有益性能和在可应用的情况下的一种或多种另外的生物活性剂的有益性能。
例如,根据本发明的各个方面的mof可以用于化妆品制剂;除臭剂;皮肤制剂如防老化皮肤制剂和在通过修剪或通过施用脱毛制剂去除毛发之前、之中或之后应用的制剂;头发制剂;脱毛制剂等。
本发明的每一个方面的进一步优选和任选的特征对应本发明的任何其它方面的优选和任选的特征。
附图说明
现在将参考下图描述非限制性的示例性实施方案,在图中:
图1显示了在no负载之前和之后的氯己定二氯化物和醋酸氯己定的uv可见光谱;
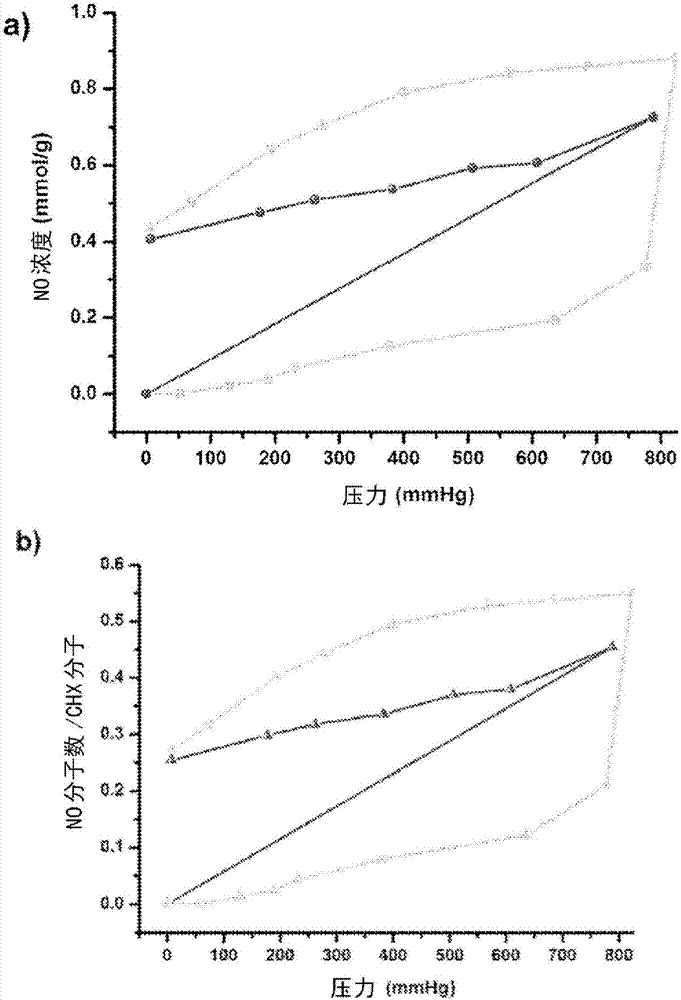
图2显示了对于醋酸氯己定的两个样品的每一个以(a)一氧化氮浓度(mmol/g)和(b)一氧化氮的分子数/氯己定分子绘制的298k的no吸附(-■-)和脱附(-□-)等温线(使用重量分析法测量);
图3显示了从醋酸氯己定在与潮湿气氛(11%rh)接触时递送的一氧化氮的化学发光分析。绘制了no浓度随时间的数据(图2a)和随时间的总no释放(图2b);
图4显示了从醋酸氯己定在与潮湿气氛(11%rh)接触时被uv光触发而递送的总一氧化氮的化学发光分析。所述样品被保持在小瓶中于室温下48小时且在辐照之前暴露于空气中;
图5显示了从氯己定no配合物在水(100%rh)中(图5a)以及被uv光触发(图5b)而递送的一氧化氮的化学发光分析;
图6显示了从氯己定二氯化物在与潮湿气氛(11%rh)接触时被uv光触发而递送的一氧化氮的化学发光分析;
图7显示了从醋酸氯己定银金属配合物在与潮湿气氛(11%rh)接触时被uv光触发而递送的总一氧化氮的化学发光分析;
图8显示了负载有4巴no的聚合物流延的氯己定(蓝色)和负载有1巴no的聚合物流延的氯己定(红色)在与潮湿气氛(11%rh)接触时的总no释放(使用化学发光分析测量),其被绘制成随时间的(a)mmol/g和(b)no分子数/氯己定(chx)分子;
图9显示了从(a)负载有4巴no的聚合物流延的氯己定和(b)负载有1巴no的聚合物流延的氯己定在与潮湿气氛(11%rh)接触时被触发uv光而递送的一氧化氮的化学发光分析;
图10显示了从负载有4巴no的聚合物流延的氯己定(上,蓝色曲线)和负载有1巴no的聚合物流延的氯己定(下,红色曲线)在与潮湿气氛(11%rh)接触时被uv光触发而递送的一氧化氮的化学发光分析;
图11显示了从环丙沙星在与潮湿气氛(11%rh)接触时被uv光触发而递送的一氧化氮的化学发光分析;
图12显示了(a)在暴露于no中之前(蓝色)和之后(红色)的醋酸氯己定以及(b)在暴露于no中之前(蓝色)和之后(红色)的环丙沙星的ft-ir光谱;
图13显示了从呋塞米在与潮湿气氛(11%rh)接触时的总no释放(使用化学发光分析测量)(被绘制成mmol/g);
图14显示了从呋塞米在与潮湿气氛(11%rh)接触时被uv光触发的一氧化氮释放的化学发光分析;
图15显示了从cpo27mg在与潮湿气氛(11%rh)接触时递送的总一氧化氮的化学发光分析(图7a)和从cpo27mg在与潮湿气氛(11%rh)接触时被uv光触发的no释放(图7b)。在两个分析之间样品被储存在暴露于空气和潮湿中的室温工作台上超过68小时;
图16显示了从cpo27ni在与潮湿气氛(11%rh)接触时递送的总一氧化氮的化学发光分析(图16a)和从cpo27ni在与潮湿气氛(11%rh)接触时被uv光触发的no释放(图16b)。在两个分析之间样品被储存在暴露于空气和潮湿中的室温工作台上超过48小时;
图17显示了从hkust-1在与潮湿气氛(11%rh)接触时递送的总一氧化氮的化学发光分析(图17a)和从hkust-1在与潮湿气氛(11%rh)接触时被uv光触发的no释放(图17b)。在两个分析之间样品被储存在暴露于空气和潮湿中的室温工作台上超过64小时;
图18显示了(a)氯己定,cpo-27mg和负载氯己定的cpo-27mg的ftir分析,以及(b)氯己定,cpo-27mg和负载氯己定的cpo-27mg的tga分析;
图19显示了来自cpo27ni和cpo27mg的氯己定释放;
图20显示了从负载氯己定的cpo27mg在与潮湿气氛(11%rh)接触时递送的总一氧化氮的化学发光分析:随时间的no浓度(图20a)和随时间的总no释放(图20b);
图21显示了醋酸氯己定,cpo27mg和负载氯己定的cpo27mg随时间的总no释放;
图22显示了从负载氯己定的cpo27mg在与潮湿气氛(11%rh)接触时被uv光触发而递送的一氧化氮的化学发光分析。在两个分析之间样品被储存在暴露于空气和潮湿中的室温工作台上超过50小时;和
图23a显示了来自以聚氨酯膜形式流延的cpo27ni的氯己定释放。图23b显示了从以聚氨酯膜形式流延的负载氯己定的cpo27ni被潮湿气氛(11%rh)触发而递送的总一氧化氮的化学发光分析。
实例性实施方案的详述
氯己定nonoate和/或n-亚硝基化合物的制备和no从其中的释放
在世界卫生组织的基本药物名单中报导了氯己定。该药物产品广泛用于消毒剂(外用于皮肤和手上)和局部应用(滴眼剂中的防腐剂,伤口敷料和防腐漱口剂中的活性物质)。此外,该生物分子也可以存在于化妆品(乳膏、牙膏和除臭剂的添加剂)。该药物主要以盐(二盐酸盐,二乙酸盐和二葡糖酸盐)的形式售卖。近年来,已经报导了不同的氯己定-金属配合物;所述药物与特定的金属(如铜和银)结合,从而提供用于氯己定的可控释放的体系,同时维持药物性能[1]。
氯己定含有伯胺和仲胺基团。本发明人已经发现,当暴露于高压力下的一氧化氮气体中时,这些胺基团能够结合no。
此外,no从氯己定的释放可以被紫外线(uv)或暴露于潮湿而触发。确实,no释放可以被这些外部刺激的任何一种或两者触发。如下详述的,在由暴露于潮湿中所引起的初始爆发之后,no释放可以通过关闭和接通uv光源而被重复触发并且停止。
有益地和出人意料地,氯己定nonoate和/或n-亚硝基化合物已经被发现在空气中是稳定的,这意味着无需特定的储存条件。
从许多不同的氯己定盐的光控制的释放是可以的。氯己定和no的组合具有协同效应,这降低了潜在的耐菌性的风险并且可以用于防控已经有抗性的微生物菌株。
氯己定-nonoate/n-亚硝基化合物的益处在于,在no释放后再生的氯己定前体是一种被充分认识的有益的生物活性剂。此外,适当的剂量、副作用和毒性被充分认识。
(1)氯己定-nonoate和/或n-亚硝基和配合物以及m-氯己定-nonoate和/或n-亚硝基盐的形成和no从其中的释放
使用以前morris[2]所报导的与mof和沸石材料相关的高温脱水和no负载技术,氯己定,其盐和配合物(前体化合物)可以被转化为其nonoate和/或n-亚硝基化合物。
也可以如lowe等[17]所概述的通常地制备氯己定-nonoate和/或n-亚硝基化合物,其中在暴露于no气氛中之前,使材料在大约室温下经受高真空。
这些技术以前已经仅仅被考虑用于以no负载mof和其它的分子筛材料,使得no吸附到骨架离子或配体上。这些方法以前未应用于“游离的”nonoate和/或n-亚硝基前体化合物。
如与醋酸氯己定(chlorhexidinediacetate),盐酸氯己定(chlorhexidinedihydrochloride)和葡萄糖酸氯己定(chlorhexidinedigluconate)相关地证实的,任何氯己定盐可以被用作原材料。
可以为了优选的no释放曲线而选择氯己定前体的特性。本发明人已经观察到在nonoate和/或n-亚硝基化合物暴露于潮湿中时的释放曲线对于已经选择的具体前体是特别敏感的。
例如,如果与潮湿或水分接触时的no的大的初始“爆发”是需要的,则例如盐酸氯己定可以是适当的。然而,醋酸氯己定在于潮湿/水分暴露时具有更平缓的释放曲线。
对于金属氯己定配合物,也已经证明nonoate和/或n-亚硝基化合物的形成。已经通过溶剂热/水热合成和机械化学合成形成了m-氯己定nonoate和/或n-亚硝基配合物。优选的方法通常通过morris等[23]所报导的低温方法进行。同样,该低温方法以前仅仅用于制备mof材料。
所用的金属可以是任何金属,但是优选具有抗微生物性质的那些如ag,ni,zn和cu。这些金属本身具有生物(例如抗菌的)活性且提供具有另外的作用方式(除了no和氯己定阴离子的活性和/或释放曲线之外)的nonoate和/或n-亚硝基盐/配合物。
除了通过暴露于潮湿空气、水分或uv光中释放no之外,也可以通过加热氯己定nonoate化合物而引发或刺激no释放。
这些材料的一个特别的特征是结合的释放触发因素可以用于提供初始的爆发和随后的持续释放。
实施例1-醋酸氯己定和二盐酸盐no负载
将50mg的醋酸氯己定水合物和盐酸氯己定的样品在室温下暴露于高真空(10-4托)中1小时。利用schlenk管线,将4大气压的no气体引入到schlenk管中,历时2小时,从而使得脱水的氯己定吸附到基团气体(radicalgas)。然后将样品暴露于真空中且用氩冲洗30分钟。然后密封容纳样品的玻璃瓶。
图1显示了所检测的纯氯己定与含有no的配合物相比的uv可见光谱。将药物暴露于一氧化氮气体将材料的颜色从白色改变为浅黄色。uv数据显示对于盐酸氯己定和二乙酸盐no配合物两者在~370cm-1的吸收带的变化。这与对于其他含有no的材料的文献报导[16]是一致的。
使用定做的重量吸附系统收集两种另外的醋酸氯己定样品(25mg)的一氧化氮吸附/脱附曲线。将每一种样品在1×10-4mbar压力暴露于高真空中过夜直至未观察到进一步的质量损失。使用水浴(0.02k的温度精度)将样品冷却至298k。
对于图2(a)和(b)的外部曲线中所示的一个样品,no是以增加的递增量引入的。在每一个no剂量之后,在进行下一次添加之前使样品的质量稳定(指示吸附完成)。继续该过程直至所引入的no压力等于大气压。在单个步骤中将第二样品暴露于1个大气压的no中,且使得其质量平衡。数据以图2(a)和(b)的内部曲线显示。在图2(a)和2(b)中所示的两个样品的脱附曲线是通过将压力以递增量降低至2×10-2mbar的最终值来测量的。
重量分析显示吸附了最大~0.9mmolno/g(图2a),这等于0.55分子no/氯己定分子(图2b)。
吸附曲线的形状显示在所施加的no压力与结合至分子的no的量之间的依赖性。在schlenk管线上通常用于负载的no的压力为在重量等温线分析过程中所得到的水平的4倍,因此本发明人会预期与氯己定配位的甚至更高的基团气体的量。通过再施加真空,no水平逐渐降低,且两个样品达到~0.4mmol/g的储存no的水平,即,约0.25分子/氯己定分子。这些数据表明,显著比例的最初储存的no已经被氯己定前体吸附。
no从样品的释放首先通过将恒定流量的潮湿氮气(11%rh)通过其上而被触发。随时间释放的no的量使用sieversnoa280i化学发光一氧化氮分析器检测直至no的排放达到低于20ppb的水平。
no的释放的初始爆发达到512ppm(图3)。样品在19小时内释放至多0.042mmol/g的一氧化氮。
在完成no释放之后,将样品保持在暴露于空气和潮湿中的室温工作台上超过48小时。将样品暴露于来自各自包含发射300-400nm且总功率为50-200w的4x15w灯泡的两个ritekelectronicsuv管型灯的uv光。这些参数不应被视为对本发明的限制。光触发的no释放立即从30ppb爆发至105ppb。在连续暴露约5min时达到约120ppb的最大值。
如图4a中所示,当关闭uv光源时立即停止排放。可以随时间重复和控制这种开-关过程。连续暴露于uv光触发了在超过4小时期间超过70ppb的一氧化氮的释放,如图4b中所示。使用上述相同的分析器记录随时间的no的释放。
实施例2-从悬浮在水中的氯己定no配合物的no释放
将100mg的醋酸氯己定的样品在室温下暴露于高真空(10-4托)中12小时。利用schlenk管线,将4大气压的no气体引入到schlenk管中,且保持2小时,从而使得脱水的氯己定吸附气体。然后将样品暴露于真空中且用氩冲洗30分钟。然后密封容纳样品的玻璃瓶。
将no负载的样品浸没于连接至no分析器的密封室内5ml的去离子水中。将连续的氮流鼓泡通过悬浮液,同时测量室气氛中存在的no的浓度。
随时间以ppm和ppb测量被水触发的no从样品的释放直至no的水平下降至低于20ppb。初始爆发的no释放达到40ppm。样品在7小时内释放至多0.035mmol/g的一氧化氮(图5a)。
在完成no释放之后立即将样品暴露于uv光中,这触发了no的进一步释放。在约10分钟的连续暴露过程中记录到约1000ppb的最大值。如图5b中所示,当关闭uv光源时立即停止排放。可以随时间重复和控制这种开-关过程。
实施例3-盐酸氯己定no负载和释放
使用盐酸氯己定作为原材料,遵循上述相同的一般方法。通过将样品暴露于恒定流量的潮湿氮气(11%rh)而获得初始爆发的no释放。材料释放少量的气体2分钟,然后停止。在将样品储存于暴露于潮湿空气中的工作台上2天之后,使用uv光触发额外的no释放。盐酸氯己定释放爆发的高达150ppb的no,历时1小时降低至75ppb。如图6中所示,可以随时间重复和控制该触发机制。
实施例4-银-氯己定配合物的no负载和释放
遵循song的程序,使用硝酸银和醋酸氯己定制备银-氯己定的样品。在表征(xrd,uvvis,sem和edx)之后,遵循以前所报导的高压程序[17]以no负载50mg样品。
通过使样品暴露于潮湿中而触发的初始爆发的no释放持续了2分钟。在如图7中所示使用uv光触发一氧化氮的另外的释放之前,将样品储存、暴露于潮湿空气中超过60小时。
银氯己定配合物释放爆发(高达175ppb)的no,其历时1小时的时间平缓地下降至100ppb。如同在上述情况中,当关闭uv光时no的释放突然停止。如图7a中所示,可以随时间重复和控制这种触发机制。如图7b中所示,即使在85小时之后也可以多次关闭和接通no的释放。
实施例5-来自含有no-配位的醋酸氯己定的聚合物膜的no释放
当其通常用于医疗装置中时,选择聚氨酯聚合物作为流延材料。醋酸氯己定(1.5g)的样品分散于聚氨酯(3g)和thf(40ml)的初步溶解的混合物。使用刮刀技术将所述混合物进行溶剂流延以制备~100μm的厚膜,所述厚膜是通过蒸发溶剂而设定的。
将聚合物膜的样品暴露于真空中过夜且使用两种不同的一氧化氮压力(1巴和4巴)进行no负载。
图8显示了在4巴和1巴负载的含负载no的氯己定膜的100mg样品的仅使用潮湿氮(湿度控制为11%rh)的no释放曲线(化学发光分析)。数据表明在4巴负载的样品在30小时的时间段内达到至多0.1mmol/g的一氧化氮(图8a),这等于0.06no分子数/氯己定分子(图8b)。还已经发现,负载有1巴的no的聚合物流延的氯己定在类似条件下没有释放出任何no(也如图8中所示)。
实施例6-来自聚合物流延的no-配位的醋酸氯己定的uv触发的no释放
在通过将样品暴露于潮湿氮中完成初始的no释放之后,将样品储存于暴露于空气和潮湿中的室温工作台上超过48小时。然后将两个样品在潮湿氮气流(湿度控制为11%rh)中暴露于uv光下。
这触发了来自包括已经负载有1巴的no的膜的样品的额外的no释放,这与其仅仅在潮湿氮中的性能正好相反。该样品在连续暴露约10分钟时达到约180ppb的最大值。
如图9a中所示,当关闭uv光源时立即停止排放。可以随时间重复和控制这种开-关过程。连续暴露于uv光触发了在超过14小时期间超过20ppb的一氧化氮的释放,如图9a中所示。
已经负载有4巴no的含氯己定的聚合物样品释放的量几乎为从1巴对应物获得的量的2倍,如图10中所示。因此在气体负载过程中使用的no的压力显著地影响了仅仅使用水和/或uv光作为触发因素从样品获得的最终释放性能。该样品在连续暴露约10分钟时达到了约300ppb的最大值,如图图9b中所示。当关闭uv光源时立即停止排放。可以随时间重复和控制这种开-关过程。
(2)no-配位的环丙沙星化合物的形成和no从其中的释放
已经发现,上述方法可以在在其结构中含有仲胺的不同药物如环丙沙星上使用。环丙沙星是用于治疗不同的细菌感染的抗生素。
实施例7
遵循上面报导的高压方法,将50mg的环丙沙星进行no负载。在暴露于恒定流量的潮湿氮气(11%rh)中之后,获得持续2分钟的小的一氧化氮初始爆发。然而,即使在将样品储存于暴露于潮湿空气中的工作台上24小时之后,使用uv光触发额外的no释放。环丙沙星释放了爆发的高达500ppb的no,历时1小时降低至约100ppb。如图11a中所示,可以随时间重复和控制该触发机制。在将样品保持暴露于潮湿空气中超过50小时之后,使用uv光作为触发因素仍然释放了额外的no,如图11b中所示。爆发的no释放达到了320ppb,历时1.5小时降低至50ppb。
ft-ir分析
对于暴露于no中后的两个样品,将no与环丙沙星分子连接的证据由ft-ir光谱中新的拉伸频率的出现来提供(图12):例如,在1040-1043cm-1的拉伸频率(n-o)(图12a),在1310-1320cm-1的拉伸频率(n-o)(图12a,b)和在1550-1500cm-1的拉伸频率(n-o)(图12a,b)。还有另外的拉伸频带:存在于全部no改性的化合物中的2270-2275cm-1,其很可能归因于n-n拉伸。
还有高于1700cm-1的小拉伸存在于每一个no配位的化合物样品中。尽管该拉伸的起因未得到充分认识,但是已经发现,其存在于含有no的化合物的其它文献报导的光谱中(参见例如j.g.nguyen,kristinek.tanabe和s.m.cohencryst.eng.comm,2010,12,2335-2338)。此外,图11b也显示了当环丙沙星暴露于no中时在3300cm-1的nh拉伸消失了。
(3)呋塞米
呋塞米是在充血性心力衰竭和水肿的治疗中使用的袢利尿剂。与一些其它的利尿剂一起,呋塞米也包括在世界反兴奋剂组织的禁止药物名单中,原因在于其声称用作其它药物的掩蔽剂。它也在世界卫生组织的基本药物(在基本的医疗卫生系统中所需的最重要的药物名单)之列。
呋塞米主要用于治疗高血压和水肿。其对于患有充血性心力衰竭所引起的水肿的大多数人而言是一线药剂。其也用于肝硬化,肾脏损害,肾病综合征,以及与适当的再水化结合用于严重的高钾血症的管理。
实施例8-呋塞米no负载和释放
将25mg呋塞米在高真空(10-4托)和室温下暴露1小时。使用schlenk管线,将4个大气压的no气体引入到schlenk管中并且保持2小时,从而使得脱水的呋塞米吸附气体。然后将样品暴露于真空下且用氩冲洗30分钟。然后密封容纳样品的玻璃瓶。
首先通过使恒定流量的潮湿氮气(11%rh)通过而触发从样品释放no。以ppm和ppb检测随时间释放的no的量直至no的排放下降至低于20ppb。
初始爆发的no释放达到512ppm(图13)。样品在19小时内释放至多0.042mmol/g的一氧化氮。
实施例9-从呋塞米no配合物的uv触发的no释放
在通过潮湿氮完成初始的no释放之后,将样品储存在暴露于空气和潮湿中的室温工作台上超过24小时。然后将样品在潮湿氮气流(湿度控制为11%rh)暴露于uv光。
呋塞米nonoate样品在连续暴露约10分钟时达到约55ppb的最大值。如图14中所示,当关闭uv光源时立即停止排放。
可以随时间重复和控制这种开-关过程。连续暴露于uv光触发了在超过7小时期间超过20ppb的一氧化氮的释放。
(4)从mof中的光触发的no释放
对于cpo-27和hkust-1型结构等(特别是具有配位不饱和的骨架金属部位的其它mof),已经证实了从mof中的uv光触发的释放。然而,该技术可以用于表现出对no的亲和势的任何mof。
遵循以前被morris[23]报导的方法制备mof。根据以前被morris[2,8]报导的高温脱水方法进行活化和no负载。然而,no负载可以通过例如如被lowe[17]描述的任何适合的方法进行,其中在暴露于高压力一氧化氮中之前,使材料在室温下经受真空。
可以为了所需的no释放曲线而选择mof。例如mg和ni-cpo-27趋向于比hkust-1释放更高量的no。
通过使材料暴露于uv光下触发了被吸附的no的释放。备选地,或除此之外,也可以通过暴露于潮湿空气和/或热中而实现no释放。
例如,在一些情况下,可以通过与水分接触而触发初始的no爆发,并且一旦一氧化氮的释放消失,可以利用uv光以通过关闭和接通uv光源选择性地触发额外的no的释放。
在该具体方法中,可以提供比以前可能的量或比例更大的量或比例的储存no的释放。尽管不希望受到理论的束缚,但是这可能是uv光触发了结合更强的(高能)no的释放的结果,所述结合更强的no通常不会通过被水置换或在常规施加的热条件下被释放。uv触发的no释放对于当独有地暴露于潮湿时表现出差的no释放的mof(例如cpo-27mg和hkust-1)具有特别的应用。这样的材料已知具有较高的no储存性能,该no以前不可能容易释放。
实施例10-cpo-27mg
将遵循morris[23]所报导的程序制备的50mgcpo-27mg的样品在室温下暴露于高真空(10-4托)1小时。然后在排空并且用氩冲洗30分钟且密封在玻璃瓶中之前,将样品暴露于4个大气压的no气体中2小时。
总的no释放:将样品暴露于恒定流量的潮湿氮气(11%rh)中且随时间检测释放的no。进行分析直至所检测的no气体水平低于20ppb。cpo-27mg仅仅释放在25小时期间至多0.05mmol/g,如图15a中所示。在完成no释放之后,将样品保持在暴露于空气和潮湿中的室温工作台上超过68小时。随后暴露于uv光中导致一氧化氮的进一步释放。光触发了缓慢爆发的no释放从80ppb到超过500ppb。当关闭uv光源时立即停止排放。如图15b中所示,可以重复这种过程。
实施例11-cpo-27ni
将遵循morris[23]所报导的程序制备的50mgcpo-27ni的样品活化并且遵循上述相同的高压技术进行no负载。
总的no释放-在样品暴露于恒定流量的潮湿氮气(11%rh)中之后,cpo-27ni在40小时期间释放总的2.8mmol/g的no,如图16a中所示。将样品保持在暴露于空气和潮湿中的室温工作台上超过2天。随后通过uv光触发进一步的no释放,如图16b中所示,这提供了高达85ppb的爆发释放和在辐照持续期间的持续释放。可以通过关闭和接通uv光而重复地触发从骨架释放一氧化氮。
实施例12-hkust-1
将遵循morris[23]所报导的程序制备的50mghkust-1的样品活化并且遵循上述相同的高压技术进行no负载。
总的no释放-通过将样品暴露于恒定流量的潮湿氮气(11%rh)中获得no的初始释放。骨架在7小时期间释放至多0.2mmol/g,如图17a中所示。在将样品储存在暴露于潮湿空气中的工作台上64小时之后,利用uv光触发额外的no释放。hkust-1释放了爆发高达65ppb以及在超过1小时期间稳定状态为60ppb的no。可以随时间重复和控制该触发机制(图17b)。
(5)负载氯己定的mof
氯己定和no-配位的氯己定已经成功地被结合至mof中并且从mof被释放。此外,mof已经被证明能够通过暴露于uv光和/或潮湿和光的组合随时间释放no。
可以根据morris[23]或lowe[17]的方法制备负载氯己定和no的mof。
在首先以前体化合物如氯己定化合物负载mof的情况下,将mof暴露于一氧化氮中可以具有双重效果:no气体结合至mof以及也结合至氯己定化合物,从而形成负载no和氯己定-no配合物的mof。
可以通过通常使用的方法,包括但不限于暴露于潮湿空气,热或uv光,从负载氯己定-no配合物的mof释放no。
光触发的no释放类似于上面对于no配合物本身和对于负载no的mof所报导的那些。两种不同类型的no结合部位的存在可以提供总no释放的增加。
实施例13-将氯己定负载到cpo-27mg和cpo-27ni中
将100mgmof(cpo-27mg或cpo-27ni)的样品与100mg醋酸氯己定混合。将混合物在110℃烘箱中脱水过夜。然后在经由橡胶隔膜引入无水乙醇(100ml)之前,将样品瓶密封并且冷却至室温。在4天之后,然后将悬浮液过滤并且用乙醇洗涤。ft-ir和tga分析证实了在骨架中氯己定的存在,如图18中所示。
实施例14-氯己定从cpo-27mg和cpo-27ni中的释放
将50mg的负载药物的mof(cpo-27mg或cpo-27ni)悬浮在50ml甲醇中。随时间对溶液取样且使用uvvis检测氯己定的浓度。图19显示了随时间药物从骨架的有效释放。药物负载的cpo-27ni显示了在第一个40小时期间达到1.3μg/ml的爆发释放,而cpo-27mg提供了随时间氯己定的更高亲和势和更高潜在的释放,其在80小时期间达到了2.5μg/ml的稳定状态。
实施例15-负载药物的cpo-27mg的no负载和释放
将50mg负载药物的cpo-27mg的样品活化并且在室温遵循上述相同的高压技术进行no负载。然后将样品暴露于恒定流量的潮湿氮气(11%rh)中。所使用的氯己定cpo-27配合物的量具有峰值为512ppm的爆发释放。负载药物的mof在45小时期间实现了0.15mmol/g的总no释放,如图20中所示。
来自纯的氯己定no配合物、纯的mof和负载药物的mof的总no释放的比较显示了在两个单独的部分上的双官能化材料的益处,如图21中所示。来自no-配位的氯己定总no释放在18小时内达到了0.03mmol/g的最大值,且来自cpo-27mg的总no释放在25小时内达到0.05mmol/g。然而,氯己定cpo-27mg配合物在40小时期间实现了0.15mmol/g的总no释放,这归因于两个部分的组合效果。
将氯己定cpo-27mg的样品保持在暴露于潮湿和空气中的室温工作台上超过40小时。然后利用uv光触发额外量的no释放,如图22a中所示。数据表明,可以通过控制暴露于uv光中的时长为至少2小时调节no剂量(图22b)。光触发的机制重复地控制了no气体随时间的释放。在初始暴露于uv光中之后,将样品再次储存在室温下,对空气和潮湿开放50小时。将样品进一步暴露于uv光中触发了额外的气体释放,如图22b中所示。来自研究量的材料的no释放在1小时期间达到了0.5ppm的稳定状态。
(6)将no配位材料结合到基体中用于应用
上述材料的每一个,包括药物nonoate和/或n-亚硝基化合物(氯己定盐和环丙沙星),mof,和负载药物的mof,可以被结合到不同的基体中。这些基体包括但不限于树脂和粘合剂(如在例如油漆、墨和涂料中使用的那些),乳膏,软膏剂,聚合物,陶瓷和玻璃,特别是在卫生保健和医疗用途(例如装置,敷料和局部治疗)中使用的那些,或者其中需要防腐/抗微生物性能的那些(例如表面上的涂层)。
可以通过任何适合的方法如,但是不限于,研磨,高速/剪切混合,挤出,电喷,流延和模塑将所述材料引入到这些基体中。可以在例如织物、塑料、金属、木材和玻璃表面上以涂层的形式使用所述材料。这可以通过以下任何适合的方法而实现:如将材料分散在树脂中以通过涂漆、浸涂、喷涂、打印等来涂覆。在适当时,也可以使用粉末涂料。可以在适当时和在必要时使用另外的试剂如分散和流变改性剂以帮助配制。
实施例16-来自含有负载氯己定的cpo-27ni的聚合物的氯己定释放
在其通常用于导管中时,选择聚氨酯聚合物作为流延材料。遵循以前morris等[23]所报导的程序,将cpo-27ni的样品进行药物负载。使用高剪切均化器将负载药物的mof悬浮在thf中,并且将其分散在预先溶解的聚氨酯中。使用刮刀技术将混合物进行溶剂流延以制备~100μm厚的膜。
将膜悬浮在适当体积的甲醇中。随时间对溶液取样,并且使用uv光谱法检测氯己定的浓度。图23a显示了来自负载的聚合物的药物释放。该释放在70小时内达到了0.12μg/ml的最大值。
实施例17-来自含有负载氯己定no配合物的cpo-27ni的聚氨酯的no释放
将负载mof的膜(如上概述制备的)脱水且遵循以前报导的程序进行负载。图23b显示了当暴露于潮湿空气中时来自在聚氨酯聚合物中流延的负载氯己定的cpo-27ni的总no释放。该样品在8小时后递送了0.25mmol/g的最大值。
参考文献
1.s.pal,e.j.yoon,y.k.tak,e.choi和j.m.song,j.am.chem.soc.,2009,131(44),16147-16155
2.r.e.morris和p.s.wheatley,angew.chem.int.ed.,2008,47,4966
3.a.c.mckinlay,b.xiao,d.s.wragg,p.s.wheatley,i.l.megson和r.e.morris,j.am.chem.soc.,2008,130,10440
4.p.d.c.dietzel,b.panella,m.hirscher,r.blom和h.fjellveg,chem.comm.,2006,9,959
5.p.d.c.dietzel,r.e.johnsen,r.blom和h.fjellveg,chem.eur.j.2008,14,2389
6.n.l.rosi,j.kim,m.eddaoudi,b.chen,m.o’keeffe和o.m.yaghi,j.am.chem.soc,2005,127,1504
7.p.k.allan,p.s.wheatley,d.aldous,m.i.mohideen,g.deweireld,s.vaesen,r.e.morris,daltontransactions,2012,41,14,4060-4066
8.r.morris,p.s.wheatley,wo2008/020218a1
9.z.bao,s.alnemrat,l.yu,i.vasiliev,q.ren,x.lu和s.deng,langmuir,2011,27,13554
10.d.j.tranchemontagne,j.r.hunt和o.m.yaghi,tetrahedron,2008,64,8553
11.h.du,j.bai,c.zuo,z.xin和j.hu,cryst.eng.comm.,2011,13,3314
12.j.-x.chen和s.-x.liu,huaxuexuebao,2004,23,2323
13.k.e.holmes,p.f.kelly和m.r.j.elsegood,daltontrans.,2004,3488
14.n.e.ghermani,g.morgant,j.d’angelo,d.desmaele,b.fraisse,f.bonhomme,e.dichi和m.sgahier,polyhedron,2007,26,2880
15.e.j.martinez,j.j.talley,k.d.jerome,t.l.boehm.wo2014012074a3
16.a.el-emam,e.glusa,j.lehmann,eur.j.med.chem.,2008,51,713-716
17.a.lowe,p.chittajallua,q.gongb,j.lib,k.j.balkusjr.,micropor.mesopor.mat.,2013,181,17-22
18.s.diring,k.kamei和s.furukawa,naturecommunications2013,2684(4)
19.b.j.heilman,s.tr.j.oliver和p.k.mascharak,j.am.chem.soc.,2012,134(28),11573-11582
20.e.j.martinez,j.j.talley,k.d.jerome,t.l.boehm,wo2014012074a3
21.j.g.nguyen,k.k.tanabe和s.m.cohen,cryst.eng.comm,2010,12,2335-2338
22.s.rojas,p.s.wheatley,r.e.morris和e.barea,cryst.eng.comm,2013,15,9364-9367
23.r.e.morris,p.s.wheatley,s.warrender,m.duncan,wo2013186542a1
24.s.pal,e.j.yoon,s.h.park,e.c.choi和j.m.song,j.antimicrob.chemother.,2010;65(10),2134-40.
25.l.k.keefer,j.l.flippen-anderson,c.george,a.p.shanklin,t.m.dunams,e.s.sagan和d.s.bohle,j.am.chem.soc.2001,13;123(23):5465-72。
- 还没有人留言评论。精彩留言会获得点赞!