一种碳二馏分前脱丙烷前加氢催化剂的制备方法与流程
本发明涉及一种炔烃选择加氢催化剂的制备方法,特别是涉及碳二馏分前脱丙烷前加氢的高抗结焦性催化剂的制备方法。
背景技术:
乙烯是石油化学工业最重要的基础原料之一,作为合成各种聚合物的单体-乙烯,绝大部分由石油烃(如乙烷、丙烷、丁烷、石脑油和轻柴油等)蒸汽裂解制得。经这种方法得到的以乙烯为主的c2馏分中还含有0.5%~2.5%(摩尔分数)的乙炔。乙炔的存在会使乙烯的聚合过程复杂化,恶化聚合物性能。当用高压法生产聚乙烯时,由于乙炔的积累,会有爆炸的危险;另外,在生产聚乙烯时,乙炔的存在还会降低聚合催化剂活性,增加催化剂的消耗。所以必须将乙烯中的乙炔降到一定值以下,才能作为合成高聚物的单体。
根据分离流程的不同,乙烯装置分为两种工艺:碳二后加氢除炔工艺及碳二前加氢除炔工艺。在碳二前加氢流程中,加氢反应器位于脱甲烷塔之前,而后加氢则采用顺序分离流程,脱除甲烷、乙烷后再进行碳二加氢反应,加氢反应器位于脱甲烷塔之后。后加氢工艺主要以lummus公司顺序分离流程技术为代表,此种工艺流程在国内早期引进的乙烯装置中较为普遍。前加氢工艺又分为前脱乙烷前加氢和前脱丙烷前加氢两种,分别由linde公司和s&w公司开发,两种流程都是在脱甲烷塔之前通过选择加氢脱除乙炔,但是在前脱丙烷前加氢工艺中,进入加氢反应器的物料不仅有c2馏份而且还有部分c3馏份,在脱除乙炔的同时需脱除大部分丙炔和丙二烯。
碳二选择加氢除炔原理:
主反应:c2h2+h2→c2h4+174.3kj/mol(1)
ch3-c≡ch+h2→c3h6+165kj/mol(2)
h2c=c=ch2+h2→c3h6+173kj/mol(3)
副反应:c2h2+2h2→c2h6+311.0kj/mol(4)
c2h4+h2→c2h6+136.7kj/mol(5)
c3h6+h2→c3h8+136.7kj/mol(6)
nc2h2→低聚物(绿油)(7)
在这些应中,反应(1)和(2)是希望发生的主反应,既脱除了乙炔、丙炔与丙二烯,又增产了乙烯和丙烯。(3)、(4)、(5)、(6)和(7)是不希望发生的副反应,造成乙烯、丙烯的损失。副反应(7),乙炔加氢二聚反应生成碳四馏分;碳四馏分聚合生成分子量较宽的低聚物,俗称“绿油”;绿油吸附在催化剂表面,最终形成结焦。结焦物阻塞催化剂孔道,使反应物不能扩散到催化剂活性中心表面,从而导致催化剂活性下降,影响催化剂运行周期和寿命。
专利us4404124通过分步浸渍法制备了活性组分钯壳层分布的选择加氢催化剂,可应用于碳二、碳三馏分的选择加氢,以消除乙烯中的乙炔和丙烯中的丙炔、丙二烯。us5587348以氧化铝为载体,调节助催化剂银与钯作用,加入碱金属、化学键合的氟制备了性能优良的碳二加氢催化剂。该催化剂具有减少绿油生成,提高乙烯选择性,减少含氧化合物生成量的特点。us5519566公开了一种湿法还原制备银与钯催化剂的方法,通过在浸渍液中加入有机或无机还原剂,制备银与钯双组分选择加氢催化剂。
以上传统的碳二加氢催化剂均采用浸渍法制备,其活性相是pd、ag双金属。此方法存在以下缺点:(1)受载体孔结构的影响,活性组分分散不能精确控制,随机性较强。(2)受浸渍液表面张力、溶剂化效应的影响,金属活性组分前驱体以聚集体形式沉积于载体表面,不能形成均匀分布。(3)碳二加氢对催化剂选择性要求较高,传统制备方法通过加大ag的量来促进其助剂作用的发挥,由此导致氢的传递受到阻碍,齐聚反应发生的可能性增大,绿油生成量增多,影响催化剂的寿命。以上三种现象的发生容易导致金属活性组分的分散性差,反应的选择性低,绿油生成量高,进而影响到催化剂的整体性能。
cn201110086174.0通过在载体上吸附特定的高分子化合物,在载体表面一定厚度形成高分子涂裹层,以带有功能基的化合物与高分子反应,使之具有能够与活性组分络合的功能基,通过活性组分在载体表面功能基上发生络合反应,保证活性组分有序和高度分散。采用该专利方法,载体吸附特定的高分子化合物,通过氧化铝的羟基与高分子进行化学吸附,载体吸附高分子化合物的量将受到氧化铝的羟基数量的限制;经过功能化的高分子与pd的络合作用不强,有时活性组分负载量达不到要求,浸渍液中还残留部分活性组分,造成催化剂成本提高。
为了提高催化剂的抗结焦性能,降低催化剂的表面结焦程度,近年来公开了采用双峰孔载体、微乳液制法负载活性组分的碳二选择加氢催化剂及制备方法。专利zl201310114077.7公开的选择加氢催化剂,其载体主要为氧化铝,具有双峰孔分布结构,其中小孔的孔径为50nm以内,大孔的孔径在60~800nm。以催化剂的质量为100%计,催化剂含有pd0.01~0.5重量%,为壳层分布,厚度为1~500um;含有ni0.2~5重量%,抗结焦组分ni通过微乳液法控制微乳液粒径大于载体小孔的粒径,使ni主要分布于载体的大孔中。专利zl201310114079.6公开了一种加氢催化剂的制备方法,催化剂载体主要为氧化铝,并具有双峰孔分布结构。催化剂含有pd和ni双活性组分,通过制备催化剂时将抗结焦组分ni以微乳液的形式进入至载体大孔中,活性组分pd主要分布于载体表面特别是小孔中。专利zl201310114371.8公开了一种适用于前脱丙烷前加氢工艺的碳二馏分选择加氢方法。该方法采用的选择加氢催化剂,其载体为氧化铝或主要为氧化铝,并具有双峰孔分化剂抗结焦性能,但催化剂载体大孔中的单组分ni,还原温度达到500℃以上,在该温度下还原,使催化剂活性组分pd聚集,大幅降低了催化剂活性。为了补偿催化剂活性损失需增加活性组分用量,从而导致催化剂选择性下降,活性组分利用率降低。
技术实现要素:
本发明的目的在于提供一种的炔烃选择加氢催化剂的制备方法,特别是碳二馏分前脱丙烷前加氢催化剂的制备方法。
本发明提供的炔烃选择加氢催化剂的制备方法,催化剂的载体为氧化铝或主要是氧化铝,并具有双峰孔分布结构,催化剂活性组分至少含有pd、au、ni、cu,其特征在于所述的活性组分pd以溶液及微乳液两种方式负载;au采用溶液法负载,与以溶液法负载的pd主要分布在载体的小孔中;ni、cu采用微乳液浸渍法负载,与以微乳液负载的pd主要分布在载体的大孔中。
在该催化剂中,炔烃的选择加氢反应发生在pd,au组成的主活性中心,ni和cu以微乳液的形式将浸渍在载体的大孔中,反应中生成的绿油在cu与ni组成的活性中心上发生饱和加氢。
对加氢反应而言,一般在催化剂应用前首先需要对加氢催化剂进行还原,保证活性组分以金属态存在,才能使催化剂具有加氢活性。因为催化剂制备过程中,活化是一个高温焙烧过程,在该过程中,金属盐分解为金属氧化物,氧化物会形成团簇,这种团簇一般是纳米尺寸的。不同的氧化物由于化学特性的不同,需要在不同的温度下进行还原。但对纳米尺寸的金属而样,200℃左右度是一个重要临界温度,超过该温度,金属粒子会有十分显著的产生聚集。因此,降低活性组分的还原温度,对加氢催化剂而言,有十分重要的意义。
本发明解决催化剂结焦的思路是:
炔烃选择加氢反应发生在组成的主活性中心,如pd、au,反应中生产的绿油等大分子,容易进入催化剂的大孔中。在催化剂的大孔中,负载了ni/cu组分,其中ni具有饱和加氢功能,绿油组分会在ni/cu组成的活性中心发生饱和加氢反应。由于双键被加氢饱和,绿油组分不能再发生聚合反应或聚合反应速率大大降低,其链增长反应终止或延缓,不能形成巨大分子量稠环化合物,容易被物料带出反应器,因此催化剂的表面的结焦程度会大大降低,催化剂的运行寿命会大幅度延长。
本发明中控制ni/cu合金定位于催化剂大孔的方法是,ni/cu以微乳液的形式负载,微乳液的粒径大于载体小孔孔径,而小于大孔的最大孔径。镍和铜金属盐包含在微乳液中,由于空间阻力的原因,难于进入尺寸较小的载体孔道中,因此主要进入载体的大孔中。
本发明并不特别限定以微乳液方式负载ni、cu、pd过程,只要是能形成大于65nm小于550nm微乳液粒径,都能使得ni、cu、pd分布在载体的大孔中。
本发明中,溶液法负载钯的过程中,采用过饱和浸渍法进行,由于小孔的虹吸作用,含有钯的溶液,更快地进入小孔中,钯是以氯钯酸离子的形式存在,由于该离子可以与载体表面的羟基形成化学键,使钯被快速的靶定,因此溶液进入孔道的速率越快,负载的速度就越快。所以在以溶液法浸渍pd的过程中,更容易负载于小孔中。
本发明中,将cu与ni一起负载,可以降低ni的还原温度,因为要单独将nio完全还原,一般需要还原温度达到450~500℃,在该温度下会引起pd的团聚,而形成cu/ni合金后,其还原温度较纯ni的还原温度可以降低100℃以上,达到350℃,从而缓解还原过程中pd的团聚。
本发明中,更优选的方式是,微乳液负载的少量pd在ni/cu合金的表面,可以进一步降低ni的还原温度,可以达到200℃以下,最低到150℃。
本发明中,采用的载体要求具有双峰孔分布结构,本发明并不特别限定双峰孔分布的大孔和小孔分布范围,可根据反应特点,如原料、工艺条件、催化剂活性组分等加以选择,特别推荐的载体是有孔径在250~550nm的大孔,小孔的孔径为50~65nm。同样的原因,本发明也不特别限定活性组分中pd、au、ni、cu的组成。载体al2o3晶型为α、θ或其混合晶型;优选的催化剂载体中氧化铝最好在80%以上。
本发明中,催化剂活性组分并不做特别限定,可根据反应特点,如原料、工艺条件等加以选择,特别推荐的催化剂:以质量为100%计,溶液负载的pd的含量0.035~0.065%,优选含量0.037~0.045%,au与溶液负载pd的重量比为1.3~3.0,优选重量比为1.5~2.5,ni的含量为0.5~8.0%,优选含量为2.0~5.8,cu与ni的重量比为0.1~0.9,优选重量比为0.3~0.8,微乳液负载的pd含量为ni+cu含量的1/150~1/250,优选为1/180~1/230。其中,微乳液负载的ni、cu、pd主要分布在载体250~550nm的大孔中。
本发明中,该催化剂在制备过程中ni/cu负载以微乳液形态进行浸渍。pd负载以溶液法及微乳液两种方法浸渍,pd、au的溶液负载则可以过饱和浸渍方法进行。
本发明并不特别限定以微乳液方式负载ni、cu、pd过程,只要是能形成大于65nm小于550nm微乳液粒径,都能使得ni、cu、pd分布在载体的大孔中。
本发明还推荐了一种微乳液负载方式,过程包括:将前驱体盐溶于水中,加入油相、表面活性剂和助表面活性剂,充分搅拌形成微乳液,其中油相为烷烃或环烷烃,表面活性剂为离子型表面活性剂和\或非离子型表面活性剂,助表面活性剂为有机醇。
本发明中,油相、表面活性剂和助表面活性剂的种类和加入量本发明也不特别限定,可根据前驱体盐、载体的孔结构来确定油相、表面活性剂和助表面活性剂的种类和加入量。
本发明推荐的油相为饱和烷烃或环烷烃,优选c6~c8饱和烷烃或环烷烃,优选环己烷、正己烷;表面活性剂为离子型表面活性剂和\或非离子型表面活性剂,优选非离子型表面活性剂,更优选是聚乙二醇辛基苯基醚或十六烷基三甲基溴化铵;助表面活性剂为有机醇,优选c4~c6醇类,更优选正丁醇和\或正戊醇。
本发明推荐的微乳液负载方式中,推荐的水相和油相重量比为3.0~4.5,表面活性剂和油相重量比为0.15~0.5,表面活性剂和助表面活性剂的重量比为1.0~1.2,控制微乳液粒径大于65nm小于550nm;优选的条件是水相和油相重量比是3.5~4.2,表面活性剂和油相重量比是0.2~0.4,控制微乳液粒径大于65nm小于550nm。微乳液粒径大于小孔的最大孔径而小于大孔的最小孔径,更有利于活性组分的负载,制备的催化剂中活性组分尤其是ni和cu的分布更均匀。
pd的溶液负载与微乳液负载ni/cu步骤次序不做限定;pd的微乳液负载在微乳液负载ni与cu步骤之后;au的溶液负载在pd的溶液负载步骤之后。在采用两个微乳液法的两个负载过程中,微乳液的粒径可以相同,也可以不同,最好相同。
本发明还提供了一种更具体的选择加氢催化剂制备方法:
(1)将ni和cu的前驱体盐溶于水中,加入计量好的油相、表面活性剂和助表面活性剂,充分搅拌形成微乳液,控制微乳液粒径大于65nm小于550nm,将载体加入到制好的微乳液中浸渍0.5~4小时后,滤除余液,在80~120℃下干燥1~6小时,在400~600℃下焙烧2~8h。得到半成品催化剂a;
(2)将pd的前驱体盐溶于水,调ph为1.8~2.8,再将半成品催化剂a加入pd的盐溶液中,浸渍吸附0.5~4h后,100~120℃干燥1~4小时,400~550℃条件下焙烧2~6h,得到半成品催化剂b;
(3)取半成品催化剂b的饱和吸水量的80~110的去离子水,加入氯金酸使其完全溶解,将半成品催化剂b浸渍在所配制的溶液中,摇晃均匀,沉化0.5~2h后,在100~120℃干燥1~4小时,400~550℃焙烧2~6小时,得到半成品催化剂c;
(4)将pd前驱体盐溶于水中,加入计量好的油相、表面活性剂和助表面活性剂,充分搅拌形成微乳液,控制微乳液粒径大于65nm小于550nm,将半成品催化剂c加入到制好的微乳液中浸渍0.5~4小时后,滤除余液,在80~120℃下干燥1~6小时,在400~600℃下焙烧2~8h,得到催化剂。
对一个样品来说,步骤(1)和步骤(4)的条件可以相同,也可以不同,最好是相同,这样可以确保pd负载在ni/cu合金的表面。
以上4个步骤中,步骤(3)w的负载是在步骤(2)溶液法pd的负载后进行;步骤4是在步骤(1)之后。
溶液法负载pd与微乳液法ni/cu的负载可以按任意次序进行。
步骤(2)中,pd的溶液法负载可采用过饱和浸渍法。
步骤(3)中,au的负载可采用过饱和浸渍法。
上述步骤(1)中的载体为氧化铝或主要是氧化铝,al2o3晶型最好为α、θ或其混合晶型。催化剂载体中氧化铝最好在80%以上,载体中还可含有其它金属氧化物如氧化镁,氧化钛等。
上述步骤(1)中的载体可以是球形、圆柱形、三叶草形、齿球形、四叶草形等。
上述步骤(1)中的载体其大孔孔体积和小孔孔体积比不限,根据活性组分负载含量决定。
上述步骤中所述的ni、cu、au和pd的前驱体盐为可溶性盐,可以是其硝酸盐、氯化盐或者其他可溶性盐。
本发明催化剂在使用前的还原温度最好是150~200℃。
此催化剂具有以下特性:在加氢反应开始时,由于钯的加氢活性高,而且主要分布在小孔中,因而乙炔的选择性加氢反应主要发生在小孔中。随着催化剂运行时间的延长,催化剂表面生成了一部分分子量较大的副产物,这些物质由于分子尺寸较大,较多的进入大孔中,而且停留时间较长,会在镍催化剂的作用下,发生双键的加氢反应,而生成饱和烃或不含孤立双键的芳香烃,不再生成分子量更大的物质。
本发明采用该方法制备的催化剂,其初始活性与较传统催化剂,活性和选择性明显提高。
本发明催化剂,即使原料中中含较多重馏分,催化剂绿油生成量大幅增加,催化剂活性及选择性仍没有下降的趋势。
附图说明
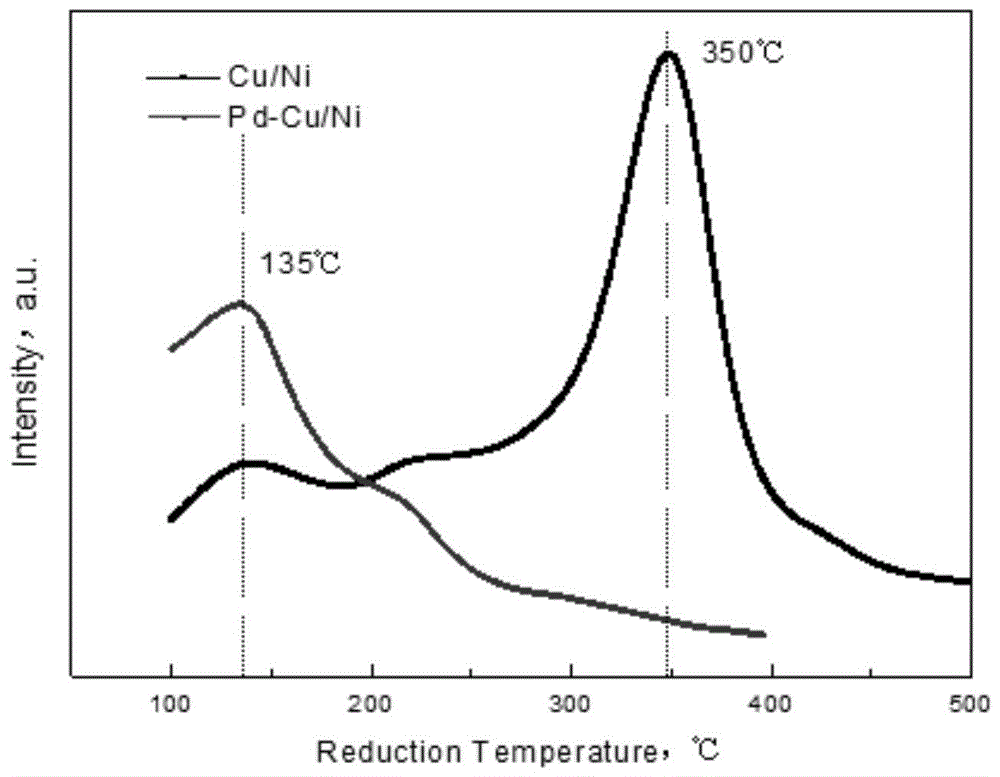
图1为实施例1中ni/cu的还原温度峰的分布图。
图2为采用前脱丙烷工艺的碳二加氢工艺流程图。
图中:1—油洗塔;2—水洗塔;3—碱洗塔;4—干燥器;5—前脱丙烷塔;6—碳二前加氢反应器;7—脱甲烷塔;8—换热器。
具体实施方式
以下对本发明的实施例作详细说明:本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和过程,但本发明的保护范围不限于下述的实施例,下列实施例中未注明具体条件的实验方法,通常按照常规条件。
分析测试方法:
比表:gb/t-5816;
孔容:gb/t-5816;
催化剂中活性组分含量:原子吸收法;
ni/cu合金的微乳液粒径分布:动态光散射粒径分析仪,在m286572动态光散射分析仪上分析;
实施例中转化率和选择性按下面公式计算:
乙炔转化率(%)=100×△乙炔/入口乙炔含量
乙烯选择性(%)=100×△乙烯/△乙炔
实施例1
载体:采用市售双峰孔分布球形氧化铝载体,直径为4mm,高温焙烧4h,称取100g。焙烧温度及载体物性指标见表1。
催化剂制备:
(1)称取一定量的硝酸镍,氯化铜溶于去离子水中,加一定量的环己烷,tritonx-100,正丁醇,充分搅拌形成微乳液,将称取的100g载体浸渍到所制备的微乳液中,浸渍1小时后,用去离子水洗涤至中性,在120℃下干燥2小时,在550℃下焙烧5h。得到半成品催化剂a。
(2)称取一定硝酸钯,溶于去离子水中,调ph为1,再将半成品催化剂a浸渍到已配制的pd盐溶液中,浸渍吸附1h后,110℃干燥2小时,380℃条件下焙烧6h,得到半成品催化剂b。
(3)称取氯金酸,用去离子水配制成得到溶液,将半成品催化剂b加入到该溶液中,摇动,待溶液全部吸收后,110℃干燥3小时,500℃条件下焙烧4h,得到所需的催化剂。
催化剂制备过程制备的微乳液的粒径及催化剂中各组分含量见表2。
使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在450℃温度,还原处理12h。
实施例2
载体:采用市售双峰孔分布球形载体,直径为4mm,其组成为氧化铝90%,氧化钛10%。经过高温焙烧焙烧4h后,称取该载体100g,载体物性指标见表1。
催化剂制备:
(1)称取一定质量硝酸镍,氯化铜溶于去离子水中,加入一定环己烷,tritonx-100,正己醇充分搅拌形成微乳液。将载体加入到制好的微乳液中浸渍1小时后,用去离子水洗涤至中性,在120℃下干燥2小时,在550℃下焙烧5h。得到半成品催化剂a。
(2)称取一定量硝酸钯溶于水中,加入一定量环己烷,tritonx-100,6.03g正己醇充分搅拌形成微乳液。将半成品催化剂a加入到制好的微乳液中浸渍4小时后,用去离子水洗涤至中性,在90℃下干燥4小时,在600℃下焙烧2h,得到半成品催化剂b。
(3)称取一定硝酸钯盐溶于水中,调ph为2,将半成品b加入pd的盐溶液中,浸渍吸附1h后,110℃干燥2小时,380℃条件下焙烧6h,得到半成品催化剂c。
(4)称取一定量氯金酸溶于去离子水中,将半成品催化剂c浸渍于配制好的溶液中,110℃干燥3小时,500℃条件下焙烧4h,得到成品催化剂。
催化剂制备过程制备的微乳液的粒径及催化剂中各组分含量见表2。
使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在300℃温度,还原处理12h。
实施例3
载体:采用市售双峰孔分布球形氧化铝载体,直径为4mm。经过高温焙烧4h后,称取该载体100g,其物性指标见表1。
催化剂制备:
(1)称取一定硝酸钯盐溶于水中,调ph为2,将载体加入pd的盐溶液中,浸渍吸附1h后,110℃干燥2小时,380℃条件下焙烧6h,得到半成品催化剂a。
(2)称取一定量氯金酸溶于去离子水中,将半成品催化剂a浸渍于配制好的溶液中,110℃干燥3小时,500℃条件下焙烧4h,得到半成品催化剂b。
(3)称取一定量硝酸镍,氯化铜溶于水中,加入一定量环己烷,tritonx-100,6.03g正己醇充分搅拌形成微乳液。将半成品催化剂b加入到制好的微乳液中浸渍4小时后,用去离子水洗涤至中性,在90℃下干燥4小时,在600℃下焙烧2h。得到半成品催化剂c。
(4)称取一定量硝酸钯溶于水中,加入一定量环己烷,tritonx-100,6.03g正己醇充分搅拌形成微乳液。将半成品催化剂c加入到制好的微乳液中浸渍4小时后,用去离子水洗涤至中性,在90℃下干燥4小时,在600℃下焙烧2h。得到成品催化剂。
催化剂制备过程制备的微乳液的粒径及催化剂中各组分含量见表2。
使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在160℃温度,还原处理12h。
实施例4
载体:采用市售双峰孔分布球形氧化铝载体,直径为4mm。经过高温焙烧4h后,称取该载体100g,其物性指标见表1。
催化剂制备:
(1)称取一定量硝酸镍,氯化铜溶于水中,加入一定量环己烷,tritonx-100,6.03g正己醇充分搅拌形成微乳液。将载体加入到制好的微乳液中浸渍4小时后,用去离子水洗涤至中性,在90℃下干燥4小时,在600℃下焙烧2h。得到半成品催化剂a。
(2)称取一定硝酸钯盐溶于水中,调ph为2,将半成品催化剂a加入pd的盐溶液中,浸渍吸附1h后,110℃干燥2小时,380℃条件下焙烧6h,得到半成品催化剂b。
(3)称取一定量氯金酸溶于去离子水中,将半成品催化剂b浸渍于配制好的溶液中,110℃干燥3小时,500℃条件下焙烧4h,得到半成品催化剂c。
(4)称取一定量硝酸钯溶于水中,加入一定量环己烷,tritonx-100,6.03g正己醇充分搅拌形成微乳液。将半成品催化剂c加入到制好的微乳液中浸渍4小时后,用去离子水洗涤至中性,在90℃下干燥4小时,在600℃下焙烧2h。得到成品催化剂。
催化剂制备过程制备的微乳液的粒径及催化剂中各组分含量见表2。
使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在170℃温度,还原处理12h。
实施例5
载体:采用市售双峰孔分布球形氧化铝载体,直径为4mm。经过高温焙烧4h后,称取该载体100g,其物性指标见表1。
催化剂制备:
(1)称取一定量硝酸镍,氯化铜溶于水中,加入一定量环己烷,tritonx-100,6.03g正己醇充分搅拌形成微乳液。将载体加入到制好的微乳液中浸渍4小时后,用去离子水洗涤至中性,在90℃下干燥4小时,在600℃下焙烧2h。得到半成品催化剂a。
(2)称取一定量硝酸钯溶于水中,加入一定量环己烷,tritonx-100,6.03g正己醇充分搅拌形成微乳液。将半成品催化剂a加入到制好的微乳液中浸渍4小时后,用去离子水洗涤至中性,在90℃下干燥4小时,在600℃下焙烧2h。得到半成品催化剂b。
(3)称取一定硝酸钯盐溶于水中,调ph为2,将半成品催化剂b加入pd的盐溶液中,浸渍吸附1h后,110℃干燥2小时,380℃条件下焙烧6h,得到半成品催化剂c。
(4)称取一定量氯金酸溶于去离子水中,半成品催化剂c浸渍于配制好的溶液中,110℃干燥3小时,500℃条件下焙烧4h,得到成品催化剂。
催化剂制备过程制备的微乳液的粒径及催化剂中各组分含量见表2。
使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在150℃温度,还原处理12h。
对比例1
载体:采用市售双峰孔分布球形氧化铝载体,直径为4mm。经过高温焙烧4h后,称取该载体100g,其物性指标见表1。
催化剂制备:
(1)称取一定量硝酸镍溶于70ml去离子水中,加入一定量环己烷,tritonx-100,正丁醇,充分搅拌形成微乳液,将载体浸渍到所制备的微乳液中,浸渍1小时后,用去离子水洗涤至中性,在120℃下干燥2小时,在550℃下焙烧5h。得到半成品催化剂a1。
(2)称取一定硝酸钯,溶于去离子水中,调ph为1,再将半成品催化剂a浸渍到已配制的pd盐溶液中,浸渍吸附1h后,110℃干燥2小时,380℃条件下焙烧6h,得到半成品催化剂b1。
(3)称取氯金酸,用去离子水配制成溶液,将半成品催化剂b1浸入到配制好的溶液中,摇动,待溶液全部吸收后,110℃干燥3小时,500℃条件下焙烧4h,得到所需的催化剂。
催化剂制备过程制备的微乳液的粒径及催化剂中各组分含量见表2。
使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在500℃温度,还原处理12h。
对比例2
载体:采用市售双峰孔分布球形载体,直径为4mm,其组成为氧化铝90%,氧化钛10%。经过高温焙烧4h后,称取该载体100g,其物性指标见表1。
催化剂制备:
(1)称取一定量硝酸镍,硝酸铜溶于去离子水中,加入一定量环己烷,14.3gtritonx-100,13.60g正己醇充分搅拌形成微乳液。将载体加入到制好的微乳液中浸渍1小时后,用去离子水洗涤至中性,在120℃下干燥2小时,在550℃下焙烧5h。得到半成品催化剂a1。
(2)称取一定量硝酸钯盐溶于水中,调ph为2,将半成品催化剂a1加入pd的盐溶液中,浸渍吸附1h后,110℃干燥2小时,380℃条件下焙烧6h,得到成品催化剂。
催化剂制备过程制备的微乳液的粒径及催化剂中各组分含量见表2。
使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在350℃温度,还原处理12h。
对比例3
载体:采用市售单峰孔分布球形氧化铝载体,直径为4mm。经过高温焙烧4h后,称取该载体100g,其物性指标见表1。
催化剂制备:
(1)称取一定氯化钯盐溶于水中,调ph为3,将已称好的载体加入pd的盐溶液中,浸渍吸附2h后,120℃干燥1小时,450℃条件下焙烧4h,得到半成品催化剂a1。
(2)称取一定氯金酸溶于去离子水中,将半成品催化剂a1浸渍于配制好的溶液中,待溶液全部吸收,100℃干燥4小时,400℃条件下焙烧6h,到所需的催化剂。
催化剂中各组分含量见表2。
使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在350℃温度,还原处理12h。
对比例4
载体:采用市售双峰孔分布球形氧化铝载体,直径为4mm。经过高温焙烧4h后,称取该载体100g,其物性指标见表1。
催化剂制备:
(1)称取一定硝酸钯盐溶于水中,调ph为2,将载体加入pd的盐溶液中,浸渍吸附1h后,110℃干燥2小时,380℃条件下焙烧6h,得到半成品催化剂a。
(2)称取一定量氯金酸溶于去离子水中,,将半成品催化剂a加入到配制好的溶液中,110℃干燥3小时,500℃条件下焙烧4h,得到半成品催化剂b。
(3)称取一定量硝酸镍,氯化铁溶于水中,加入一定量环己烷,tritonx-100,6.03g正己醇充分搅拌形成微乳液。将半成品催化剂b加入到制好的微乳液中浸渍4小时后,用去离子水洗涤至中性,在90℃下干燥4小时,在600℃下焙烧2h。得到成品催化剂。
催化剂制备过程制备的微乳液的粒径及催化剂中各组分含量见表2。
使用前放置于固定床反应装置中,用摩尔比为n2:h2=1:1的混合气体,在160℃温度,还原处理12h。
表1实施例及对比例催化剂载体物性
表2实施例及对比例催化剂活性组分含量
将上述催化剂在固定床单段反应器进行性能评价。反应条件:空速10000h-1,压力3.0mpa。反应物料组成见表3。
表3反应物料组成
催化剂评价结果见表4。催化剂1、2、3分别来自实施例1、2、3;对比1、2、3、4分别来源于对比例1、2、3、4。
表4催化剂评价结果
当然,本发明还可有其它多种实施例,在不背离本发明精神及其实质的情况下,熟悉本领域的技术人员可根据本发明作出各种相应的改变和变形,但这些相应的改变和变形都应属于本发明权利要求的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!