一种制备碘七氧化五铋/锰锌铁氧体复合磁性光催化材料的方法与流程
本发明涉及一种制备碘七氧化五铋/锰锌铁氧体(bi5o7i/mnxzn1-xfe2o4)复合磁性光催化材料的方法,属于无机环境光催化材料技术领域。
背景技术:
碘七氧化五铋(bi5o7i)作为新型的纳米光催化材料,属于富氧型铋基卤氧化物,主要有正交相的α-bi5o7i和单斜相的β-bi5o7i,且α-bi5o7i比β-bi5o7i的光催化活性能更优异。bi5o7i导带底部由bi6p轨道组成,价带顶部主要由bi6s、o2p和i5p轨道组成。bi5o7i具有高活性光催化活性能主要是因为bi6s和o2p轨道会形成大量分散的杂化价带,有利于光生空穴的迁移和发生氧化反应,而将i5p轨道引入到价带中后进一步分散了价带并增加了光生空穴的迁移率。bi5o7i较高的导带位置有利于o2被还原为o2-,且多bi和多o结构使其反应速率进一步提升。其制备方法包括:水热法、共沉淀法、焙烧法等。光催化剂在反应体系中均匀分布于液体中,难以分离和回收再利用是制约光催化剂应用进程的主要因素。复合磁性光催化剂通过外加磁场实现催化剂的回收再利用,克服了常规过滤回收方式工艺复杂、能耗较大且回收率低的缺点。
锰锌铁氧体(mnxzn1-xfe2o4)属于软磁铁氧体材料,与传统金属软磁材料(fe3o4)相比具有高饱和磁化强度、高磁导率、低矫顽力、低损耗、生产成本低、产品稳定性强等优点,因此以mnxzn1-xfe2o4为磁性基体制备的复合光催化材料具有稳定的磁性,便于催化剂的回收和循环使用。目前,常见的mnxzn1-xfe2o4制备方法包括共沉淀法、水热法、溶胶-凝胶法、煅烧法[56]等。
目前,对于bi5o7i的研究主要集中在提高其活性,而研究如何制备bi5o7i的复合磁性光催化剂的报道较少。如“journalofmaterialsscience:materialsinelectronics”2018年第29卷中的“enhancedphotocatalyticperformanceofz-schemecu2o/bi5o7inanocomposites”(对比文件1),采用焙烧法制备出纯bi5o7i,然后再复合得到cu2o/bi5o7i复合光催化剂。该方法的不足之处在于:(1)制备bi5o7i时是将碘化钾(ki)的乙二醇溶液缓慢滴加到五水硝酸铋(bi(no3)3·5h2o)的乙二醇溶液中并在高压釜反应24h(120℃)形成前驱体,然后再洗涤、焙烧制得,前驱体在有机溶剂乙二醇体系中反应生成,不仅成本高、能耗高,而且会产生高浓度有机废水;(2)制备的bi5o7i催化活性不高,120min对罗丹明b的降解率仅为52%,与氧化亚铜的复合物cu2o/bi5o7i的降解率为95.5%;(3)光催化剂难以实现回收再利用,运行成本较高。
又如发明专利“一种锰锌铁氧体-氧化铋磁性光催化剂的制备方法”(公开号:cn104437536a)(对比文件2),以焙烧法制备锰锌铁氧体,然后采用浸渍焙烧法制备锰锌铁氧体/氧化铋复合磁性光催化剂。该方法的不足之处在于:(1)锰锌铁氧体是在1200℃焙烧3h制得,能耗高;(2)利用焙烧法制备的锰锌铁氧体样品颗粒尺寸本身较大,比表面积较小,不利于锰锌铁氧体与氧化铋的充分结合,无法保证结合的稳定性;(3)采用焙烧法制备复合磁性光催化剂,导致复合磁性光催化剂比表面积较小,不利于光催化降解过程中催化剂与有机污染物的充分接触和反应。
技术实现要素:
本发明的目的是针对bi5o7i催化活性不高及难以回收的问题,提出一种bi5o7i/mnxzn1-xfe2o4复合磁性光催化剂的制备方法,制备方法简单、成本低。制备的bi5o7i/mnxzn1-xfe2o4复合磁性光催化剂在模拟太阳光照射下具有较高的光催化效率,且便于通过外加磁场从液相体系中分离和回收,回收后的催化剂仍具有较高的光催化活性。该方法既简易高效的实现了资源再利用,又避免了催化剂可能带来的二次污染。
本发明bi5o7i/mnxzn1-xfe2o4的制备方法如下:
(1)mnxzn1-xfe2o4的制备
采用水热法制备mnxzn1-xfe2o4,按照摩尔比zno:mno:fe2o3=13.3:32.8:53.9,分别称取0.9652g硫酸锌、1.386g硫酸锰、5.3884g硫酸铁,加入50ml去离子水,超声振荡使其溶解得到混合溶液;在磁力搅拌的作用下,向混合溶液中滴加一定浓度的naoh,调节溶液的ph为13,继续搅拌15min;将搅拌后的溶液转移至100ml的反应釜中,在200℃下反应5h,反应完成后冷却、抽滤,分别用蒸馏水和乙醇洗涤7次,在80℃下干燥12h,最后研磨得到mnxzn1-xfe2o4。
(2)bi5o7i/mnxzn1-xfe2o4复合磁性光催化材料的制备
分别量取5ml的乙二醇和35ml蒸馏水混合得到混合液a,称取0.97g的bi(no3)3·5h2o加入到a中,超声5分钟,得到悬浊液b;称取0.332g的ki加入到悬浊液b中,搅拌5分钟,得到悬浊液c;称取占复合物质量比例为5%~15%的第(1)步制备的mnxzn1-xfe2o4加入悬浊液c中,搅拌60分钟后转移到100ml反应釜内胆中,在160℃下反应12h,室温下冷却、抽滤,用蒸馏水洗涤多次,并于80℃下烘干5h;将烘干样研磨后放入100ml陶瓷坩埚,置于马弗炉中,在480℃焙烧2h,得到bi5o7i/mnxzn1-xfe2o4样品。
本发明采用上述技术方案,主要有以下效果:
(1)本发明方法制备的bi5o7i/mnxzn1-xfe2o4复合磁性光催化剂具有较高的光催化活性,在模拟太阳光氙灯照射下,0.1g制备的bi5o7i/mnxzn1-xfe2o4复合磁性光催化分散于100ml浓度为10mg/l的罗丹明b溶液中,光照120min后对罗丹明b的降解率达到98.7%(优于对比文件1制备的cu2o/bi5o7i复合光催化剂)。
(2)本发明方法制备的bi5o7i/mnxzn1-xfe2o4复合磁性光催化剂在外加磁场作用下的回收率高达90.1%,且3次重复使用后的降解率仍达到87.2%。
(3)本发明方法制备的bi5o7i/mnxzn1-xfe2o4复合磁性光催化剂,其平均晶粒尺寸为71.3nm,比表面积为7.07m2/g,其制备操作简单,所需设备少,能耗低。
附图说明
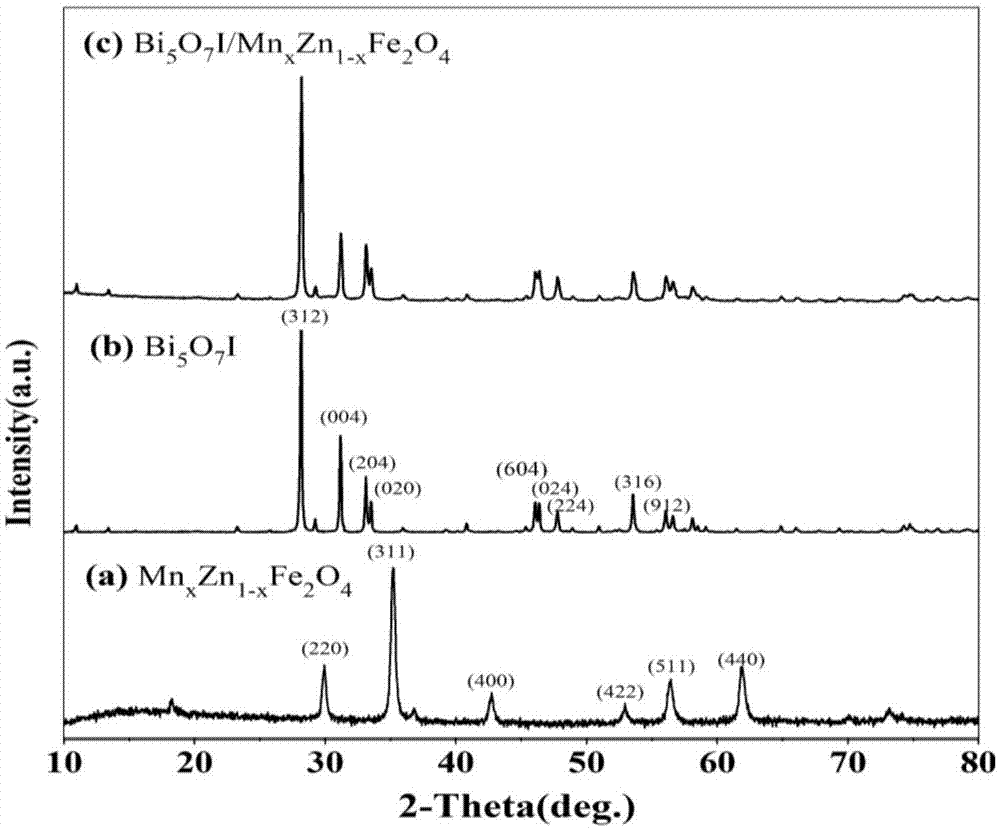
图1为mnxzn1-xfe2o4、bi5o7i和bi5o7i/mnxzn1-xfe2o4的x射线衍射图谱。
图2为mnxzn1-xfe2o4、bi5o7i和bi5o7i/mnxzn1-xfe2o4的红外光谱图。
图3为mnxzn1-xfe2o4和bi5o7i/mnxzn1-xfe2o4的磁性样品的磁滞回归线图。
具体实施方式
下面结合具体实施方式,进一步说明本发明。
实施例1
一种制备bi5o7i/mnxzn1-xfe2o4复合磁性催化材料的制备,具体步骤如下:
(1)mnxzn1-xfe2o4的制备
采用水热法制备mnxzn1-xfe2o4,按照摩尔比zno:mno:fe2o3=13.3:32.8:53.9,分别称取0.9652g硫酸锌、1.386g硫酸锰、5.3884g硫酸铁,加入50ml去离子水,超声振荡使其溶解得到混合溶液;在磁力搅拌的作用下,向混合溶液中滴加一定浓度的naoh,调节溶液的ph为13,继续搅拌15min;将搅拌后的溶液转移至100ml的反应釜中,在200℃下反应5h,反应完成后冷却、抽滤,分别用蒸馏水和乙醇洗涤7次,在80℃下干燥12h,最后研磨得到mnxzn1-xfe2o4。
(2)bi5o7i/mnxzn1-xfe2o4复合磁性光催化材料的制备
分别量取5ml的乙二醇和35ml蒸馏水混合得到混合液a,称取0.97g的bi(no3)3·5h2o加入到a中,超声5分钟,得到悬浊液b;称取0.332g的ki加入到悬浊液b中,搅拌5分钟,得到悬浊液c;称取占复合物质量比例为5%的第(1)步制备的mnxzn1-xfe2o4加入悬浊液c中,搅拌60分钟后转移到100ml反应釜内胆中,在160℃下反应12h,室温下冷却、抽滤,用蒸馏水洗涤多次,并于80℃下烘干5h;将烘干样研磨后放入100ml陶瓷坩埚,置于马弗炉中,在480℃焙烧2h,得到bi5o7i/mnxzn1-xfe2o4样品。
实施例2
一种制备bi5o7i/mnxzn1-xfe2o4复合磁性催化材料的制备,具体步骤如下:
(1)mnxzn1-xfe2o4的制备
同实施例1中(1)。
(2)bi5o7i/mnxzn1-xfe2o4复合磁性光催化材料的制备
分别量取5ml的乙二醇和35ml蒸馏水混合得到混合液a,称取0.97g的bi(no3)3·5h2o加入到a中,超声5分钟,得到悬浊液b;称取0.332g的ki加入到悬浊液b中,搅拌5分钟,得到悬浊液c;称取占复合物质量比例为10%的第(1)步制备的mnxzn1-xfe2o4加入悬浊液c中,搅拌60分钟后转移到100ml反应釜内胆中,在160℃下反应12h,室温下冷却、抽滤,用蒸馏水洗涤多次,并于80℃下烘干5h;将烘干样研磨后放入100ml陶瓷坩埚,置于马弗炉中,在480℃焙烧2h,得到bi5o7i/mnxzn1-xfe2o4样品。
实施例3
一种制备bi5o7i/mnxzn1-xfe2o4复合磁性催化材料的制备,具体步骤如下:
(1)mnxzn1-xfe2o4的制备
同实施例1中(1)。
(2)bi5o7i/mnxzn1-xfe2o4复合磁性光催化材料的制备
分别量取5ml的乙二醇和35ml蒸馏水混合得到混合液a,称取0.97g的bi(no3)3·5h2o加入到a中,超声5分钟,得到悬浊液b;称取0.332g的ki加入到悬浊液b中,搅拌5分钟,得到悬浊液c;称取占复合物质量比例为15%的第(1)步制备的mnxzn1-xfe2o4加入悬浊液c中,搅拌60分钟后转移到100ml反应釜内胆中,在160℃下反应12h,室温下冷却、抽滤,用蒸馏水洗涤多次,并于80℃下烘干5h;将烘干样研磨后放入100ml陶瓷坩埚,置于马弗炉中,在480℃焙烧2h,得到bi5o7i/mnxzn1-xfe2o4样品。
实验结果
实施例2制备的bi5o7i/mnxzn1-xfe2o4复合磁性光催化材料催化降解活性最佳。为了方便对比,制备了bi5o7i样品。bi5o7i制备方法为实施例2步骤(2)中不加入mnxzn1-xfe2o4。
bi5o7i的xrd图谱如图1(b)所示,所有衍射峰均能指标化为正交晶系的bi5o7i(jcpdsno.:40-0548),表明成功制备出了bi5o7i晶体,晶胞参数为
mnxzn1-xfe2o4的xrd图谱如图1(a)所示,从图中可以看出所有衍射峰均能指标化为立方晶系的mnxzn1-xfe2o4(jcpdsno.:74-2399),晶胞参数为
bi5o7i/mnxzn1-xfe2o4的xrd图谱如图1(c)所示,与纯bi5o7i的xrd图谱对比发现,bi5o7i/mnxzn1-xfe2o4的主要衍射峰与bi5o7i基本一致,表明mnxzn1-xfe2o4的复合并没有改变bi5o7i晶体的偏好生长方向以及晶体结构。并且mnxzn1-xfe2o4的部分衍射峰与bi5o7i的衍射峰发生了重合。
图2(a)为mnxzn1-xfe2o4的红外光谱图,其中3434cm-1处的吸收峰属于mnxzn1-xfe2o4表面吸附水的-oh伸缩振动峰,2360cm-1附近出现的吸收峰为样品表面吸附水的-oh弯曲振动峰,568cm-1处出现的峰对应于为zn-o键的振动,1399cm-1处出现的峰对应于fe-o-fe键的伸缩振动。
图2(b)为bi5o7i的红外光谱图,其中491cm-1处的吸收峰归属于bi5o7i结构中的bi-o键的伸缩振动峰,610cm-1处的吸收峰属于bi-o键的弯曲振动峰,846cm-1、1389cm-1处的吸收峰属于bi-o键的振动峰。
图2(c)为bi5o7i/mnxzn1-xfe2o4的红外光谱图,由图可知,bi5o7i中bi-o键的弯曲振动峰(610cm-1)蓝移至609cm-1;mnxzn1-xfe2o4中吸附水的-oh振动峰消失,主要原因是经过了焙烧过程;mnxzn1-xfe2o4中zn-o键的振动峰(568cm-1)在图2(c)中出现(569cm-1);且fe-o-fe键的伸缩振动(1399cm-1)蓝移至1395cm-1,说明bi5o7i和mnxzn1-xfe2o4发生了复合且存在相互作用。
mnxzn1-xfe2o4的磁性参数测试如图3(a)所示,其饱和磁化强度为69.8emu/g,矫顽力为42oe。
bi5o7i/mnxzn1-xfe2o4的磁性参数如图3(b)所示,其饱和磁化强度为3.8emu/g,矫顽力为126.7oe,表明复合光催化剂bi5o7i/mnxzn1-xfe2o4具有较强的磁性。
光催化实验表明,在模拟太阳光氙灯照射下,用0.1g制备的复合磁性光催化剂降解100ml浓度为10mg/l的罗丹明b溶液,光照120min对罗丹明b的降解率达到98.7%,在外加磁场下对光催化剂的磁回收率为90.1%,重复使用3次后的降解率为87.2%,说明采用本发明制备的bi5o7i/mnxzn1-xfe2o4复合磁性光催化剂具有较高的光催化活性和稳定性。
- 还没有人留言评论。精彩留言会获得点赞!