硬模板法制备多孔富氮氮化钴的方法及应用与流程
本发明属于催化剂制备技术领域,涉及一种硬模板法制备多孔富氮氮化钴,并应用于析氧反应催化。
背景技术:
氢气(h2)是目前最清洁、最有前景并且可持续发展的新能源之一,它通过电解水产生,电化学分解水得到h2和o2是目前最有效的途径之一,但是,在电催化水分解过程中,阳极上发生的析氧反应(oer)涉及复杂的四电子转移以及多步中间体过程往往使得其反应动力学迟滞,这就要求极高的过电位,从而导致全水分解效率降低。基于贵金属钌(ru)和铱(ir)的电催化剂(如:ruo2、iro2)是目前最高效的析氧反应(oer)催化剂,然而,这些贵金属元素在地球上的储量是极其稀少并且价格也十分昂贵,因此并不能满足大规模利用和推广的需求。而地球储量丰富的元素制备的析氧反应(oer)催化剂具有价格低廉、析氧反应(oer)催化活性高以及优异的长期稳定性,所以这些地球储量丰富的非贵金属催化剂在新能源领域已经展现出替代贵金属的潜能,但是,大多数地球储量丰富的电催化剂由于其绝缘体或者半导体特征阻碍了电催化剂到集电器之间的电子传输,这极大的阻碍了析氧反应(oer)效率。因此需要进一步探究地球储量丰富元素的催化剂如何改善其析氧反应(oer)中电子的传导。
近期,具有金属性的氮化钴被证明作为一种新型的高性能的析氧反应(oer)电催化剂,并且地球储量丰富。然而,化学反应的本质主要是化学键的断裂和重建,在同样反应条件下,需要更低活化能(决速活化自由能)的催化反应能够优先诱发催化反应的发生,从而具有高的反应速。但是,大家通常都会忽略氮化钴优异析氧反应(oer)催化性能最主要的因素就是热力学优势。
技术实现要素:
为达到上述目的,本发明提供一种硬模板法制备多孔富氮氮化钴的方法,解决了现有技术中大多数地球储量丰富的电催化剂由于其绝缘体或者半导体特征阻碍了电催化剂到集电器之间的电子传输,极大的阻碍了析氧反应(oer)效率的问题。
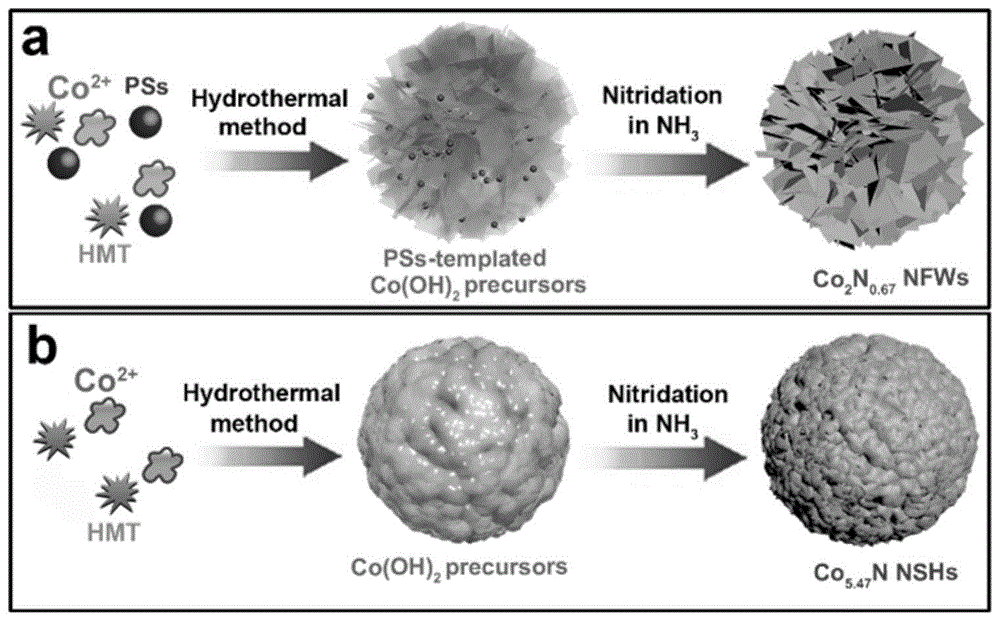
为解决上述技术问题,本发明所采用的技术方案是,以廉价的硝酸钴通过水热法,低温干燥,最后在氨气氛围条件下,管式炉的炉温范围为350-450℃,加热保持1.5-2.5小时即可制得co5.47n纳米微球(co5.47nnshs)材料。同时作为对照,使用聚苯乙烯磺酸钠小球为模板,制备得到多孔富氮co2n0.67纳米花(co2n0.67nfws)材料,两种不同氮化钴催化剂的制备过程如图1所示,制备过程按以下步骤进行:
步骤s1,制备co(oh)2nfws前驱体:
水热法合成,不使用聚苯乙烯磺酸钠小球作为模板,按比例将2.5-5mmolco(ch3coo)2·4h2o、5-10mmol六甲基四胺加入到去离子水中搅拌,将上述混合溶液加入到聚四氟乙烯内衬高压斧中,置于高温烘箱中,反应完全以后,待高压斧和产物均冷却到室温,将产物用去离子水和无水乙醇分别清洗,再过夜烘干即可得到co(oh)2nfws;
步骤s2,将制得的co(oh)2nfws放入管式炉中,在氨气氛围下,350-450℃保持1.5-2.5h,冷却到室温取出即可。
进一步的,所述步骤s1中,使用聚苯乙烯磺酸钠小球作为模板通过水热法合成,来制备co(oh)2nfws前驱体,按比例将2.5-5mmolco(ch3coo)2·4h2o、5-10mmol/l聚苯乙烯磺酸钠小球、5-10mmol六甲基四胺加入到去离子水中搅拌,其他操作保持一致。
进一步的,所述步骤s1中,co(oh)2nfws前驱体,聚苯乙烯磺酸钠小球为模版的,使用pss-co(oh)2标示。
进一步的,所述步骤s1中,是在40-80ml去离子水中搅拌1-3h;混合溶液加入到40-100ml聚四氟乙烯内衬高压斧中,置于高温烘箱中,在110-130℃下保持10-13h;将产物用去离子水和无水乙醇分别洗3-6次,40-80℃下过夜烘干。
进一步的,所述步骤s2中,管式炉升温速率为5-10℃min-1。
进一步的,所述一种硬模板法制备多孔富氮氮化钴的方法制备的多孔富氮氮化钴应用于析氧反应催化。
关于技术方案中出现的相关操作、数值范围做以下说明:步骤s1中“2.5-5mmolco(ch3coo)2·4h2o、5-10mmoll-1聚苯乙烯磺酸钠小球(pss)、5-10mmol六甲基四胺”,所选材料物质的量的范围是经过实验研究并结合实际实验室设备仪器可以实现的最佳范围得出的,如果范围过大除了做各个组对照实验,实验室的相关仪器并不能满足大容量范围,如果数值范围过小,实验中容易带来一些系统误差,干扰后续实验结果分析,本发明所选的这一范围既可以满足实验统计学的意义,同时又能够很好的做到实验级别的重复操作;步骤s2中“350-450℃保持1.5-2.5h”,经实验研究发现氨气在350-450℃温度范围可以很好将金属氢氧化物或者氧化物转化为氮化物,如果温度过于高会腐蚀前驱体,如果温度过于低,则不能成功得到氮化物,350-450℃保持1.5-2.5h,该时间范围下刚好可以得到形貌结构好的氮化物,如果保持时间太短,则得不到纯相的氮化物,如果保持时间过于长,则得到的氮化物表面相貌容易被氨气大量腐蚀;“混合溶液加入到40-100ml聚四氟乙烯高压斧中,置于高温烘箱中”,通过高温烘箱提供的热量加入高压斧,高压斧中的高温高压使得高压斧中混合溶液中碱分解得到氢氧根与溶液中的金属离子结合发生化学反应,得到氢氧化物前驱体,40-100ml聚四氟乙烯,这一范围下得到的产物在后续处理中结合实验室具体设备仪器,可操作性更强,如果偏小,得到的产物量太少,对于下一步的氮化产率低,如果过大,后续处理过程将浪费大量的时间以及实验试剂;在110-130℃下保持10-13h,经实验研究发现,前驱体碱溶液在此温度范围刚好能够分解产生目标产物所需氢氧根,此时间范围刚好能够保证反应进行充分完全,如果温度过于小,碱溶液不能完全分解为氢氧根,使得最终产物产率低,大量浪费实验原料,如果温度过于高,实际实验中并不需要这么高温度反应就可以发生,不仅浪费能源,同时温度太高会有实验安全隐患,如果时间太短,则反应不完全,这样会造成反应效率低,反应产物产率低,如果反应时间过长,高压斧长时间保持高压,实验不仅不安全,而且大量浪费不必要的时间以及能源;将产物用去离子水和无水乙醇分别洗3-6次,40-80℃下过夜烘干,用去离子水洗去产物中一些无机盐副产物,用乙醇洗是洗去产物中一些有机的副产物,洗3-6次是为了保证能够充分将一些副产物洗净,保证产物的纯度,由于前期使用去离子水、乙醇洗终产物,所以需要在40-80℃下过夜烘干终产物,如果洗的次数太少,会洗不净副产物,使得终产物纯度不够,洗得次数太多会大量流失产物,最终得到的产物产率低且量少,如果温度太低,不能有效烘干洗涤以后的产物中带有的微量水和大量的乙醇,如果温度过于高会使得终产物有变质的风险;40-80ml去离子水中搅拌1-3h,这一数值范围刚好可以使得前驱体溶液搅拌达到最佳的均一状态有利于接下来的水热反应,如果搅拌时间过短,则搅拌得到的前驱体溶液不能充分混合不均一,不利于下一步的水热反应,如果搅拌时间过长,溶液已经达到均一状态了,数值偏大是过渡消耗实验时间,耗时耗力;管式炉升温速率为5-10℃min-1,所选择数值范围刚好可以使得管式炉在安全范围内进行反应,同时使得氮化反应刚好在最优状态进行,如果选择数值偏小,会使得升温速度过慢,前驱体在氮化过程中经历太长时间的温度梯度变化,可能得到各种杂相物质,如果所选择数值范围偏大的话,升温速度过快,会使得氮化得到的物质形貌坍塌过于快,最终得到的纳米材料的形貌太差。
本发明的有益效果是:
1.材料成本低,与传统析氧反应催化剂材料贵金属相比,该材料地球储量丰富,价格低廉,来源广泛易得,且实验结果与贵金属材料之一的氧化铱相比更优异,具体表现为相比于氧化铱在碱性介质中的析氧反应,有更低的起始电位和η10(电流密度10macm-2时的电位值),更高的长期稳定性。
2.材料制备方法简单,采用水热法制备氮化钴前驱体这一步引入聚苯乙烯磺酸钠小球(pss)作为硬模板,使得制备的氮化钴前驱体通过氨解步骤不仅有效去除模板得到多孔结构,使得氨解能够充分掺氮得到富氮氮化钴,还能保持前驱体的片状堆积得到纳米花结构,该材料用于析氧反应催化,表现出优异的催化活性,与传统制备方法相比,制备程序大大简化,制备时间大大节约,制备条件更加温和。
3.氮化钴材料由于将氮元素引入到过渡金属钴(co)中,可以使过渡金属co的d-带电子密度增加并且d-带收缩,从而使得氮化钴电子结构类似于贵金属pd和pt,态密度跨过费米能级,所以,氮化钴也具有类似于金属的导电性,同时氮的引入占据了主金属co的间隙位置形成间质合金,从而使得氮化钴有比主金属co高的耐腐蚀性,这使氮化钴能够很好的适用于酸性或者碱性介质中电催化水分解中的析氧反应。
4.电化学析氧反应测试:制备得到的co2n0.67纳米花(nfws)用于析氧反应催化,具有低的起始电位、η10(电流密度10macm-2时的电位值)、小的塔菲尔斜率值、小的电荷传质电阻、大的双电层电容值、大的催化剂单原子的催化效率以及优异的热力学优势,并且具有优异的长期稳定性,既有效节约了成本,又提高了电化学析氧反应的催化性能。
5.采用聚苯乙烯小球作为硬模板,制备三维多孔结构富氮氮化钴具有以下优势:方法简单、重现性好、氮化钴形貌均一和性能稳定,在去除聚苯乙烯硬模板后,得到的材料保持了硬模板的孔道结构,因此得到三维多孔结构的氮化钴。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1是两种不同氮化钴催化剂的制备过程图。
图2a是co2n0.67nfws的x-射线多晶衍射仪谱图。
图2b是co2n0.67nfws的扫描电子显微镜图。
图2c是co2n0.67nfws上单元纳米片npts的高倍透射电子显微镜图。
图2d是co2n0.67nfws氮气吸附-脱附图(其中插图为co2n0.67nfws的孔径分布曲线图)。
图2e是co2n0.67nfws的x-射线能谱仪(edx)元素分布图。
图2f是co2n0.67nfws的x-射线能谱仪(edx)元素分布图。
图2g是co2n0.67nfws的x-射线能谱仪(edx)元素分布图。
图2h是co2n0.67nfws的x-射线能谱仪(edx)元素分布图。
图3a是几种催化剂样品的循环伏安法扫描曲线(lsv)图。
图3b是几种催化剂样品通过电活性面积归一化以后的循环伏安法扫描曲线(lsv)图。
图3c是图a中对应循环伏安法扫描曲线(lsv)的塔菲尔斜率图。
图3d是三种样品在电位为1.6vvs.rhe时的电化学交流阻抗谱图。
图3e是co2n0.67nfws与co5.47nnshs的双电层电容谱图。
图3f是三种催化剂对应的交叉频率图。
图4a是o基中间体在co3n[101]面上的吸附过程图。
图4b是o基中间体在co3n[101]面上的吸附过程图。
图4c是o基中间体在co3n[101]面上的吸附过程图。
图4d是o基中间体在co4n[111]面上的吸附过程图。
图4e是o基中间体在co4n[111]面上的吸附过程图。
图4f是o基中间体在co4n[111]面上的吸附过程图。
图4g是co3n和co4n析氧反应(oer)过程中的自由能谱图。
图4h是co3n和co4n析氧反应(oer)过程中的自由能谱图。
图5是co2n0.67nfws和co5.47nnshs在电位为1.6vvs.rhe时的稳定性图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
步骤s1,制备co(oh)2nfws前驱体:
co(oh)2nfws前驱体不使用聚苯乙烯磺酸钠小球(pss)作为模板通过水热法合成,按比例将3.5mmolco(ch3coo)2·4h2o、7mmol六甲基四胺(hmt)加入到60ml去离子水中搅拌2h,将上述混合溶液加入到80ml聚四氟乙烯内衬高压斧中,置于高温烘箱中,在120℃下保持12h,反应完全以后,待高压斧和产物均冷却到室温,将产物用去离子水和无水乙醇分别洗4次,60℃下过夜烘干即可得到co(oh)2nfws;同时采用同样的方法来制备co(oh)2nfws前驱体但使用聚苯乙烯磺酸钠小球(pss)作为模板,按比例将3.5mmolco(ch3coo)2·4h2o、7mmol/l聚苯乙烯磺酸钠小球(pss)、7mmol六甲基四胺(hmt)加入到60ml去离子水中搅拌2h,其他步骤保持一致;
步骤s2,将不使用聚苯乙烯磺酸钠小球(pss)和使用聚苯乙烯磺酸钠小球(pss)两种方法制得的co(oh)2nfws前驱体(聚苯乙烯磺酸钠小球(pss)为模板的,使用pss-co(oh)2标示)放入管式炉中,管式炉升温率为5℃min-1,在氨气氛围下,400℃保持2h,冷却到室温取出即可;
步骤s3,将催化剂负载在旋转环盘电极(rde)上(玻碳电极,d=5mm,玻碳电极的几何表面积a=0.19625cm2)制备得到工作电极;在修饰电极前,首先将旋转环盘电极(rde)分别按照以下顺序用1、0.3、0.05mm的氧化铝粉末仔细抛光打磨,之后再依次使用稀hno3溶液、乙醇和去离子水洗涤;将制备好的催化剂粉末(3mg)分散到nafion(0.1wt%)水溶液(1ml)中超声60min;然后将30μl(包含90μg催化剂)上述分散液缓慢滴加到洁净的旋转环盘电极(rde)表面(负载量约0.459mgcm-2),并在红外灯下干燥备用;同时也使用商业铂碳催化剂(pt/c、20wt%铂负载量)修饰电极作为对照样;在本工作中,室温下,在标准三电极电解槽中进行电化学测试;旋转环盘电极(rde)修饰电极作为工作电极,铂丝电极作为对电极,ag/agcl电极(饱和kcl溶液)作为参比电极;本工作中析氧反应(oer)测试的电解液为1.0mkoh溶液;用标准的2273稳压电化学工作站分别测试交流阻抗(eis)、循环伏安曲线(cv)以及循环伏安法扫描曲线(lsv);本工作中的所有电势均是参照于可逆氢电极(rhe);计算公式为erhe=eag/agcl+0.0591ph+0.198v,其中erhe为参照的可逆氢电极的电势值,eag/agcl为参照ag/agcl电极测试得到的电势值,ph为电解液的ph值;所涉及的电流密度(j)为工作电极上记录的电流与玻碳电极几何面积的比值;起始电位(eonset)是oer/her(上述已经提到的参照于可逆氢电极的电位)电流与基线的交点对应的电位数值;电解液的阻抗与循环伏安法扫描曲线(lsv)用来做电解池本身电阻的补偿(ir矫正),本工作中所有的循环伏安法扫描曲线(lsv)都经过电解池本身电阻的补偿(ir矫正);析氧反应(oer)的测试:以电势值(evs.rhe)为纵坐标,log|j(macm-2)|为横坐标,选择合适的电位区间作图就可得到tafel曲线,通过塔菲尔公式η=blog(|j|)+a,其中η为过电势,得到的tafel斜率值b,此处a为具体到一个方程的坐标值,不同的催化剂具有不同数值,该数值,可用来评估纳米材料催化剂的析氧反应(oer)的动力学;
催化剂单原子的催化效率使用交叉频率值表示(tofs),使用以下公式来计算:tof=j×a/(4×f×n),其中,j代表电流密度,a表示玻碳电极的几何面积(0.19625cm2),f表示法拉第效率(96485cmol-1),n代表分散在玻碳电极上的co的物质的量(mol);这里,首先使用x-射线衍射仪(xrd)表征,得到元素的含量比例,然后计算出来不同修饰电极上co的摩尔数;
步骤s4,所有的密度泛函理论(dft)计算都是通过perdew–burke–ernzerhof(pbe)计算,使用castep程序,离子核中心使用ultrasoftpseudopotentials来描述其可转移性,以及降低平面波数来获得轨道的扩展能;通过x-射线衍射仪(xrd)测试,co2n0.67的x-射线衍射仪(xrd)标准卡片信息(jcpdsno.06-0691)显示其主要co的氮化物为co3n与co单质形成的合金,co5.47n的x-射线衍射仪(xrd)标准卡片信息(jcpdsno.41-0943)显示其主要co的氮化物为co4n与co单质形成的合金;于是可以推测:co2n0.67用于析氧反应(oer)催化的主要有效成分为co3n,co5.47n用于析氧反应(oer)催化的有效成分主要为co4n;根据x-射线衍射仪(xrd)信息显示,co2n0.67的最强吸收峰晶面为101晶面,co5.47n最强吸收峰晶面为111晶面;于是,构筑co3n(3×4×3)和co4n(3×3×3)晶胞做密度泛函理论(dft)计算;在coxn(x=3,4)上反应步骤的自由能使用以下公式来表示:δg=δe+δzpe–tδs,在这里,δe来源于密度泛函理论(dft)计算的反应能,δzpe是源于
实施例2
将实施例1步骤s1修改为,制备co(oh)2nfws前驱体:co(oh)2nfws前驱体不使用聚苯乙烯磺酸钠小球(pss)作为模板通过水热法合成,按比例将2.5mmolco(ch3coo)2·4h2o、5mmol六甲基四胺(hmt)加入到40ml去离子水中搅拌1h,将上述混合溶液加入到60ml聚四氟乙烯内衬高压斧中,置于高温烘箱中,在110℃下保持11h,反应完全以后,待高压斧和产物均冷却到室温,将产物用去离子水和无水乙醇分别洗3次,40℃下过夜烘干即可得到co(oh)2nfws;同时采用同样的方法来制备co(oh)2nfws前驱体但使用聚苯乙烯磺酸钠小球(pss)作为模板,按比例将2.5mmolco(ch3coo)2·4h2o、5mmol/l聚苯乙烯磺酸钠小球(pss)、5mmol六甲基四胺(hmt)加入到40ml去离子水中搅拌1h;将实施例1步骤s2中的管式炉升温率改为10℃min-1;其他步骤不变。
实施例3
将实施例1步骤s1修改为,制备co(oh)2nfws前驱体:co(oh)2nfws前驱体不使用聚苯乙烯磺酸钠小球(pss)作为模板通过水热法合成,按比例将5mmolco(ch3coo)2·4h2o、10mmol六甲基四胺(hmt)加入到80ml去离子水中搅拌3h,将上述混合溶液加入到100ml聚四氟乙烯内衬高压斧中,置于高温烘箱中,在130℃下保持13h,反应完全以后,待高压斧和产物均冷却到室温,将产物用去离子水和无水乙醇分别洗6次,80℃下过夜烘干即可得到co(oh)2nfws;同时采用同样的方法来制备co(oh)2nfws前驱体但使用聚苯乙烯磺酸钠小球(pss)作为模板,按比例将5mmolco(ch3coo)2·4h2o、10mmol/l聚苯乙烯磺酸钠小球(pss)、10mmol六甲基四胺(hmt)加入到80ml去离子水中搅拌3h,其他步骤不变。
实施例4
将实施例1步骤s2中管式炉炉温改为350℃,其他步骤不变。
实施例5
将实施例1步骤s2中管式炉炉温改为450℃,其他步骤不变。
实施例6
将实施例1步骤s2中管式炉炉温保持时间改为1.5h,其他步骤不变。
实施例7
将实施例1步骤s2中管式炉炉温保持时间改为2.5h,其他步骤不变。
实施例8
将实施例1步骤s3中“将30μl(包含90μg催化剂)上述分散液缓慢滴加到洁净的旋转环盘电极(rde)表面(负载量约0.459mgcm-2)”修改为“将20μl(包含60μg催化剂)上述分散液缓慢滴加到洁净的旋转环盘电极(rde)表面(负载量约0.306mgcm-2)”其他步骤不变。
实施例9
将实施例1步骤s3中“将30μl(包含90μg催化剂)上述分散液缓慢滴加到洁净的旋转环盘电极(rde)表面(负载量约0.459mgcm-2)”修改为“将40μl(包含120μg催化剂)上述分散液缓慢滴加到洁净的旋转环盘电极(rde)表面(负载量约0.611mgcm-2)”其他步骤不变。
对照组1
将实施例1步骤s1修改为,制备co(oh)2nfws前驱体:co(oh)2nfws前驱体不使用聚苯乙烯磺酸钠小球(pss)作为模板通过水热法合成,按比例将2mmolco(ch3coo)2·4h2o、4.5mmol六甲基四胺(hmt)加入到30ml去离子水中搅拌3h,将上述混合溶液加入到35ml聚四氟乙烯内衬高压斧中,置于高温烘箱中,在100℃下保持9h,反应完全以后,待高压斧和产物均冷却到室温,将产物用去离子水和无水乙醇分别洗2次,30℃下过夜烘干即可得到co(oh)2nfws;同时采用同样的方法来制备co(oh)2nfws前驱体但使用聚苯乙烯磺酸钠小球(pss)作为模板,按比例将2mmolco(ch3coo)2·4h2o、4mmol/l聚苯乙烯磺酸钠小球(pss)、4.5mmol六甲基四胺(hmt)加入到30ml去离子水中搅拌3h;将实施例1步骤s2中的管式炉升温率改为3℃min-1;其他步骤不变。
对照组2
将实施例1步骤s1修改为,制备co(oh)2nfws前驱体:co(oh)2nfws前驱体不使用聚苯乙烯磺酸钠小球(pss)作为模板通过水热法合成,按比例将6mmolco(ch3coo)2·4h2o、12mmol六甲基四胺(hmt)加入到90ml去离子水中搅拌0.5h,将上述混合溶液加入到35ml聚四氟乙烯内衬高压斧中,置于高温烘箱中,在140℃下保持9h,反应完全以后,待高压斧和产物均冷却到室温,将产物用去离子水和无水乙醇分别洗2次,30℃下过夜烘干即可得到co(oh)2nfws;同时采用同样的方法来制备co(oh)2nfws前驱体但使用聚苯乙烯磺酸钠小球(pss)作为模板,按比例将6mmolco(ch3coo)2·4h2o、11mmol/l聚苯乙烯磺酸钠小球(pss)、12mmol六甲基四胺(hmt)加入到90ml去离子水中搅拌0.5h;将实施例1步骤s2中的管式炉升温率改为4℃min-1;其他步骤不变。
对照组3
将实施例1步骤s1修改为,制备co(oh)2nfws前驱体:co(oh)2nfws前驱体不使用聚苯乙烯磺酸钠小球(pss)作为模板通过水热法合成,按比例将6mmolco(ch3coo)2·4h2o、12mmol六甲基四胺(hmt)加入到90ml去离子水中搅拌3h,将上述混合溶液加入到110ml聚四氟乙烯内衬高压斧中,置于高温烘箱中,在140℃下保持14h,反应完全以后,待高压斧和产物均冷却到室温,将产物用去离子水和无水乙醇分别洗7次,90℃下过夜烘干即可得到co(oh)2nfws;同时采用同样的方法来制备co(oh)2nfws前驱体但使用聚苯乙烯磺酸钠小球(pss)作为模板,按比例将6mmolco(ch3coo)2·4h2o、11mmol/l聚苯乙烯磺酸钠小球(pss)、12mmol六甲基四胺(hmt)加入到90ml去离子水中搅拌3h;将实施例1步骤s2中的管式炉升温率改为11℃min-1;其他步骤一致。
对照组4
将实施例1步骤s2中管式炉炉温改为340℃,其他步骤不变。
对照组5
将实施例1步骤s2中管式炉炉温改为460℃,其他步骤不变。
对照组6
将实施例1步骤s2中管式炉炉温保持时间改为1h,其他步骤不变。
对照组7
将实施例1步骤s2中管式炉炉温保持时间改为3h,其他步骤不变。
对照组8
将实施例1步骤s3中“将30μl(包含90μg催化剂)上述分散液缓慢滴加到洁净的旋转环盘电极(rde)表面(负载量约0.459mgcm-2)”修改为“将15μl(包含55μg催化剂)上述分散液缓慢滴加到洁净的旋转环盘电极(rde)表面(负载量约0.290mgcm-2)”其他步骤不变。
对照组9
将实施例1步骤s3中“将30μl(包含90μg催化剂)上述分散液缓慢滴加到洁净的旋转环盘电极(rde)表面(负载量约0.459mgcm-2)”修改为“将45μl(包含125μg催化剂)上述分散液缓慢滴加到洁净的旋转环盘电极(rde)表面(负载量约0.650mgcm-2)”其他步骤不变。
实施例1数据分析
实施例1在数据分析中取得的效果最优,相关数据分析结果叙述如下:
图2a为x-射线多晶衍射仪谱图,通过图2a可以看出,co2n0.67nfws的x-射线衍射仪(xrd)谱图显示所有峰信息对应着纯co2n0.67物相(jcpdsno.06-0691),这也表明了pss-co(oh)2nfws前驱体成功转化为co2n0.67nfws,同时,根据x-射线衍射仪(xrd)输出的jcpds卡片信息显示co2n0.67归属于p63/mmc空间群的六方晶系。使用扫描电子显微镜(sem)和透射电子显微镜(tem)来表征co2n0.67nfws的形貌。通过图2bsem图可以看出co2n0.67nfws是由大量的纳米片(npts)堆积而成,纳米片的厚度约为20nm。从图2c中可以清晰看出单元npts的多孔结构。氮气吸附-脱附图2d显示co2n0.67nfws具有高的比表面积(25.40m2g-1),并且为多级孔结构且孔尺寸分布范围在2-200nm(图2d)。co2n0.67nfws的高分辨tem(hrtem)图2c中的插图可以观察到晶格间距约0.204nm,对应着co2n0.67的101晶面间距。x-射线能谱仪(edx)元素分布图显示co2n0.67nfws中co、n和o的元素分布(图2e-2h)。除了co和n元素分布信息,少量o元素也被检测到,表明co2n0.67nfws中具有高纯度的co2n0.67物相,表面少量被氧化。
图3中,为了证实co2n0.67nfws能否作为高效的析氧反应(oer)催化剂,对其进行了一系列电化学测试,电化学测试条件如下:1.0mkoh、o2饱和、三电极体系。图3a显示的是不同样品在扫速为5mvs-1时的循环伏安法扫描曲线(lsv),本工作中所有图片中循环伏安法扫描曲线(lsv)均是经过电解池本身电阻的补偿(ir矫正)。从图3a可以得到,co2n0.67nfws做析氧反应(oer)催化具有更低的起始电位值(ηonset)和η10(电流密度10macm-2时的电位值)值,分别为221mv和258mv,而co5.47nnshs分别为296mv和359mv以及ruo2分别为278mv和354mv均远高于co2n0.67nfws。为了研究氮化钴用于析氧反应(oer)的本征催化活性,使用电活性面积归一化测试所得的析氧反应(oer)电流,归一化以后,co2n0.67nfws依然显示出比co5.47nnshs更优的析氧反应(oer)催化性能(图3b)。co2n0.67nfws的塔菲尔斜率值(tafelslope)为52.22mvdec-1,低于co5.47nnshs(73.69mvdec-1)(图3c),表明co2n0.67nfws对于析氧反应(oer)催化具有更优异的动力学优势。此外,电化学交流阻抗(eis)图显示co2n0.67nfws电荷-转移电阻值为26ω,比co5.47nnshs(58ω)低(图3d),证明co2n0.67nfws的电荷转移过程更高效。从图3e中可以得到,co2n0.67nfws的cdl值为249.1mfcm-2,是远远高于co5.47nnshs(100.4mfcm-2),表明co2n0.67nfws具有更高的电活性面积。此外,co2n0.67的tof值(1.16s-1,电位值为1.55v时(vs.rhe))也大于co5.47n(0.24s-1)(图3f),表明co2n0.67的单原子催化效率更高,证明co2n0.67具有更加优异的本征析氧反应(oer)催化活性。
图4中,为了明确co2n0.67和co5.47n的析氧反应(oer)活性的本质,将两种催化剂的具体析氧反应(oer)路径使用密度泛函理论(dft)计算其反应决速步骤的热力学能。co2n0.67和co3n均属于p63/mmc(194)空间群的六方晶系,co5.47n和co4n均属于fm-3m(225)空间群的面心立晶系。x-射线衍射仪(xrd)结果也显示co2n0.67nfws和co5.47nnshs最强吸收峰晶面分别是(101)和(111)晶面。因此,co3n的(101)和co4n的(111)晶面作为暴露晶面分别作为co2n0.67和co5.47n的密度泛函理论(dft)计算模型。吸附在co3n或者co4n上的氧中间体(oh*、o*和ooh*)的理想结构是由perdew–burke–ernzerhof(pbe)方法来构筑。在co3n(101)晶面的表面(图4a-c),oh-优先吸附在临近n的co-co键
图5中,析氧反应(oer)催化剂另外一个重要的参数就是长期稳定性,本工作中使用计时电流法来测试催化剂的长期稳定性。如图5所示,可以清晰看到,15h以后,co2n0.67nfws的计时电流值还保持为原始值的91.6%,是明显优于co5.47nnshs(60.9%,图5)。尤其在刚开始的前0.2h,co2n0.67nfws的电流值几乎保持不变,这也证明了co2n0.67nfws高的稳定性。
实施例2分析
图2a为x-射线多晶衍射仪谱图,通过图2a可以看出,co2n0.67nfws的x-射线衍射仪(xrd)谱图显示所有峰信息对应着纯co2n0.67物相(jcpdsno.06-0691),这也表明了pss-co(oh)2nfws前驱体成功转化为co2n0.67nfws,同时,根据x-射线衍射仪(xrd)输出的jcpds卡片信息显示co2n0.67归属于p63/mmc空间群的六方晶系。使用扫描电子显微镜(sem)和透射电子显微镜(tem)来表征co2n0.67nfws的形貌。通过图2bsem图可以看出co2n0.67nfws是由大量的纳米片(npts)堆积而成,纳米片的厚度约为20nm。从图2c中可以清晰看出单元npts的多孔结构。氮气吸附-脱附图2d显示co2n0.67nfws具有高的比表面积(19.05m2g-1),并且为多级孔结构且孔尺寸分布范围在1.5-150nm(图2d)。co2n0.67nfws的高分辨tem(hrtem)图2c中的插图可以观察到晶格间距约0.206nm,对应着co2n0.67的101晶面间距。x-射线能谱仪(edx)元素分布图显示co2n0.67nfws中co、n和o的元素分布(图2e-2h)。除了co和n元素分布信息,少量o元素也被检测到,表明co2n0.67nfws中具有高纯度的co2n0.67物相,表面少量被氧化。
图3中,为了证实co2n0.67nfws能否作为高效的析氧反应(oer)催化剂,对其进行了一系列电化学测试,电化学测试条件如下:1.0mkoh、o2饱和、三电极体系。图3a显示的是不同样品在扫速为5mvs-1时的循环伏安法扫描曲线(lsv),本工作所有图片中循环伏安法扫描曲线(lsv)均是经过电解池本身电阻的补偿(ir矫正)。从图3a可以得到,co2n0.67nfws做析氧反应(oer)催化具有更低的起始电位值(ηonset)和η10(电流密度10macm-2时的电位值)值,分别为276mv和322mv,而co5.47nnshs(370mv和448mv)高于co2n0.67nfws。为了研究氮化钴用于析氧反应(oer)的本征催化活性,使用电活性面积归一化测试所得的析氧反应(oer)电流,归一化以后,co2n0.67nfws依然显示出比co5.47nnshs更优的析氧反应(oer)催化性能(图3b)。co2n0.67nfws的塔菲尔斜率值(tafelslope)为65.28mvdec-1,低于co5.47nnshs(92.11mvdec-1)(图3c),表明co2n0.67nfws对于析氧反应(oer)催化具有更优异的动力学优势。此外,电化学交流阻抗(eis)图显示co2n0.67nfws电荷-转移电阻值为33ω,比co5.47nnshs(73ω)低(图3d),证明co2n0.67nfws的电荷转移过程更高效。从图3e中可以得到,co2n0.67nfws的cdl值为187mfcm-2,是远远高于co5.47nnshs(75.3mfcm-2),表明co2n0.67nfws具有更高的电活性面积。此外,co2n0.67的tof值(0.87s-1,电位值为1.65v时(vs.rhe))也大于co5.47n(0.18s-1)(图3f),表明co2n0.67的单原子催化效率更高,证明co2n0.67具有更加优异的本征析氧反应(oer)催化活性。
图4中,为了明确co2n0.67和co5.47n的析氧反应(oer)活性的本质,将两种催化剂的具体析氧反应(oer)路径使用密度泛函理论(dft)计算其反应决速步骤的热力学能。co2n0.67和co3n均属于p63/mmc(194)空间群的六方晶系,co5.47n和co4n均属于fm-3m(225)空间群的面心立晶系。x-射线衍射仪(xrd)结果也显示co2n0.67nfws和co5.47nnshs最强吸收峰晶面分别是(101)和(111)晶面。因此,co3n的(101)和co4n的(111)晶面作为暴露晶面分别作为co2n0.67和co5.47n的密度泛函理论(dft)计算模型。吸附在co3n或者co4n上的氧中间体(oh*、o*和ooh*)的理想结构是由perdew–burke–ernzerhof(pbe)方法来构筑。在co3n(101)晶面的表面(图4a-c),oh-优先吸附在临近n的co-co键
图5中,析氧反应(oer)催化剂另外一个重要的参数就是长期稳定性,本工作中使用计时电流法来测试催化剂的长期稳定性。如图5所示,可以清晰看到,15h以后,co2n0.67nfws的计时电流值还保持为原始值的68.7%,是明显优于co5.47nnshs(45.7%,图5)。尤其在刚开始的前0.1h,co2n0.67nfws的电流值几乎保持不变,这也证明了co2n0.67nfws高的稳定性。实施例2分析中提到的图并非是指本发明的说明书附图,本发明的说明书附图是针对实施例1的附图。
实施例3分析
在制备前驱体过程中采用高浓度范围制备,高浓度范围制备不仅仅是耗费材料,同时制备后处理过程中也是大量损耗样品,反而降低制备得到样品的产率,既不经济也不符合科学配比制备的原则。
实施例4分析
当将前驱体制备的温度改为350℃低温氮化以后,会发现低温氮化氮化不完全,不能有效将氮掺杂进去co前驱体得到纯物相的氮化钴。
实施例5分析
当氮化温度过高时,大量氨气分解会过渡腐蚀样品,样品形貌会一定程度的坍塌,影响后续的电催化效率。
实施例6分析
当氮化时间为1.5h时,由于氮化时间太短,并不能充分将前驱体氮化彻底,得到的氮化钴的物相并不是纯相,是杂相物质,影响后续的电化学催化效率以及分析催化本征活性。
实施例7分析
当氮化时间为2.5h以后,由于氮化的时间过于长,会使得最终得到的氮化钴产物形貌结构坍塌严重,影响后续产物的催化性能,得到的催化性能比最优样品差。
实施例8分析
将催化剂的负载量“改为20μl(包含60μg催化剂)上述分散液缓慢滴加到洁净的旋转环盘电极(rde)表面(负载量约0.306mgcm-2)”由于催化剂的负载量太小,催化过程中催化的活性位点数目不足,所以不能有效达到最优催化效率。
实施例9分析
将催化剂的负载量改为“将40μl(包含120μg催化剂)上述分散液缓慢滴加到洁净的旋转环盘电极(rde)表面(负载量约0.611mgcm-2)”催化剂的负载量太大,部分催化活性位点被覆盖,不仅仅得不到最优的催化效率,同时还大量浪费催化剂材料。
对照组1分析
选择极端小的数据范围制备合成前驱体以及氮化制备样品,由于水热过程中碱液浓度不足,所有会导致水热形成氢氧化物前躯体的产率很低,形貌大量杂质,导致后续氮化也形成大量杂质,使得催化效率大大降低。
对照组2分析
选择极端大范围数值合成前驱体,水热过程中碱溶液浓度过高,会抑制正向反应形成前驱体氢氧化物的速率,不仅仅大量浪费实验原材料,反而降低了产物的产率,氮化过程中形成过多杂质,降低后续催化效率。
对照组3分析
此组实验条件下得到的前驱体产物由于烘干温度过高,会出现一定程度的变质,导致后续形成氮化物过程中有杂相物质出现。
对照组4分析
当再过低降低氮化温度的时候,氮化不彻底,就不可避免的出现杂相,后续的催化效率降低。
对照组5分析
当氮化温度极端高的时候,导致氨气分解过快,氮化过渡,会使得氮化物形貌坍塌,不能保持好的纳米结构,降低后续析氧反应催化效率。
对照组6分析
氮化时间极端短,氮化不完全,几乎得不到纯相的氮化钴物质,没办法进行后续的催化反应。
对照组7分析
氮化温度极端长,会使得氮化物最终氮化时间过长,样品容易被腐蚀,形貌坍塌,降低后续析氧反应催化效率。
对照组8分析
催化剂负载量出现极端小的情况,不能将旋转环盘电极(rde)完全修饰覆盖完全,催化活性位点数目太少,不能得到优异的催化效果。
对照组9分析
催化剂负载量极端高的情况,电极表面覆盖大剂量的催化剂,会使得催化活性位点部分被覆盖,并不能有效将所有催化活性位点发挥作用,反而抑制了部分的催化活性位点,因此不仅仅浪费催化剂材料,反而还降低催化剂催化析氧反应的催化效率。
实验结论
1.本发明成功合成并证明了co2n0.67nfws作为一种性能优异的地球储量丰富的非贵金属电催化剂并且可以高效促进析氧反应(oer)催化。聚苯乙烯磺酸钠小球(pss)模板不仅能够帮助有效构筑三维多级孔结构,而且可以促进co2n0.67物相的形成。密度泛函理论(dft)计算表明临近n的co原子可以通过掺杂n来激活,从而作为析氧反应(oer)的活性位点,不同于先前报道的氮化钴氮含量高会使得氮化物导电性降低从而降低析氧反应(oer)活性,而催化反应中导电性仅仅是其中的一项因素,更重要的是其反应中的动力学和热力学优势。
2.本发明证明了co2n0.67比co5.47n在碱性溶液中具有更高的析氧反应(oer)活性,通过密度泛函理论(dft)计算,在co2n0.67表面比co5.47n表面具有更高的热力学优势。本发明不仅合成了新的具有高析氧反应(oer)活性的co2n0.67nfws材料,而且还证明富氮诱导形成更多的活性位点co,从而使得富氮的氮化钴具有更高的热力学优势促进析氧反应(oer)活性,为将来有效设计高效的应用于能源的催化剂提供思路。
以上所述仅为本发明的较佳实施例而已,并非用于限定本发明的保护范围。凡在本发明的精神和原则之内所作的任何修改、等同替换、改进等,均包含在本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种四氧化三钴及其制备方法与流程
- 一种钒铁钴‑硅钙合金负载纳米硫化锌‑氧化钛的耐热铸铁用复合变质剂及其制备方法与制造工艺
- 一种钴碳孔状纳米复合物氧还原电催化剂及其制备方法和应用与制造工艺
- 一种以金属醇盐为前驱体制备球形钴酸锌/碳复合材料的方法与制造工艺
- 一种基于四氧化三钴阵列的负载型贵金属催化剂的制备和用途的制造方法与工艺
- 一种基于Au@Ag异质结纳米棒的玉米赤霉烯酮免疫传感器的构建的制造方法与工艺
- 四氧化三钴/石墨烯复合材料(Co3O4/N‑RGO)的制备方法及应用与制造工艺
- 一种石墨相碳化氮纳米片/四氧化三钴纳米片鱼鳞状结构复合纳米材料及制备方法和应用与制造工艺
- 一种小粒径球形碳酸钴的制备方法与制造工艺
- 一种深度除去氯化铵溶液中钴离子的方法与制造工艺