一种用于双电子、可逆氧气还原反应的催化剂的制作方法
本申请的主题要求2013年11月1日所提交的美国临时专利申请第61/898,532号的优先权,本申请引用上述美国临时专利申请的全部内容。
政府利益
本发明是依据美国国家科学基金会授予的根据授权/合同号EPS1004083的政府支持作出的。政府对于本申请具有某些权利。
技术领域
本发明涉及一种用于双电子、可逆氧气还原反应的催化剂。还涉及包含该催化剂的空气电极和电化学蓄电池,以及制备该催化剂的方法和使用该催化剂制备过氧化氢的方法。
背景技术:
由于与使用化石燃料相关的环境问题以及对能源安全的需要,发展替代和可再生能源令人关注。包括风能、水力发电、太阳能、生物质燃料、地热能及核能的几种不同类型的可再生能源已经被开发。然而,这些能源的一部分并不恒定和/或易运输,如风能和太阳能。因此,往往有必要或便于将能源转换成电能来运输,之后再作储存,例如热量通过蓄热器储存或化学能储存在蓄电池或电容器中。
此外,汽车产业做出诸多研究,尝试使用清洁技术代替内燃机以减少污染。可能的替代装置包括两种不同类型的电化学装置,即燃料电池(fuel cells)和蓄电池(batteries)。燃料电池可以通过与氧气发生电化学反应将环保燃料(如,氢)中的化学能转换成电能。但是,为了让此种方法具有可行性,仍需要研发和/或建立燃料生产的新方法以及燃料储存和全国运输的系统。
蓄电池和超电容器将电能作为化学能和电场能传输并储存。特别地,蓄电池将电能作为化学氧化还原能储存并在使用时将此化学能又转化成电能。化学能可以储存在蓄电池负极的活性材料中。放电时,电子通过外围电路从负极游离到正极为负荷通电,并在离子通过中介电解质(intervening electrolyte)或电解质从负极侧直接流入到正极侧的同时完成放电反应。在可再充电蓄电池(也称之为二次蓄电池)中,电子和离子在再充电期间在相反的方向上游离。考虑到所建立的输电网络系统的存在,假设可以改善这些装置的成本、安全性、能量密度和/或可再充电次数,那么在汽车产业中蓄电池(如,可再充电蓄电池)的使用对于取代内燃机来说会是一个有吸引力的选择。
由于锂空气蓄电池的高理论能量密度(即为12kWh/kg),其目前是许多科学研究的主题。放电时,锂金属阳极释放锂离子和电子。该电子经过外围电路,且锂离子从电解质游离到阴极。氧气结合电子和锂离子完成反应,并生成氧化锂或过氧化锂(即Li2O和Li2O2)。再充电时,氧化锂或过氧化锂分解生成氧气且锂离子返回到阳极并还原成金属锂。蓄电池放电循环期间,氧电极具有多孔性,从而如存储Li离子与O2反应所生成的固体产物(即Li2O和Li2O2),且通常包括促进反应的催化剂。根据电解质的类型,已经推荐了四种不同类型的锂空气蓄电池:质子惰性、水成、固态及混合水/质子惰性。尤其是与质子惰性锂空气蓄电池相关的一个问题是驱动产物返回为氧气和金属锂的过电压很大,通常大于1.5V。
因此,需要不断改善电化学系统,例如改善金属空气蓄电池,以及能够在这些系统中使用的强力双观能团催化剂(即能够降低充电过电压和放电过电压两者的催化剂),从而提高效率和再充电速率。例如,为帮助提高系统效率,需要既可促进氧气还原反应(ORR)又可促进析氧反应(OER)的可逆催化剂。
过氧化氢是一种重要的日用化学品。过氧化氢可当作漂白剂(如在造纸工业中)使用,并且可当作一种清洁和消毒剂使用。还发现过氧化氢可在化妆品工业中用作推进剂。多数过氧化氢目前是通过蒽醌氧化过程生成,其产生了大量的化学废物。此外,由于难以小规模地实施蒽醌氧化过程,所生产的过氧化氢通常需要运输和存储,这就会引起安全问题。也可通过氢氧直接或电化学合成以更小的规模生成过氧化氢。必要时,可通过允许在现场生成过氧化氢,这些方法将会降低存储和运输的需求。直接合成法将牵扯其自身安全问题(如,由于氢气和氧气混合形成的可燃性)。近期作出诸多研究以改善用于过氧化氢电化学合成的ORR催化剂,但依然需要附加且高性能的用于生成过氧化氢的ORR催化剂。
技术实现要素:
依据目前所揭露主题的一些实施例,提供一种用于双电子、可逆氧气还原反应的催化剂。在一些实施例中,所述催化剂包括:(a)金,及(b)钴配位络合物或其聚合物。在一些实施例中,所述钴配位络合物包括一种由四配位基有机螯合配体所螯合的钴离子。在一些实施例中,所述四配位基有机螯合配体为N,N'-双(亚水杨基)乙二胺或其衍生物。
在一些实施例中,所述钴配位络合物存在于与所述金接触的溶液中。在一些实施例中,所述钴配位络合物存在于覆盖金结构表面的涂层中。在一些实施例中,所述金存在于颗粒中。在一些实施例中,所述颗粒为纳米颗粒,任选地,其中所述纳米颗粒具有1nm到20nm的直径范围。
在一些实施例中,所述钴配位络合物具有式(I)的结构:
其中:每个R1、R2、R3、R4、R5、R6、R7及R8分别独立地表示选自下面组中的基团:氢、烷基、环烷基、芳烷基、芳基、杂芳基、烷氧基、芳氧基、芳烷氧基、硫代烷基、硫代芳烷基、硫代芳基、氨基烷基、氨基芳烷基、氨基芳基及导电聚合物,并且R为亚烷基或亚芳基。在一些实施例中,R1、R2、R3、R4、R5、R6、R7及R8的其中一个或多个是选自下面组中的导电部分:苯硫基、吡咯基、-NHC6H5、-SC6H5、-CH=CH-C6H5、-C≡CH及导电聚合物。在一些实施例中,R2、R3、R6及R7的其中一个或多个包括苯硫基,任选地,其中R3和R7均为苯硫基。在一些实施例中,R为亚苯基或任选地取代的乙烯。
在一些实施例中,所述催化剂包括钴配位络合物的聚合物,其中,所述聚合物由所述钴配位络合物的个别单体单元的有机螯合配体之间的键形成,任选地,其中所述聚合物通过电聚合制备。在一些实施例中,所述聚合物表现为覆盖含金结构表面的涂膜。
依据目前所公开的主题的一些实施例,提供一种包含于此处公开的催化剂的空气电极。依据目前所公开的主题的一些实施例,提供一种包含于此处公开的催化剂的电化学蓄电池,任选地,其中,所述电化学蓄电池为金属空气蓄电池或燃料电池。
依据目前所公开的主题的一些实施例,提供一种进行双电子氧气还原的方法。在一些实施例中,所述方法包括于此处公开的在存在催化剂的情况下氧气接触水,任选地,其中,所述还原为可逆或接近可逆。
依据目前所公开的主题的一些实施例,提供一种生成过氧化氢的方法。在一些实施例中,所述方法包括于此处公开的在存在催化剂的情况下氧气接触水溶液,以提供过氧化氢。
依据目前所公开的主题的一些实施例,提供一种制备双电子氧气还原的催化剂的方法。在一些实施例中,该方法包括:(a)提供一种溶液,其包括钴配位络合物,任选地,其中所述钴配位络合物包括一种由四配位基有机螯合配体所螯合的钴离子,任选地,其中所述四配位基有机螯合配体为N,N'-双(亚水杨基)乙二胺或其衍生物;(b)提供一种结构,其包括一个金表面;(c)将所述结构与步骤(a)中的溶液接触;(d)电聚合所述钴配位络合物以形成覆盖至少一部分金结构的电聚合膜。
目前所公开的主题的一个目的在于提供双电子氧气还原的催化剂(如,可逆氧气还原),以及提供电极和电化学蓄电池,其包括催化剂、生产催化剂的方法及使用催化剂析氧及/或提供过氧化氢的方法。
目前所公开的主题的一个目的已在上文阐明,且其已通过本公开的主题全部或部分地实现,随着后续说明,结合如下述的最优说明的附图,其他目的将更加清晰。
附图说明
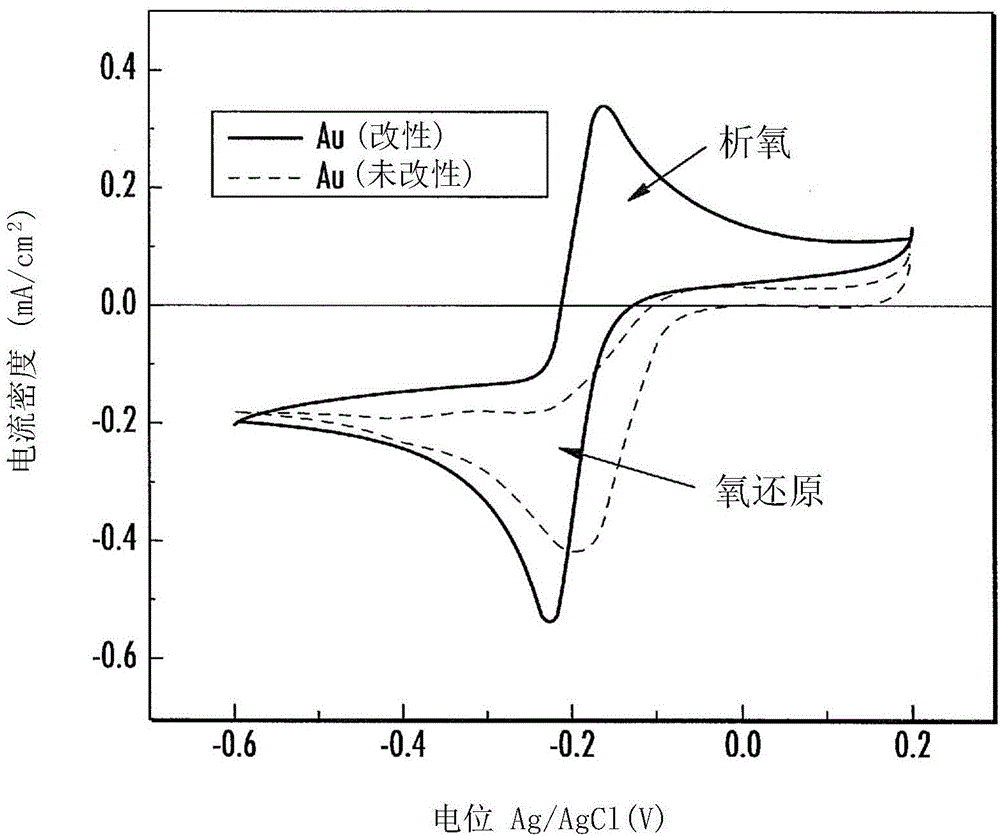
图1为展示通入氧气饱和的1摩尔(1M)氢氧化钾(KOH)电解质中常规(未改性)金(Au)电极(虚线)与钴salen改性金电极(实线)的循环伏安(CV)比较的图表。扫描速率为每秒50毫伏(mV/s)。电极表面积为0.19625平方厘米(cm2)。参比电极为氯化银(AgCI),对电极为金丝。
图2为展示用于1摩尔(1M)氢氧化钾(KOH)电解质中以及包括1摩尔KOH和2毫摩尔(mM)钴salen中旋转圆盘多晶金电极上的氧气还原铁弗的图表。扫描速率为每秒10毫伏(mV/s);电极每分钟转数(rpm)为1000;电极表面积为0.19625平方厘米(cm2);参比电极为氯化银(AgCI),对电极为金丝。
图3A为展示金(Au)表面的外亥姆霍兹层(OHL)中结合钴salen的伸展氧分子(O2)能够传入表面上的内亥姆霍兹层(IHL)的示意图。该示意图也展示了结合OHL和IHL的水分子(H2O)以及氢氧离子(OH)。
图3B为展示电子如何通过隧道现象从金表面转移到金表面上结合钴salen的氧分子(O2)的示意图。该示意图也展示了金表面的结合外亥姆霍兹层(OHL)和内亥姆霍兹层(IHL)的水分子(H2O)和氢氧离子(ˉOH)。
图4A为展示3、10、20及30周期后,通入氧气饱和的包括1摩尔(1M)氢氧化钾(KOH)的电解质中具有游离型钴salen的金电极催化活性的循环伏安(CV)比较图表。扫描速率为每秒50毫伏(mV/s)。参比电极为氯化银(AgCI),对电极为金丝。
图4B为展示1、30、50、100、150、200及250周期后,通入氧气饱和的包括1摩尔(1M)氢氧化钾(KOH)的电解质中在表面上具有电聚合钴salen的金电极催化活性的循环伏安(CV)比较图表。扫描速率为每秒50毫伏(mV/s)。参比电极为氯化银(AgCI),对电极为金丝。
图5为通入氧气饱和的1摩尔(1M)氢氧化钾(KOH)的电解质中金电极上的电聚合噻吩改性钴salen的循环伏安(CV)。扫描速率为每秒50毫伏(mV/s)。参比电极为氯化银(AgCI),对电极为金丝。
图6为展示在1摩尔(1M)氢氧化钾(KOH)的电解质中使用金/钴salen催化剂(旋转圆盘金电极上的钴salen膜(RRDE))的氧气还原反应(ORR)电流密度和两个电极ORR选择性的关系图表。参比电极为可逆氢电极(RHE)。
图7A为比较传统生产过氧化氢催化剂(即黑炭),金/钴salen催化剂及质子交换膜燃料电池催化剂(即碳上的铂(Pt))的氧气还原反应(ORR)电流密度(毫安/平方厘米(mA/cm2))的图表。使用旋转圆盘电极进行ORR:每秒10伏特(mV/s);每分钟1000转数(rpm);在1摩尔(1M)氢氧化钾(KOH)电解质中使用可逆氧电极(RHE)。
图7B为图7A所描述的催化剂的塔弗曲线的曲线图。
图8A为根据目前所公开的主题的一个实施例展示电解池生成过氧化氢(H2O2)的示意图。左手边的电池为一种电解槽,包括一个阳极(包括镍网析氧催化剂),一个阴离子交换膜及一个空气电极(阴极),其中空气电极包括一个包括目前所公开的主题的一种催化剂的气体扩散电极。过氧化氢可通过阴极去除。右手边的电池为一种燃料电池,包括一个与阳极及阴极相关的氢源,阴极包括一个包括目前所公开的主题的一种催化剂的气体扩散电极。碱性水电解质流经去除可在电解质中溶解的过氧化氢的电池。
图8B为电流效率与图8A的电解槽的电解池工作电压的图表。
图9A为根据目前所公开的主题的一个实施例展示可逆锌空气电池的示意图。所述电池包括一个置于多孔锌电极和空气电极之间的阴离子交换膜。所述空气电极与一层金/钴salen催化剂相连。含水电解质(未示出)可与催化层和阴离子交换膜接触。所述电池被配置成将过氧化氢水溶液(含水过氧化氢)输送到电池外,以进行储存并在再充电期间返回到电池中。
图9B为图9A所描述的锌空气电池的充电(圆圈)和放电(正方形)极化曲线的图表。
图10A为根据目前所公开的主题的一个实施例展示锂空气电池放电图的示意图。所述电池包括一个作为系统一部分的分离电解质结构,该系统进一步包括一个用于在电池外部分离与储存过氧化锂(Li2O2)的装置。所述电池也包括一个与流动含水电解质和空气电极以及置于含水电解质与锂阳极之间的锂离子(Li+)导电固态膜接触的催化层。
图10B为展示图10A中锂空气电池充电图的示意图。
图11为根据目前所公开的主题的另一实施例展示锂空气电池结构的示意图。图示了电池放电的阴离子流动。尽管多孔载体包含有机电解质,但锂离子(Li+)可从受保护的锂阴极流出,并也可通过固态电解质流向碱性水电解质。所述碱性水电解质与空气电极的气体扩散层(GDL)相关的催化层接触。空气电极的氧气通过催化剂分解成氢过氧化物(HO2ˉ)。所述碱性电解质可通过电池中的进出口流过电池(见粗灰色箭头)。同样图示了集电器和电流的电路。
图12为根据目前所公开的主题的另一实施例展示锂空气电池结构的示意图,其中提供了一种在受到外部碰撞事故时将电池爆炸危险控制到最低的安全规程。
图13为展示1摩尔O2饱和的KOH溶液中ORR循环伏安图(金/碳-a)催化剂(a.不存在钴salen,b.在制备墨水中存在钴salen)的图表。
图14为展示尤其是氧气还原峰的缩小的扫描范围中金电极(poly)上不同扫描速率的循环伏安图的图表。电解质:1摩尔KOH+1毫摩尔钴salen,电流为校准掉额外离子电阻后的。
具体实施方式
现在将更全面地描述目前所公开的主题。然而,目前所公开的主题可以许多不同的形式体现,并不应被解释成限于以下所阐明的实施例和所附示例中。而是,提供这些实施例以使本申请将是透彻且完整的,并且将实施例的范围更充分地传达给本领域技术人员。
本文列出的所有参考文献包括但不限于其所有专利、专利申请和公开,并且特此通过引用包含在本文内的科技期刊的整体或技术方法、技术和/或本文所用的成分,从某种程度上说,它们为此补充、解释、提供背景。
I.定义
尽管以下术语坚信为本领域普通技术人员之一所熟知,但仍提出以下定义以方便解释目前所公开的主题。
除非另外限定,这里使用的所有术语(包括技术术语和科学术语)具有依照目前所公开的主题所属的技术领域内的普通技术人员所通常理解的相同的含义。
依照长期存在的专利法惯例,术语“一”、“一个”和“该”在此申请中(包括权利要求)使用时指的是“一个或多个”。
描述两个或多个项目或条件时,术语“和/或”指的是所有命名项目或条件均存在或适用的情况,或者指的是其中只有一个(或少于全部)命名项目或条件存在或适用的情况。
尽管本公开支持指代仅替代和“和/或”的定义,但权利要求中使用的“或”习惯用于表示“和/或”,除非明确表示为指的是仅替代或替代互相排斥。
此处所使用的“又一个”可表示至少一个第二个或更多。
术语“包括”与“包括”、“包含”同义或“以...为特征”是包括的或开放式的,且不排除其他不排除其他未陈述的元素或方法步骤。“包括”是一个用于权利要求语言中的技术术语,其表示所命名元素是必不可少的,但是也可添加其他元素并仍构成权利要求范围内的构想。
此处所使用的短语“包括”排除任何未在本权利要求中规定的元素、步骤或成分。当短语“包括”出现在权利要求主体的条款中时,而不是立即紧随序言出现时,其仅限制该条款中所阐明的元素;其他原理作为一个整体并不被排除在权利要求外。
此处所使用的短语“基本包含”限制规定材料或步骤请求,加上那些并不会实质性影响所请求主题的基本和新颖特点的范围。
关于术语“包括”、“包含”及“基本包含”,其中此处只使用三个术语的其中一个,目前所公开和请求主题可包括使用其他两个术语中的任何一个。
除非另有说明,所有表示温度、pH、重量百分比数量的数字以及在本规范和权利要求中阐明使用的数字应当理解为在所有实例中被术语“大约”所修改。相应地,除非表示相反,本规范和所附权利要求中所阐明的数字参数是近似的,其因寻求目前所公开的主题所获得的所需属性而变化。
当指示一个值时,此处所使用的术语“约”意味着包括从指定量的一个示例±20%或±10%中,在另一示例±5%中,在另一示例±1%中并且在再一示例±0.1%中的变量,因为这些变量适于进行本公开的方法。
此处所使用的术语“烷基”指的是C1-20包括线性(即“直链”)、支链、饱和、或至少部分地饱和,并且在一些实例中完全不饱和(即烯基、炔基)烃链,其包括如甲基、乙烷基、丙烷基、异丙基、丁基、异丁基、邻丁基、戊烷基、己基、辛基、乙烯基、丙烯基、丁烯基、戊烯基、己烯、辛烯基、丁间二烯基、丙炔基、丁炔基、戊炔基、己炔基、庚烯基和丙二烯基。“支链”指的是烷基可连接线性烷基链,其中低级烷基比如甲基、乙烷基或丙烷基。“低级烷基”指的是具有1到8个碳原子(即C1-8烷基)的烷基,如,1、2、3、4、5、6、7或8个碳原子。“高级烷基”指的是具有10到20个碳原子的烷基,如,10、11、12、13、14、15、16、17、18、19或20个碳原子、
任选地,烷基可被一个或多个烷基取代基所取代(一个“取代的烷基”),其可以是相同的或不同的。术语“烷基取代基”包括但不限于烷基(饱和的或不饱和的)、取代烷基(如,卤代和完全卤代烷基,比如但不限于-CF3)、环烷基、卤素、硝基、羟基、羰基、羧基、酰基、烷氧基、芳氧基、芳烷氧基、硫代烷基、硫代芳烷基、硫代芳基、氨基(如,氨基烷基、氨基芳烷基、氨基芳基等)、磺酰基和亚磺酰基。
此处所用术语“芳基”指的是芳香族取代基,其可为单个芳环或多个稠合在一起共价连接或连接至常见基团的芳环,比如但不限于亚甲基或乙烯部分。该常见连接基团也可为如苯甲酮中的羰基或如二苯醚中的氧。因此,芳基的示例包括但不限于苯基、萘基、联苯和二苯醚等。芳基包括杂芳基,其中芳环或多环包括杂环原子(如,氮、氧、硫或硒)。示例性杂芳基包括但不限于呋喃基、吡啶基、嘧啶基、咪唑基、苯并咪唑基、苯并呋喃基、苯并噻吩基、喹啉基、异喹啉基和噻吩基。
该芳基可选择性地由一个或多个芳基取代基取代(取代的芳基),其可相同也可不同,其中“芳基取代基”包括烷基(饱和或不饱和的),取代的烷基(如,卤代烷基和全卤代烷基,比如但不限于-CF3)、环烷基、芳基、取代的芳基、芳烷基、卤代、硝基、羟基、酰基、羧基、烷氧基(如,甲氧基)、芳氧基、芳烷氧基、硫代烷基、硫代芳烷基、硫代芳基、氨基(如,氨基烷基、氨基芳烷基、氨基芳基等)、磺酰基和亚磺酰基。
“亚烷基”指的是具有1到20个碳原子的直链或支链二价脂肪族烃基,如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20个碳原子。该亚烷基可为直链、支链或环状。该亚烷基也可选择性地不饱和和/或由一个或多个“烷基取代基”取代。任选地,可沿着所述亚烷基插入一个或多个氧原子、硫原子或者取代或未取代的氮原子(此处也可指“烷氧基烷基”),其中氮取代基为先前所述的烷基。示例性亚烷基包括亚甲基(-CH2-)、乙烯(-CH2-CH2-)丙烯(-(CH2)3-)、亚环己基(-C6H10-)、-CH=CH-CH=CH-、-CH=CH-CH2-、-(CH2)q-N(R)-(CH2)r-,其中每个q和r独立地为从0至20的整数,例如0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20,且R为氢或低级烷基;亚甲二氧基(-O-CH2-O-);及亚乙二氧基(-O-(CH2)2-O-)。亚烷基可具有2到3个碳原子并且可进一步具有6-20个碳原子。
术语“亚芳基”指的是二价芳基,例如,二价苯基或萘基。该亚芳基可选择性地由一个或多个芳基取代基取代和/或包括一个或多个杂原子。
“芳烷基”指的是芳基-烷基-或烷基-芳基-,其中芳基和烷基如上所述,并且可包括取代的芳基、杂芳基和取代的烷基。因此,“取代的芳烷基”指的是包括一个或多个烷基或芳基取代基的芳烷基。示例性芳烷基包括苯甲基、苯乙基和萘甲基。
“环”和“环烷基”指的是具有3到10个碳原子的没有芳基的单个或多个环,如3、4、5、6、7、8、9或10个碳原子。环烷基可饱和或部分饱和。环烷基也可选择性地由此处限定的烷基取代基取代。任选地,可沿着所述环烷基链插入一个或多个氧原子。代表性的单环环烷基环包括环戊基、环己基和环庚基。多环环烷基环包括金刚烷基、八氢萘、萘烷、莰烷、降金刚烷基。
“烷氧基”和“烷氧基”指的是烷基-氧-基团,其中如上所述烷基包括取代的烷基。此处所用术语“烷氧基”指的是例如,甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基、叔丁氧基和正戊氧基。术语“烷氧基”可与“烷氧基”换用。
“芳氧基(Aryloxyl)”和“芳氧基(aryloxy)”指的是烷基-氧-基团,其中如上所述芳基包括取代的芳基。此处所用术语“芳氧基”指的是例如苯氧基和烷基、取代的烷基或烷氧基取代苯氧基。
“芳烷氧基(Aralkyloxyl)”、“芳烷氧基(aralkoxyl)”和“芳烷氧基(aralkoxy)”指的是芳烷基-氧-基团,其中芳烷基如上所述。一个示例性芳烷氧基为苄氧基。“取代的芳烷氧基”指的是芳烷氧基,其中芳烷基的烷基和或芳基部分可由一个或多个烷基或芳基取代基取代。
术语“乙烯基”指的是基团-CH=CH2,可选性地,其中烷基取代基可取代氢原子的一个或多个。因此,乙烯基指的是取代的或为取代的乙烯基。“乙烯基苯基”指的是基团-CH=CH-苯基或-CH=CH-C6H5。
此处所用的术语“卤代”、“卤化物”或“卤素”指的是弗代、氯代、苯代和碘代基团。
术语“氨基”指的是基团-N(R)2,其中每个R为独立地氢、烷基、取代的烷基、芳基、取代的芳基、芳烷基或取代的芳烷基。术语“氨基烷基”和“烷基氨基”指的是基团-N(R)2,其中每个R为氢、烷基、取代的烷基,以及其中至少一个R为烷基或取代的烷基。“芳基胺”和“氨基芳基”指的是基团-N(R)2,其中每个R为氢、烷基、取代的烷基,以及其中至少一个R为烷基或取代的烷基,例如,苯胺(即-NHC6H5)。
术语“硫代烷基”指的是基团-SR,其中每个R选自氢、烷基、取代的烷基、芳烷基、取代的芳烷基、芳基和取代的芳基。相似地,术语“硫代芳烷基”和“硫代芳基”指的是-SR基团,其中R为分别为芳烷基和芳基。
“配位化合物”是混合物,其中金属离子和双电子供给、配体或螯合基团之间存在配位键。因此,配体或螯合基团通常为双电子供给、分子或具有可向金属离子供给的未共用电子对的分子离子。
术语“配位键”指的是电子对供给和金属离子上配位点之间的相互作用,其于电子对供给和金属离子间产生一个吸引力。使用该术语并于意欲限定,在此等配位键中也可依据金属离子和电子对供给的特性分为具有或多或少的共价特性(如果不是完全共价的话)。
此处所用的术语“配体”通常指的是一类,例如分子或离子,其例如以某种方式,例如,结合与另一类相互作用。更具体地,此处所用的“配体”指的是分子或离子,其与溶液中的分子离子结合以此形成“配位化合物”。请查看Plenum中水溶液中金属络合物Martell,A.E.,and Hancock,R.P专题:纽约(1996),其以引用的方式并入于此。术语“配体”和“螯合基团”可换用。有机配体具有两个或两个以上的基团,其具有分离的未共用电子对,例如,亚烃基或亚芳基。具有未共用电子对的基团包括但并不限于杂环化合物中的-CO2H、-NO2、-B(OH)2、-SO3H、PO3H、磷酸酯和杂原子(如,氮)。
此处所用的“四配位基”指的是螯合配体,其与金属离子形成四个配位键。
此处所用的“双官能团”催化剂可催化氧气还原反应和析氧反应和/或可降低充电过电压和放电过电压。
此处所用术语“可逆”指的是化学和电化学可逆。对目前所公开的主题的化学可逆催化剂来说,氧化峰电流密度与还原电流密度相同(即峰还原电流密度与峰氧化电流密度的比值为1),并且所有被还原的氧可重新被氧化。电化学可逆性指的是在工作电极和溶液中的氧化还原物质之间发生的电子转移速率,尤其是指电子转移快速发生。电子转移速率常数(即速率常数ks大于0.020cm/s)可指示电化学可逆性。
术语“接近可逆”指的是目前所公开的主题的催化剂化学上、电化学上或两者都接近可逆。就化学可逆性而言,峰还原电流密度与峰氧化电流密度的比值介于1.001和1.11和/或90%或更多(90、95、96、97、98或99%)之间或更多被还原的氧可重新被氧化。就电化学可逆性而言,速率常数ks介于0.20cm/s和0.00005cm/s之间。而且,CV的形状可指示更高可逆的例子。参照图14,峰分离为36mV,这最有可能为两个电极氧化还原反应。其次,在两个电极氧化还原的示例过程中,分离峰值电位差36mV≈58mV/2展示这是接近可逆的例子。继续查看图14,氧化峰电流略小于还原峰电流,峰还原电流密度与峰氧化电流密度的比值为1:0.81.这也证明了这是准可逆的例子,并且阴极还原产物具有EC机理。
继续查看图14,CV中的扫面速率变化率是一个基本量但是很有用。通过改变扫描速率,本实验更改了电子传递和反应物传送速度之间的平衡。可展示可逆氧化还原过程和准可逆氧化还原过程以非均质性电子传递速度的区别。对可逆的例子来说,不管扫描速率是多少,非均质性电子传递速率足够快能够达到平衡。因此,CV的峰值电位可能不会随着扫描速率的变化而变化,其为验证氧化还原对是否电化学可逆的一个标准。从图14,我们可以看出峰值电位分离几乎没有移动,扫描速率增长了一个量级。根据这个标准,这是可逆的例子。
术语“电化学电池”指的是通过化学反应可产生电能或使用电能能促进化学反应的系统。在目前所公开的主题的一些实施例中,电化学电池可包括至少两个电极和一个电解质。示例性电话电池包括但是并不限于金属空气电池(如,金属空气蓄电池),比如锌空气电池、铝空气电池、镁空气电池、糖空气电池等,其使用空气电极(即氧气还原电极);燃料电池(如,氧氢燃料电池、直接甲醇燃料电池等),电解槽(如,生成过氧化氢的电解槽)以及电化学生物传感器(如,酶传感器、氧传感器、蛋白质传感器等)。
术语“电解质”指的是能够传导离子的溶液。一般地,电解质为液体或固体(如,凝胶、聚合物或陶瓷)。液态电解质可含水或基于有机溶剂,并且进一步包括一个或多个离子传导盐。
术语“蓄电池”指的是包括一个或多个能够将储存的化学能转换为电能的电化学电池或电池。术语“一次蓄电池”指的是一次性使用的蓄电池,其中在放电过程中电极材料不可逆更改;而术语“二次蓄电池”指的是能够多次放电和再充电的蓄电池,并且其中在再充电过程中,可储存原始的电极材料。
II.总则
金属空气蓄电池为人熟知已长达一个世纪,但是近期对新型高能量密度电池的需求也受到了关注。金属空气蓄电池将氧气用作阳极活性材料,能够将高能量密度与缩小的尺寸及减轻的重量结合起来。金属空气蓄电池的实例包括例如,锂空气蓄电池、镁空气蓄电池和锌空气蓄电池。
金属空气蓄电池可通过空气电极(其可为例如,气体扩散电极(GDE))中的氧气进行氧化还原反应及金属电极中包含的金属的氧化还原反应进行充电/放电。因此,金属空气蓄电池可包括一个空气电极,其可包括能够收集空气电极电流的空气电极集电器;一个阴极,其包含阴极活性材料(即金属或金属合金)且其可包含能够收集阴极电流的阴极电极集电器;以及一个或多个电解质,其置于空气电极和阴极之间。与其他蓄电池不同,阳极活性材料不储存在蓄电池内。然而当蓄电池暴露于外界环境中,氧气可例如通过氧扩散膜和多孔空气电极进入蓄电池。一般来说,催化剂与空气电极相连,例如,在空气电极的表面上,以催化氧气的还原。
对可再充电蓄电池来说,广泛发展到如今的空气电极种类包括传统的含水空气电极和质子惰性空气电极。传统的含水空气电极基于四个电子不可逆氧气还原反应(ORR)化学过程:
使用这种空气电极化学过程的再生燃料电池具有如下的电池放电电压和充电电压:
电池放电电压:0.9V电池再充电电压:1.43V
主要在锌空气电池和再生燃料电池系统中研究和使用传统的含水空气电极。对这种类型的可再充电空气电极来说,其难点是由于化学过程的不可逆性质造成充电电压和放电电压之间存在一个巨大差值,因而很难寻找能够降低该电压差而依旧保持稳定且不贵的双官能团催化剂。虽然已经实现了一些改善,该系统效率以合理的充电和放电速率依旧在50%到65%。
质子惰性空气电极可提供宽的电化学窗并保持电池结构简单。然而,在氧气还原过程中,生成的过氧化锂和超氧化锂(LiO2)对电解质和粘合剂材料有很强的反应性,并且伴随计划的空气电极化学过程会发生很多副反应。此外,氧化剂放电产物为绝缘的电子和锂离子。因此,高电流密度放电会引起堵塞问题且损坏多孔载体结构。因此,对这种空气电池的进一步使用将关注低功率密度和长期大规模能量存储应用。此外,在非水电解质中,已证的可逆空气电极化学过程为先前唯一的电子反应并且放电产物为过氧化物,其未释放该空气电极的全电位。
依据一些实施例,目前所公开的催化剂可催化可逆的双电子ORR:
基于这种空气电极化学过程的再生燃料电池具有如下的电池充电电压和放电电压:
电池放电电压:0.83V电池充电电压:0.83V
“可逆”意味着化学和电化学均可逆。因此,该系统电压效率可达很高的比率。进一步地,该催化剂分别在两个电极ORR和两个电极OER上具有很高的动力学性能。并且,水溶液中生成的过氧化氢可提供各种先前不可能的空气电池特性。因为在水溶液中生成过氧化氢,因此分离能量密度和功率密度是可能的。
过氧化氢(H2O2)是一种重要的日用化学品。因为过氧化氢“绿色环保”的特性,因此对其需求越来越多。然而,目前主要通过氢蒽醌氧化过程(AO)以工业规模生产过氧化氢,其不被视为绿色环保的方法。该AO过程涉及溶于有机溶剂混合液中的烷基蒽醌前驱物的连续氢化和氧化。该过程生成了大量的化学废物并且很难小规模生产。此外,大量过氧化氢的运输、存储和处理牵涉危害且比较贵。因此,过氧化氢直接当场合成法才令人满意。
自从1914年汉高和韦伯首次申请了从H2和O2直接合成H2O2的专利应用,对此研究已长达一个世纪。然而,由于安全问题(H2体积超出4-94%,H2-O2混合物易燃),该研究所取进展甚微。可通过稀释H2/O2给进流远离爆炸极限或通过使用膜型反应器分离两种气体来避免爆炸危害;但是这两种方法反应率低且不适用于商业目的。在另一方面,通过近期对燃料电池的研究,进一步研发中部具有分离器的O2和H2的反应效率。相比常规传统的直接合成法,电化学直接合成法具有很多优势。直接合成和电化学直接合成的AO过程的比较如表1所示。
表1化学直接合成和电化学直接合成的AO过程的比较。
依据其他电极(即非空气电极)中进行的哪个化学过程,电化学直接合成可具有两种生成过氧化氢的模式:燃料电池模式和电解模式。燃料电池直接合成可视作基于膜化学反应器的高级版本。不仅两种反应物通过电解质分离,而且电解质内的物质传输也比简单的膜反应器快。这是因为反应物的传输不受空气扩散通过膜以完成反应的限制。对燃料电池系统来说,部分化学反应也通过直接转化过程产生电,其比通过热处理更有效率。伴随燃料电池研究而研发的GDE也提供使用从大气中获取空气中的氧,其在常规的化学直接合成中很难实现。因此,燃料电池直接合成对于现场生成过氧化氢来说不失为一个好方法。
另一电化学过氧化氢直接合成方法涉及使用析氧反应电极,以替换燃料电池的氢电极且在电解模式中操作电池。这在使用电比使用氢更成本有效的地方更有益。燃料电池和电解直接合成得益于使用高级基于催化剂的空气电极,以生成过氧化氢。
已在电化学现场生成过氧化氢的工业和学术研究上作出诸多研究。例如,描述的过程使用由碳粉/PTFE复合材料或GDEs覆盖的一堆网状玻璃态碳制成的阴极。这些研究已在解决水溶液中给进电池的氧的低溶解度的问题上取得巨大进步。然而,因为与由于使用了低性能的催化剂的以其他方法生产的过氧化氢相比,以这些方法生产的过氧化氢的价格相对较高,因而商业化尝试失败。基于低性能催化剂的电化学系统的系统效率低。对于生产过氧化氢的燃料电池系统来说,这意味着在相同量的反应物下将生成更少的电能,因为在过电压上损失了更多的能量以驱动动力,这将增加过氧化氢的成本。对电解池来说,这意味着需要更多的能量来生成过氧化氢。而且对两种生产方法来说,为保持相同的过氧化氢生产速率需要更大的电池。这是因为低效率运行点也会引起相对较低的工作电流密度,其反过来使得生成每单位过氧化氢的硬件成本更高。
对两个电极氧气还原来说,具有更活跃性能和高分离性的催化剂在进一步研发金属空气电池和燃料电池直接合成过程用于生产过氧化氢上更有优势。然而,一般来说,已知的氧气还原反应的电催化涉及具有四电子氧气还原的相当高的动力学性能也涉及具有双电子氧气还原的相对低的动力学性能。尽管已对探索高级的催化剂作出了一些研究,但依然将碳视为最常用的催化剂。作为很多不同种类的电化学系统中已知的催化剂载体,用于双电子ORR生产过氧化氢的碳具有较低的电化学动力学性能。具有稍许改善性能的其他催化剂难以克服碳在成本上的优势。
目前所公开的主题至少部分地基于对用于双电子ORR的新型催化剂的发现,双电子ORR包括金(Au)和钴salen(或其衍生物和/或聚合物)。如实例中所述,由金和钴salen制成的催化剂与碳相比具有较高的动力和初始电位,可促进氧气还原,同时在广泛的电位范围内仍保持对双电子氧气还原超过90%的选择性。例如参见图6和7A。具有简单电化学电池结构的初始过氧化氢实验表明在相同的试验条件下,目前所公开的催化剂具有高于常规碳/PTFE复合材料两倍的电流,而未损失任何电流效率。这意味着在相同的电压和试验条件下,目前所公开的催化剂可生成近两倍的过氧化氢。此外,不仅发现双电子ORR上的碳有了很大改进,而且如图7B所示,目前所公开的催化剂的动力学性能与Pt不相上下,Pt为高性能燃料电池系统中最常用的ORR催化剂。
高性能催化剂的价值不仅为催化剂本身,而且也提供了高性能燃料电池流场设计的使用。近期燃料电池研究中,高电流密度燃料电池的流场设计近来有明显的改进。然而,如果基于低性能碳ORR催化剂建立过氧化氢生产燃料电池/电解槽,那些创新都无用。基于目前所公开的高性能双电子ORR催化剂,流场设计的现有知识可适于建立更高性能和更高效的燃料电池/电解槽来生产过氧化氢。如下进一步所述,使用如图8A所示的电解池,目前所公开的催化剂的过氧化氢生产电流效率在广泛的电位范围内超过90%。
III.双电子、可逆ORR催化剂
因此,在一些实施例中,目前所公开的主题的提供一种用于双电子氧气还原的催化剂。该催化剂可逆或接近可逆。该催化剂包括钴配位络合物和金。该钴配位络合物包括由有机螯合配体,例如,四配位基有机螯合配体或其聚合物所螯合的钴离子。
在一些实施例中,所述有机螯合配体为N,N'-双(亚水杨基)乙二胺或其衍生物。因此,在一些实施例中,钴配位络合物为N,N'-双(亚水杨基)乙二胺(II)(即钴salen,其也称为亚乙基双(水杨基亚)钴(II)):
在一些实施例中,钴配位络合物可包括N,N'-双(亚水杨基)乙二胺的衍生物。在一些实施例中,所述钴配位络合物具有式(I)的结构:
其中:
R1、R2、R3、R4、R5、R6、R7及R8各自独立地选自包括下列各项的组中:H、烷基、环烷基、芳烷基、芳基、杂芳基、烷氧基、芳氧基、芳烷氧基、硫代烷基、硫代芳烷基、硫代芳基、氨基烷基、氨基芳烷基、氨基芳基及导电聚合物;并且
R为亚烷基或亚芳基。
在一些实施例中,R1-R8中的一个为导电部分,比如为导电聚合物(即形成自导电聚合物或低聚物的单价基团)。本领域已知的多个导电聚合物包括但不限于聚乙炔、聚亚苯基次亚乙烯基、聚吡咯、聚噻吩(如,3,4-乙烯二氧噻吩(PEDOT))、聚苯胺和聚亚苯基硫化物。合适的导电部分也可选自基于这些聚合物的单体的基团,比如苯硫基(即衍生自噻吩的单体取代基)、吡咯基、-NHC6H5、-SC6H5;-CH=CH-C6H5和-C≡CH。在一些实施例中,导电部分或导电聚合物也可由配位化合物的亚烷基或亚芳基R取代。在一些实施例中,R2、R3、R6及R7的其中一个或R3和R7为/均为导电部分或聚合物。
在一些实施例中,RrRs的一个或多个为烷氧基、烷氧基取代的苯基、苯硫基和苯并噻吩基。例如,R1-R8的一个或多个可以为甲氧基。
在一些实施例中,配位化合物为噻吩改性钴salen,其中R1-8的一个或多个为苯硫基。在一些实施例中,R2、R3、R6及R7的一个或多个为苯硫基。在一些实施例中,R3和R7为苯硫基。因此,在一些实施例中,所述钴配位络合物为具有该结构的噻吩改性钴salen:
R基团亚烃基或亚芳基可任选地被取代和/或包括一个或多个杂原子(如,杂亚芳基)。在一些实施例中,R为亚烃基,其任选地具有一个或多个插入到碳链(如R可包括一个或多个O-CH2CH2-O-单元)的氧原子、杂亚芳基(如,二价的吡啶基)、亚苯基或环亚烃基(如,二价的环己基)。在一些实施例中,R为乙烯或任选地由乙烯,例如。基团-CH(R9)CH(R10)-取代,其中R9及R10可独立地选自氢、烷基、环烷基、芳烷基、芳基、杂芳基、烷氧基、芳氧基、芳烷氧基、硫代烷基、硫代芳烷基、硫代芳基、氨基烷基、氨基芳烷基、氨基芳基,或其中R9及R10共同选自亚烃基或亚芳基。因此,在一些实施例中,所述钴配位络合物具有其中一个结构:
在一些实施例中,所述钴配位络合物可为聚合物,例如包含多个单体单元的聚合物,每个聚合物基于钴配位络合物,其中在配位化合物单体单元的螯合配体中的原子(如,碳原子)间以及附加单体单元螯合配体中的原子(如,碳原子)间形成共价键(如,σ键)。在一些实施例中,所述聚合物由钴配位络合物(如,钴salen或其衍生物)的电聚合溶液提供。
可使用任意适合的包含金的结构,只要该结构的表面的至少一部分包括金。因此,所述金可为松散形式的金,比如多微孔金或任意松散金。所述金可为多晶或具有单个或主要结晶取向(如,金(100)或金(111))。所述金可为存在于(如,空气或其他电极的)表面上的金层或膜。例如,所述金可通过电镀或任意其他合适的(技术)覆盖在载体材料上,其包括另一元素或多种元素。在一些实施例中,所述载体材料可为碳或另一材料,例如,存在于空气电极和/或燃料电池的表面上。合适的碳载体材料包括但不限于人造石墨、天然石墨、无定形碳、硬碳、软碳、乙炔黑、中间相炭微球、炭黑、科琴黑(Kejen black)、中孔炭、多孔碳基、碳纳米管、碳纳米纤维、碳纸、碳布及石墨烯。
所述金可以颗粒的形式存在,其任选地包括载体材料,除所述金以外只要所述颗粒包括金表面,以及其可具有任意合适的形状或多种形状,其包括但是不限于球形、碟形、锥形、圆柱形、锥体形、棱镜形、立方体形、长方体形等。所述颗粒可为规则或不规则。在一些实施例中,所述颗粒为微-或纳米颗粒。在一些实施例中,纳米颗粒具有平均直径,其为100nm或更小,50nm或更小,或20nm或更小(如,1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20nm)。
在一些实施例中,所述金纳米颗粒具有金表面并且进一步包括一种载体材料,例如碳载体材料。所述碳载体材料可为但不限于炭黑或一般用于空气电极的其他碳材料中的一个。在一些实施例中,所述金纳米颗粒包括1wt%与30wt%之间的金(如,1、2、4、6、8、10、12、14、16、18、20、22、24、26、28和30wt%的金)。
钴化合物可存在于溶液(如,电解质溶液)中,其与所述金接触或以固体形式存在,例如,非共价固定在金表面上。在一些实施例中,所述钴化合物以薄膜的形式存在,通过烘干金表面上的钴化合物的溶液生成。在一些实施例中,所述钴配位络合物在金表面上聚合(如,电聚合)。
在一些实施例中,所述催化剂复合材料包括金与钴配合化合物的重量比为2:1到1:30之间。在一些实施例中,金与钴配位络合物的重量比为1:5到1:15之间(如,1:5、1:6、1:7、1:8、1:9、1:10、1:11、1:12、1:13、1:14及1:15)。在一些实施例中,金与钴配位络合物的重量比为1:10。
在一些实施例中,目前所公开的主题提供一种用于可逆、双电子氧气还原的制备催化剂的方法,其中所述方法包括:
(a)提供一种溶液,其包括钴配位络合物,其中所述钴配位络合物包括一种由四配位基有机螯合配体所螯合的钴离子。
(b)提供一种包括金表面的结构;以及
(c)将所述结构与来自步骤(a)的溶液接触。
在一些实施例中,所述四配位基有机螯合配体为N,N'-双(亚水杨基)乙二胺或其衍生物。
在上述方法的一些实施例中,所述方法进一步包括:(d)电聚合所述钴配位络合物以在结构表面(即,覆盖至少一部分金表面)上形成电聚合膜。任选地,遵循以下步骤(c),可烘干步骤(a)中的所述溶液以形成非聚合配位化合物的膜。
在一些实施例中,包括钴配位络合物的所述溶液可使用有机溶剂、例如,但不限于乙腈(ACN)、二甲基亚砜(DMSO)、二甲基甲酰胺(DMF)、N-甲基-2-吡咯烷酮(NMP)及二甲氧基乙烷(DME)或另一质子惰性溶剂制备。在一些实施例中,所述溶液进一步包括一种盐,例如,但不限于四丁基溴化铵(TBAB)或四丁基溴化铵四氟硼酸盐。
在一些实施例中,所述钴配位络合物可为任意具有上述式(I)或其聚合物的结构的化合物。在一些实施例中,所述钴配位络合物可选自钴salen、钴萨罗汾及噻吩改性钴salen。在一些实施例中,包括金表面的所述结构为纳米颗粒,例如,金纳米颗粒或金/碳纳米颗粒。在一些实施例中,所述结构为电极,例如空气电极,其中所述金为存在于所述空气电极表面上的层或涂层。
IV.电化学电池、电池组件和系统
在一些实施例中,目前所公开的主题提供了一种电化学电池的组件,包括一中催化剂,该催化剂包括金及钴配位络合物(如,salen钴配合物或其衍生物及/或聚合物)。例如,所述电池组件可为包括催化剂的电解质、包括催化剂的电极(如,金或空气电极)或载体,例如包括催化剂的膜或颗粒结构,可插入电解溶液或在电极表面上分层。
在一些实施例中,目前所公开的主题提供了一种包括催化剂的空气电极。任何合适的空气电极都可适用,且可包裹一个或多个含有催化剂层。空气电极可包括具有一个表面的碳材料(如,多孔碳材料)。在一些实施例中,表面可包裹一层薄惰性材料。层可为连续或非连续。在一些实施例中,惰性材料层的厚度为0.1nm到100nm之间。在一些实施例中,可在材料(如,惰性材料或空气电极的其他碳材料)表面增加一层多孔材料(如,碳纸或碳布)作为气体扩散层。
可增加一层薄金属或含金属的纳米颗粒(如金属氧化物或金属/碳纳米颗粒)作为上述任何或所有其他表面层的外敷层。例如,金属或含金属的纳米颗粒可包括目前所公开的催化剂的金成分。所述纳米颗粒,如存在,其平均颗粒大小在1到100nm之间(或在1nm到50nm之间,或1nm到20nm之间)。所述金属或金属纳米分子颗粒之后可包裹一层钴配位络合物的薄膜和/或聚合物。
所述碳材料科为任何可用于空气电极的碳材料。所述材料包括,但不限于人造石墨、天然石墨、无定形碳、硬碳、软碳、乙炔黑、中间相炭微球、炭黑、科琴黑、中孔炭、多孔碳基、碳纳米管、碳纳米纤维及石墨烯。“惰性材料”表示在含氧大气中稳定(即,所属材料在环境条件下不与氧气发生反应)。所述材料也对空气电极所接触的任何电解质稳定。适用的惰性材料包括,但不限于金属氧化物、金属卤化物、氟氧化物、金属磷酸盐、金属硫酸盐、非金属氧化物及非金属元素(如,硅)。
所述空气电极可包括多个碳材料薄层。其也可包括将碳材料聚集在一起或维持碳材料与集电器接触的粘合剂,其可作为所述空气电极的一部分。使用的粘合剂包括,但不限于聚(丙烯腈)、聚(偏二氟乙烯)、聚乙烯醇、聚乙烯、聚苯乙烯、聚氧化乙烯、聚四氟乙烯、聚酰亚胺、丁苯橡胶、羧甲基纤维素、明胶或这些材料的任意共聚物或混合物。
所述包裹碳的材可通过多种沉积方法制备,包括薄层和/或空气电极催化剂的沉积。这些方法包括,但不限于原子层沉积(ALD)、溅镀、喷射、喷雾、共沉淀、化学气相沉积(CVD)、物理气象沉积(PVD)或电子束沉积(EBD)。在一些实施例中,通过电聚合在金或含金纳米颗粒层上增加一层钴配位络合物薄膜。
在一些实施例中,所述空气电极具有空气电极集电器,用于收集空气电极的电流。在一些实施例中,所述空气电极集电器可具有多孔结构或致密结构(只要其具有合适的电子电导率)。在一些实施例中,考虑到空气(氧气)的理想扩散,可使用多孔结构。多孔结构的示例包括如网状结构(其中结构纤维整齐排列)、非织造纤维结构(其中结构纤维随机排列)和三维网状结构(具有独立孔或连通孔)。空气电极集电器的材料示例包括但不限于不锈钢、镍、铝、铁、钛、铜等金属;碳纤维、碳布和碳纸等碳材料;以及氮化钛等具有高电子电导率的陶瓷材料。在一些实施例中,所述集电器的厚度可在10到1000微米间。在一些实施例中,所述空气电极可包括在电池中,且所述电池壳作为所述空气电极集电器。
在一些实施例中,目前所公开的主题提供了一种电化学电池,包括一中催化剂,该催化剂包括金及钴配位络合物(如,salen钴配合物或其衍生物及/或聚合物)。在一些实施例中,目前所公开的主题提供了一种燃料电池或包括所述催化剂的电池(如,金属空气电池)。
在一些实施例中,目前所公开的主题提供了一种空气电池,其中包括负极(如,锂或锌电极等金属电极)、空气电极和至少一种配置在电极之间的电解质。在一些实施例中,所述空气电极可为上述空气电极。可使用任何适用的电解质或电解质组合。例如,所述电解质可为非质子(即,有机物)、含水和/或固态(如,凝胶或聚合物)。在一些实施例中,所述电池壳包括至少一种与空气电极接触的含水电介质。在一些实施例中,所述固态电解质的厚度在0.1到10μm之间,在一些实施例中,在3到5μm之间。
所述负极可包括具有释放和储存金属离子(导电离子)能力的负极活性材料。所述负极活性材料可具有收集负极电流的负极集电器。所述负极活性材料无特定限制,只要其可以释放和存储导电离子种类(如,金属离子)即可。例如,在一些实施例中,所述负极可为金属电极,其包括的材料从以下物质中选择,包括但不限于金属、金属合金、金属氧化物、金属硫化物和金属氮化物,其中包含导电离子种类的金属离子。在一些实施例中,碳材料可作为负极活性材料使用。在一些实施例中,金属或金属合金可作为负极活性材料使用。适用的金属包括但不限于锂(Li)、钠(Na)、钾(K)、镁(Mg)、钙(Ca)、铝(Al)、锌(Zn)和铁(Fe)。在一些实施例中,所述负极为锂或锌电极。尤其是,作为锂空气电池的负极活性材料,如锂金属;锂铝合金、锂锡合金、锂铅合金和锂硅合金等锂合金;氧化锂;钛酸锂等锂复合氧化物;硫化锂锡和硫化锂钛等锂硫化物;氮化锂钴、氮化锂铁和氮化锂锰等氮化锂皆可使用。
所述负极可包括至少一种负极活性材料。在一些实施例中,其也可包括用于固定负极活性材料的粘合剂。例如,当箔式金属或合金用作负极活性材料时,负极可形成只具有负极活性材料的形式。但是,当使用粉状负极活性材料时,负极可形成包括负极活性材料及粘合剂的形式。此外,所述负极可包括导电材料。所用粘合剂和导电材料的种类和量可与上述空气电极所使用的相同。
在一些实施例中,所述负极可具有负极集电器。负极集电器的材料无特定限制,只要其具有导电性即可。例如,铜、不锈钢及镍等材料皆可使用。箔片、平板和网形等都可作为负极集电器的形状。在一些实施例中,电池的外壳硬件可作为负极集电器。
包括载体电解质盐和有机溶剂的非水相(即有机)电解溶液可作为电解溶液使用。所述有机溶剂无特定限制。例如,所述有机电解质可包括,但不限于碳酸丙烯酯(PC)、碳酸亚乙酯(EC)、碳酸亚乙烯酯、碳酸二甲酯(DMC)、碳酸甲乙酯(EMC)、碳酸二乙酯(DEC)、甲基丙基碳酸酯、异丙基甲基碳酸酯、丙酸乙酯、丙酸甲酯、γ-丁内酯、乙酸乙酯、乙酸甲酯、四氢呋喃、2-甲基四氢呋喃、乙二醇二甲醚、乙二醇二乙醚、ACN、DMSO、二乙氧基乙烷;DME以及四乙二醇二甲醚(TEGDME)。
此外,离子液体可用于或用作有机电解溶液。例如,适用的离子液体包括但不限于N,N,N-三甲基-N-丙基铵双(三氟甲磺酰)胺(TMPA-TFSA]、N-甲基-N-丙基吡咯烷双(三氟甲烷-磺酰)胺(PP13-TFSA)、N-甲基-N-丙基吡咯烷双(三氟甲烷磺酰)胺(P13-TFSA)、N-甲基-N-丁基吡咯烷双(三氟甲烷磺酰)胺(P14-TFSA)和N,N-二乙基-N-甲基-N-(2-甲氧乙基)铵双(三氟甲烷磺酰)胺(DEME-TFSA)等脂类季铵盐;以及1-甲基-3-乙基咪唑四氟硼酸盐(EMIBF4)、1-甲基-3-甲基咪唑双(三氟甲烷磺酰)胺(EMITFSA)、1-烯丙基-3-乙基咪唑溴化物(AEImBr)、1-烯丙基-3-乙基咪唑四氟硼酸盐(AEImBF4)、1-烯丙基-3-乙基咪唑双(三氟甲烷磺酰)胺(AEImTFSA)、1,3-联丙烯咪唑溴化物(AAlmBr)、1,3-联丙烯咪唑四氟硼酸盐(AAImBF4)和1,3-联丙烯咪唑双(三氟甲烷磺酰)胺(AAlmTFSA)等烷基咪唑季铵盐。在一些实施例中,所述有机电解质包括AcN、DMSO、DME、PP13-TFSA、P13-TFSA、P14-TFSA、TMPA-TFSA和/或DEME-TFSA。
任何在电解质溶剂中具有溶解度,且拥有理想金属离子导电性的载体电解质盐均可接受。在一些实施例中,可使用含具有欲使之导电的金属离子的金属盐。例如,在锂空气电池中,锂盐可作为载体电解质盐食用。可使用LiPF6、LiBF4、LiCIO4、LiAsF6、LiOH、LiCl、LiNO3和Li2SO4等无机锂盐。此外,也可使用CH 3CO2Li、锂二乙二酸硼酸盐(LiBOB)、LiN(CF3SO2)2(即LiTFSA)、LiN(C 2F 5SO2)(即LiBETA)和LiN(CF3SO2)(C4F9SO2)等有机锂盐。在非水电解溶液中,由于无特定限制,载体电解质盐的浓度可进行设定,如,在范围0.5M到3M中。
在一些实施例中,所述电解质为含水电解质,其中含有水分。所述含水电解质也可包括载体电解质盐。在一些实施例中,所述含水电解质为碱性。在一些实施例中,所述含水电解质的pH为9,或为12,或更高(如,在9到14之间或在12到14之间)。在一些实施例中,pH为13。所述含水电解质的pH可进行调节,如,通过加入适当的金属氢氧化物(如KOH、NaOH、LiOH等)。所述含水电解质也可包括其他在水中具有溶解度,且具有理想离子导电性的载体盐。可使用含具有欲使之导电的金属离子的金属盐。例如,对于锂空气电池,可使用LiCl、LiNO3、Li2SO4和CH3COOLi等锂盐。
所述电解溶液可以浸渍在分隔器和多孔结构中的状态纳入电池,该分隔器具有绝缘性,可确保空气电极和负极之间的绝缘性,该多孔结构可保留电解溶液。聚烯烃(包括聚乙烯和聚丙烯)等绝缘树脂和玻璃可作为分隔器的材料。此外,所述分隔器的多孔结构可为网状结构(其中结构纤维整齐排列)、非织造纤维结构(其中结构纤维随机排列)和三维网状结构(具有独立孔或耦合孔)。所述分隔器的厚度可为10到500微米。
电解质凝胶可通过凝胶化上述电解溶液得到。例如,作为凝胶化非水相电解溶液的方法,聚氧化乙烯(PEO)、聚丙烯腈(PAN)、聚偏二氟乙烯(PVDF)或聚甲基丙烯酸甲酯(PMMA)等聚合物可加入非水相电解溶液。可形成电解质凝胶,例如,在所述聚合物与上述电解溶液混合后,通过在基础材料上浇注并干燥进行混合物涂层,干燥的混合物从基础材料上脱离,并按照需要将其切割成片。
可按照导电金属离子适当选择固体电解质,无特定限制。例如,对于锂空气电池,表示为LiaXbYcPdOe(其中X表示从B、Al、Ga、In、C、Si、Ge、Sn、Sb和Se等元素中选择的至少一种;Y表示从Ti、Zr、Ge、In、Ga、Sn和Al等元素中选择的至少一种;a到e满足关系:0.5<a<5.0,0≤b<2.98,0.5≤c<3.0,0.02<d.≤3.0,2.0<b+d<4.0及3.0<e≤12.0)的LISICON(即,锂超离子导体)氧化物;LixLa1-xTiO3等钙钛矿氧化物;Li4XO4-Li3YO4等LISICON氧化物(其中X表示从Si、Ge和Ti等元素中选择的至少一种,Y表示从P、As和V等元素中选择的至少一种)以及Li3DO3-Li3YO4(其中D表示B,Y表示从P、As和V等元素中选择的至少一种);以及Li7La3Zr2O12等Li-La-Zr-O基石榴石氧化物等均可使用。所述固体电解质可进行模铸,如轧制或与溶剂混合制备悬浮液,涂布及干燥。
目前所公开的电池也可具有除上述空气电极、电解质和负极外的其他组成构件。通常,所述电池壳具有容纳空气电极、负极和电解质的功能。所述电池壳的形状无特定限制。例如,硬币形、平板形、圆柱形和叠层形都可使用。所述电池壳可为开放透气型或密封不透气型,只要其可以向空气电极提供氧气。所述开放透气型电池壳的结构使至少一个空气电极能够充分与空气接触。例如,所述电池壳可包括与空气电极相连的进氧孔。所述进氧孔可包括一个能够选择性输送氧气的氧气输送膜。在一些实施例中,可使用聚硅氧烷基膜。另一方面,在密封不透气型电池壳中,具有一根氧气(空气)输入管和一根输出管。
在一些实施例中,目前所公开的主题提供了一种作为示例性电化学电池的锂空气电池,包括锂电极、多孔载体(包括有机电解质);固态电解质、含水电解质、含催化剂的层和空气电极。该电池的一种可能配置如图11所示。如图11所示,所述电池可包括受保护的锂电极,其与多孔载体材料接触。所述多孔载体材料包括设置在阳极和固态电解质之间的有机电解质。所述有机电解质可包括DMSO和LiClO4(作为示例);但也包括上述上述另一有机电解质和/或盐。在一些实施例中,所述固态电解质为一种LISICON材料。
在一些实施例中,所述多孔载体的厚度为200微米。在一些实施例中,所述固态电解质的厚度为3微米到5微米之间。但是,所述载体材料和固态电解质可具有任何适当及预期的厚度。在一些实施例中,所述载体材料和固态电解质可在维持机械稳定性的前提下尽可能薄。
再次参考图11,所述固态电解质反过来配置在多孔载体材料和含水电解质(如,碱性含水电解质)的中间。因此,锂电极中的锂可被还原为锂阴离子,并通过多孔载体和固态电解质传递至含水电解质,同时锂电极在含水电解质中受到防水保护。
所述含水电解质也和与空气电极的气体扩散层(GDL)相接触的催化层接触。所述GDL为多孔结构(如,碳布或纸),可将氧气(O2)从空气电极传递至催化层,在其中还原为氢氧化物。在一些实施例中,所述含水电解质的pH等于或高于13。在一些实施例中,所述含水电解质可进一步包括硅酸钠或等过氧化物稳定剂,或EDTA或DTPA等过渡金属离子络合剂或螯合剂,或另一种氨基多羧酸,或一种磷酸盐。如图11所示,所述锂空气电池具有流动电解液的特征,该空气电池系统包括针对含水电解质的一个入口和一个出口,因此其可以流过电池,将电池放电期间形成的过氧化锂转移,如,其可存储在固相中。因此,在一些实施例中,所述电池与一个或多个储存和/或分离装置,容器或槽(在其中可将过氧化锂与含水电解质溶液分开和/或储存)连接(即,通过该入口和出口)。所述电池也可包括与锂阳极及空气电极相连的集电器和承载电流的电路。在一些实施例中,所述一个或多个储存和/或分离装置配置为存储排放产物,例如但不限于空气电极排放产物(作为浓缩液或固体)。在一些实施例中,所述存储装置或槽用于在电池放电时保护空气电极排放产物。为更好地进行保护,可进行分离过程,将排放产物与主体电解质相分离,从而保持排放产物为固相或浓缩液相。
图10A和10B进一步说明了目前所公开的主题的一实施例,其中包括流动电解液锂空气电池。
图10A展示了放电期间的锂空气电池和流动含水电解质。所述电池包括锂阳极、锂离子导电固态膜、含水电解质、催化层和空气电极。放电期间,在含水电解质中形成过氧化锂(Li2O2)。在所述电池的出口处,电解质可从电池流向分离槽,其中,可对电解质进行冷却,使过氧化锂发生沉淀。剩余含水电解质可通过与浓缩LiOH接触的出口离开分离槽,使碱性电解质通过电池入口循环返回电池。由于在分离/储存装置中分离不完全,所述电解质中可能仍包括一些Li2O2。图10B展示了充电期间的相同锂空气电池。在一些实施例中,所述一个或多个储存和/或分离装置配置为存储排放产物,例如但不限于空气电极排放产物(作为浓缩液或固体)。在一些实施例中,所述存储装置或槽用于在电池放电时保护空气电极排放产物。为更好地进行保护,可进行分离过程,将排放产物与主体电解质相分离,从而保持排放产物为固相或浓缩液相。
所述电池的能量密度可通过将储存的能量除以锂金属的质量以及在分离/储存装置中储存过氧化锂所需的水量计算。假设每个锂离子需要1当量的H2O进行溶解,且沉淀出一摩尔可储存的Li2O2,则能量密度可为3277瓦时(Wh)/kg。如果需要2当量的H2O,则能量密度可为1903Wh/kg。如果需要3或4当量的H2O,则能量密度可分别为1341或1035Wh/kg。
现在参考图12,其提供了一种在受到外部碰撞时,最大限度上控制电池爆炸事故的安全规程。当受到外部碰撞时,薄固态Li+传导电解质可能破裂。如果锂金属与含水电解质接触,会产生大量氢气,由此导致潜在的爆炸风险。以上所示特别安全规程可最大程度上降低该事故风险。当遭遇外部意外时,电解质入口开始将惰性气体,而非通常的含水电解质送入电池中。同时,电池的出口仍然将含水电解质排出电池,从而在最大程度上减少可与锂金属接触的水量。输入的氧气也从阴极侧切断,剩余的氧气与氮气一起从电池排出。整个程序可降低含水锂空气电池的潜在爆炸风险。
因此,继续参考图12,依据一些实施例,在事故发生时,电池经配置可提供如下一项或多项:(i)电解质入口将惰性气体,而非含水电解质输入电池;(ii)电池的电解质出口将含水电解质排出电池,在最大程度上减小可与金属(任选地为锂金属)接触的水量;以及(iii)从阴极侧切断输入的氧气,剩余的氧气与氮气一起从电池排出。任选地,(i)、(ii)和/或(iii)可在同时发生。.
在一些实施例中,目前所公开的主题提供了一种锌空气电池。在一些实施例中,所述锌空气电池可包括上述空气电极、锌电极和一种或多种电解质。在一些实施例中,所述锌空气电池包括锌电极(如,多孔锌电极)、阴离子交换膜和与含催化剂的层相连的空气电极。例如,如图9A所示,阴离子交换膜可与锌电极的表面及含催化剂的层接触,所含催化剂的层可与阴离子交换膜、空气电极和/或与空气电极相连的气体扩散层接触。氧气可从空气电极向催化层输送,并还原为过氧化物。所述电池系统可进一步包括将过氧化氢水溶液输出电池,以便分离和/或储存过氧化氢(如,从空气电极的出口)。因此,在一些实施例中,所述空气电极与含有过氧化氢水溶液的储存槽流体连通。其他排放产物,即由氧化锌和碱形成的锌酸盐可通过在锌电极表面上使用膜和/或更换新电极活性材料形成能够保存的固体络合物。
在一些实施例中,所述过氧化氢水溶液包括过氧化物稳定剂。在一些实施例中,所述过氧化物稳定剂可为硅酸钠或过渡金属离子络合剂或螯合剂。在一些实施例中,所述过氧化物稳定剂为乙二胺四乙酸(EDTA)、二乙烯三胺五乙酸(DTPA)或一种磷酸盐。在一些实施例中,至少一个出口或至少一个入口连接到一个或多个储存和/或分离槽。在一些实施例中,所述一个或多个储存和/或分离装置配置为存储排放产物,例如但不限于空气电极排放产物(作为浓缩液或固体)。在一些实施例中,所述存储装置或槽用于在电池放电时保护空气电极排放产物。为更好地进行保护,可进行分离过程,将排放产物与主体电解质相分离,从而保持排放产物为固相或浓缩液相。在一些实施例中,将过氧化氢从电池输出,以便进行储存、纯化和/或稳定。
在一些实施例中,目前所公开的主题提供了一种可用于燃料电池或电解槽模式的电化学电池,并包括目前所公开的催化剂。一种模式(即燃料电池)输出氢气,另一种输入电。所述催化剂可为上述空气电极的一部分。在一些实施例中,目前所公开的主题可提供包括目前所公开的催化剂的一种燃料电池(即,作为气体扩散电极的一部分,在一些实施例中,可具有通过电聚合式(I)的钴配位络合物制备的改进的Au网或Au纳米颗粒催化剂),并可进一步包括在产生过氧化氢时有用的氢源。参见右手侧的图8A。碱性水电解质可流过电池。溶解在电解质中的过氧化物可从电池带出,如粗箭头所示。在阳极和阴极处发生的反应如下:阳极:
H2+2OHˉ→2eˉ+2H2O
阴极:O2+2H2O+2eˉ→2OHˉ+H2O 2
左手侧的图8A展示了含有目前所公开的催化剂的电解槽(即作为气体扩散电极的一部分,在一些实施例中,可具有通过电聚合式(I)的钴配位络合物制备的改进的Au网催化剂)。其进一步使用镍网作为析氧催化剂(即,作为阳极的一部分)及阳极和阴极之间的阴离子交换膜。可通过阴极的出口将过氧化氢除去(如,以水溶液形式)。在阳极和阴极处发生的反应如下:
阳极:4OHˉ→4eˉ+2H2O+O2
阴极:O2+4H2O+2eˉ→2OHˉ+H2O2
V.双电子、可逆氧气还原的进行方法
在一些实施例中,目前所公开的主题提供了一种进行双电子氧气还原的方法,其中该方法包括在存在含金和钴配位络合物的催化剂的前提下,将氧气与水接触。在一些实施例中,所述还原为可逆或接近可逆(化学上和电化学上)。在一些实施例中,所述钴配位络合物包括一种由四配位基有机螯合配体所螯合的钴。在一些实施例中,所述有机螯合配体为N,N'-双(亚水杨基)乙二胺或其衍生物。在一些实施例中,所述配位化合物具有上述式(I)或其聚合物的结构。在一些实施例中,所述钴配位络合物为钴salen,噻吩改性钴salen或双水杨醛邻苯二胺合钴。
在一些实施例中,金与钴配位络合物的重量比为任意合适比例,可为但不限于1:5、1:6、1:7、1:8、1:9和1:10。在一些实施例中,所述钴配位络合物存在于与所述金接触的水溶液中。在一些实施例中,所述钴配位络合物存在于覆盖金结构表面的涂层中,例如但不限于大颗粒金、金膜或含金纳米颗粒。在一些实施例中,所述钴配位络合物在金表面上聚合(如,电聚合)。在一些实施例中,所述催化剂存在于电化学电池中,如,作为空气电极的一部分。
在一些实施例中,氧气与催化剂的接触在氧饱和的碱性水溶液中进行。所述水溶液也可包括一种或多种电解质载体盐。所述碱性溶液的pH至少可为9,或在9到14之间。在一些实施例中,pH在12到14之间(如,12、12.5、13、13.5或14)。在一些实施例中,pH为13。在一些实施例中,所述碱性溶液中包括KOH、NaOH或LiOH等金属氢氧化物。在一些实施例中,所述金属氢氧化物浓度为0.1M或更高。在一些实施例中,所述金属性氧化物浓度为1.0M或3.0M。因此,在一些实施例中,所述接触在氧饱和的碱性环境中进行。
在一些实施例中,所述接触还可在硅酸钠或过渡金属离子螯合剂等过氧化物稳定剂存在的情况下进行。在一些实施例中,所述过氧化物稳定剂为乙二胺四乙酸(EDTA)、二乙烯三胺五乙酸(DTPA)等氨基多羧酸或一种磷酸盐。
在一些实施例中,所述接触在-20℃到80℃的温度下进行。在一些实施例中,所属接触在20℃到80℃的温度下进行。在一些实施例中,所属接触在20℃到50℃的温度下进行。(如,20、25、30、35、40、45或50℃)。在一些是示例中,所属接触在20℃到35℃的温度下进行。
在一些实施例中,还原产生过氧化氢。因此,在一些实施例中,目前所公开的主题提供了一种在存在含金和钴配位络合物的催化剂以提供过氧化氢的前提下,将氧气与水溶液(如,碱性水溶液)接触的方法。在一些实施例中,所述催化剂包括金和式(I)的钴配位络合物或其聚合物。在一些实施例中,所述钴配位络合物为钴salen,噻吩改性钴salen或双水杨醛邻苯二胺合钴。
在一些实施例中,所述催化剂存在于含有氧原的燃料电池中。在一些实施例中,所述催化剂存在于进一步包括锌电极、空气电极和阴离子交换膜的电化学电池中,其中所述阴离子交换膜存在于锌电极和空气电极之间,所述催化剂与所述空气电极和/或所述空气电极的气体扩散层接触,任选地,其中锌电极为多孔。
在一些实施例中,所述催化剂存在于进一步包括氢电极或析氧电极的电化学电池中。在一些实施例中,在所述两电极之间采用碱性电解质,任选地,其可为阴离子交换膜或含水碱性电解质。在一些实施例中,可储存、纯化和/或稳定化过氧化氢。
示例
以下所包括的示例,用于为本领域内普通技术人员实施目前所公开的主题的典型实例提供指导。鉴于本发明及本领域技术的一般水平,本领域技术人员应理解以下示例的目的仅为提供示范,在不偏离目前所公开的主题范围的前提下,可对其作出许多更改、修改或变更。
示例1
材料和方法
电极制备:
在Pine Research Instrumentation(美国北卡罗莱纳州达勒姆市)购买了三种5mm直径圆盘电极,用于下述研究:多晶金(Pine Research Instrumentation零件编号AFE2A050AU,在室温下使用;零件编号AFE5TO50AUHT,在高温下使用);玻璃碳(零件编号AFE2A050GC),在室温下使用;以及多晶铂(零件编号AFE5TO50PTHT),在高温下使用。所述多晶金属电极通过使用5μm的氧化铝粉抛光,再用0.05μm的粉末抛光进行机械清洁(比勒公司,德国杜塞尔多夫市)。然后,经过机械清洁的金属电极通过循环伏安测量法进行电化学清洁,其中扫描范围为2.2V,扫描速率为50mV/s,在0.1M HClO4酸性电解质中进行,以清除共价键合化合物。所述电极也在正常电化学窗口中进行多个扫描循环,直至标准循环伏安特性稳定。所述玻璃碳圆盘进行机械清洁,但不进行电化学清洁。
此外,使用了两个RRDE电极(同样购自Pine Research Instrumentation,美国北卡罗来纳州达勒姆市):具有5mm直径金插入圆盘(零件编号AFED050P040AU)和铂环(零件编号AFE6R1PT;6.5mm内径;7.5外径;收集效率25%)的金圆盘RRDE;以及具有直径为5.61mm的玻璃碳圆盘以及内径6.25、外径7.92mm、收集效率37%的环的玻璃碳圆盘RRDE(零件编号AFE7R9GCPT)。两个RRDE电极都按照所述圆盘电极的清洁方法进行清洁,圆盘电极和环形电极分开进行电化学清洁程序。
改性电极制备:
制备三种改性电极:钴salen改性多晶金电极、Pt/C催化剂改性玻璃碳电极;以及Au/C催化剂改性玻璃碳电极。尤其是,对于一些研究,在含钴salen的电极上进行实验。但是,采用薄膜改性方法制造拟稳钴salen改性电极。按照本方法制备清洁的多晶金圆盘电极(多晶金RDE或RRDE),然后将15μl的2mM钴salen溶液沉积在所述金电极的表面上。所述电极干燥30分钟后,在其表面上形成一层薄膜。
所述Pt/C催化剂和Au/C改性玻璃碳电极通过在玻璃碳电极表面上沉积12μl油墨制备。
所述油墨使用5%NAFIONTM溶液(Sigma Aldrich,美国密苏里州圣路易斯市),在离聚物与催化剂比例为30/70的甲醇中制备。所述30%Pt/C催化剂(BASF,美国新泽西州弗洛勒姆帕克)具有直径为2nm的纳米Pt颗粒。所述Au/C催化剂通过两种不同的方法制备,即Turkevich方法和磁控溅射,使用两种不同的纳米颗粒大小,分别称为Au/C-a和Au/C-b。
按照第一种方法,将氯金酸的水溶液(H[AuCI4],400ml H2O中含88.4mg)加热至70℃。然后将柠檬酸三钠溶液(4ml H2O中含212mg)加入,以开始还原氯金酸,将混合物在70℃下搅拌3小时。溶液冷却后,加入炭黑(XC-72,Cabot Corporation,美国马萨诸塞州波士顿)的乙醇分散液,将混合物搅拌20分钟,然后进行离心。用乙醇和水清洗固体,然后真空干燥,以便得到完美的粉状催化剂。由起始材料计算得到的金-碳比例为30%(重量比)。使用谢乐公式和X射线衍射(XRD)数据得到的纳米颗粒大小为18nm。
此外,所述Au/C催化剂可通过磁控溅射制备,即先将炭黑和两根包裹的搅拌棒放入一套连接到真空电机的不锈钢杯中。在14W的功率下,溅射高纯度金靶材(99.99%)160分钟,即在氩(Ar)气中对持续旋转的炭黑进行直流磁控溅射。使用电感耦合等离子体(ICP)光电直读光谱仪测定的金负载量为4.77%,平均颗粒大小(通过扫描透射电子显微镜(STEM)数据测定)为1.8nm。
电化学测量:
电化学测量在与周围环境密封隔绝的标准三室电化学电池中进行。针对RDE研究,使用旋转圆盘圆环电极装置(Pine Research Instrumentation,美国北卡罗莱纳州达勒姆市)。使用恒电位仪(SP-200,Bio-Logic SAS,法国克莱)对三电极电池系统进行测量,使用多通道恒电位仪(VMP3,Bio-Logic SAS,法国克莱)对RRDE四电极系统进行测量。金线对电极通过多孔玻璃熔块与工作电极分开。Hg/HgO参比电极或双功能饱和Ag/AgCI参比电极用于在碱性电解质中进行测量,饱和Hg/HgSO4参比电极用于在酸性电解质中进行测量。在一些实例中,通过在电池上喷射H2,并在Pt工作电极处测量开路电位将所述参比电极校准为可逆氢电极(RHE)。对于RDE实验,与用于覆盖电极入口和紧密包围旋转电极轴的轴承插接。O2和Ar环境通过由在电池气体出口的水或油起泡器产生的微正压维持。
对于碱性电解质电化学测量,碱性溶液由氢氧化钾、氢氧化锂(均购自Sigma Aldrich,美国密苏里州圣路易斯市)和超纯水制成。具有水套的玻璃电化学电池(零件编号RRPG085,Pine Research,美国北卡罗莱纳州达勒姆市)用于温控实验,热水浴的环形器用于控制温度。EDTA(Sigma-Aldrich,美国密苏里州圣路易斯市)作为过氧化物稳定剂使用。对于有机电极电化学测量,所述玻璃电化学电池在烘箱中干燥以除去水分,有机电解质使用乙腈(牌,美国宾夕法尼亚州森特瓦利)和四丁基溴化铵(TBAB,Sigma-Aldrich,美国密苏里州圣路易斯市)制备。钴salen(Sigma-Aldrich,美国密苏里州圣路易斯市)用于含水和有机电解质中。
在每次电化学测量前,电位在开路电压(OCV)下保持30分钟。对于循环伏安(CV)研究,在OCV电位保持后,电位从ODV电位扫描到较低的电位,然后再返回。
通过绘制log(电流密度)与电位在相关RDE研究动力学区域的曲线得到铁弗曲线。HCD(高电流密度)范围铁弗斜率通过从0.1mA/cm2到1mA/cm2的范围中选择数据点计算。LCD(低电流密度)范围铁弗斜率通过从0.01mA/cm2到0.1mA/cm2的范围中选择数据点计算。
示例2
钴Salen改性金电极上的氧气还原
将多晶Au电极按示例1中所述进行机械和电化学清洁。然后在纯碱性电解质溶液(1M氧饱和的KOH)中对其进行测试。将10μl的1M KOH+2mM钴salen溶液在电极表面上沉积,然后干燥30分钟,形成薄膜。将所述薄膜改性电极放回碱性电解质溶液中。图1展示了以钴salen薄膜进行改性前后,电极ORR CV的对比。
图1展示了CV表明改性Au电极(实线CV)进行了双电子的可逆氧气还原反应。在实线CV(循环伏安法)中,氧气还原发生在阴极扫描中,析氧发生在阳极扫描中。对于未改性的Au电极(虚线),在阴极扫描中有氧气还原,但在阳极扫描中无析氧。
图2展示了RDE实验数据的铁弗曲线,以便描绘改性和未改性Au电极在碱性氧饱和电解质中电进行化学反应的动力学。图2中的曲线显示改性电极相关的斜率值较低(HCD范围为56mV/十进位(dec),在LCD范围为36mV/dec),表明改性电极的氧气还原较快。
之前已经有理论表明,在裸金表面上ORR的速度限制步骤是首个电子传递(参见Zurilla,R.W.,Electrochim.Acta(1978),125,1103;and Erickson,H.,etal..Electrochim.Acta(2009),54,7483-7489),其可与氧在金表面上的化学吸附有关:O2+eˉ→O2ˉ(ads).因此,不受任何理论限制,钴salen薄膜改性Au电极的速度提升被认为与增强了在Au表面上的氧化学吸附(与氧和钴salen的可逆键合相关)有关。进一步认为,钴salen与金表面通过物理吸附(即,通过范德华力吸附)发生作用,而非紧密相连,如化学吸附在Au上,同样不受任何理论限制,其表明钴salen位于金表面的外亥姆霍兹层(OHL)中。当氧分子与钴salen键合时,与常规氧相比,其具有较低的电子密度和更好的电子受体。根据计算,同钴salen化合物键合的氧分子长度范围为1.28到1.29埃,与氧和Au化学吸附的分子长度相同。因此,O2与钴salen可进行预交互作用伸展分子,使其更容易化学吸附到Au上。参见图3A。或者,钴可成为反应中心,并通过在Au表面上进行隧道电子转移结束首个电子反应。参见图3B。
示例3
pH和温度的作用
温度:
金电极在氧饱和的含钴salen碱性电解质(1M KOH+2mM钴salen)中的催化活性在三个不同温度下测量:25℃、50℃和70℃。与在25℃下时相比,所述催化剂在50℃和70℃下具有较高的还原电流密度。该较高还原电流的出现至少是由于氢过氧化物向氢氧化物的进一步还原所引起。同样,所述催化剂在70℃下具有比50℃更加陡峭的还原曲线,表示在Au表面上氢过氧化物的化学吸附在高温下受到促进。此外,随温度的升高,阳极峰电流密度降低,这被认为是由于过氧化物产生的减少。由于随着温度的升高,纯多晶金电极产生过氧化物的催化活性也大幅改变,金/钴salen化合物的这一相似数据间接表明Au主要负责所述催化剂催化活性的可逆性质(钴salen被认为负责反应物的集中和反应动力学)。
pH:
为确定pH对Au/钴salen催化剂催化活性的作用,使用钴salen薄膜改性多晶金电极在具有不同KOH浓度的氧饱和碱性电解质中进行了一系列CV研究,其中所用KOH分别为:0.01M KOH、0.1M KOH、1M KOH和3M KOH。根据CV的形状,可确定所述催化剂在1M KOH和3M KOH中效果良好。对于0.1M和0.01M KOH溶液,可逆性随KOH浓度降低而下降。该结果可能与当碱浓度降低时,接近电极表面的双层成分的改变有关,以及与对该双层结构敏感的过氧化物氧化机制有关。或者/此外,该结果可能与羟基浓度降低时,氢过氧化物离子分解率的改变有关。
示例4
过氧化物稳定剂的作用
在目前所公开的Au/钴salen体系的氧化还原过程中,可通过电化学氧气还原,然后在碱性环境中化学分解产生氢过氧化物。为降低电池再充电过电位及提高再充电速度,可保留过氧化物。由于过氧化物自分解可与由过渡金属离子引起的芬顿过程有关(参见Croft etal..J.Chem.Soc.Perkin Trans.(1992)2,153-160),过渡金属离子功能抑制剂可作为过氧化物稳定剂使用。例如,一种常用的化合物为硅酸钠,可稳定含有或不含过渡金属离子的过氧化物。DTPA和EDTA等过渡金属络合剂或螯合剂也可作为过氧化物稳定剂使用。
因此,EDTA被选为示范电解质添加剂,以稳定过氧化物,钴salen薄膜改性多晶金电极的CV在含1M KOH和1M EDTA的氧饱和电解质中进行。还原电流密度的峰值与氧化电流密度的峰值比率为1:0.99,表明在氧气还原过程中产生的过氧化物基本上全部重新氧化。这进一步表明对于Au/钴salen催化剂,氧气还原过程为纯两个电子还原过程,因为还原产物可在无需较高过电位的情况下氧化。
示例5
钴Salen的电聚合
由于salen的电聚合电位超过含水电化学窗口,钴salen的电聚合在有机电解质环境中进行。因此,在乙腈中制备含有2nM钴salen和50mM TMBAF4的溶液,并且在多晶金电极中进行电聚合(扫描速率50mV/sec)。作为比较,也在高密度热解石墨(BHPG)电极中对钴salen进行电聚合。对于BHPG电极,钴salen的氧化电聚合电流-电位曲线显示在1.35V处出现峰。此外,在重复扫描中,Co(ll/lll)峰在0.4V处减弱,Co(l/ll)峰在-1.2V处增强。形成绿膜。在金电极CV12个循环图示曲线中,Co(ll/lll)峰转为正向,且比BHPG电极的电聚合曲线更加陡峭,表明Co2+氧化变得更加困难。不受任何理论限制,该转变也可表示表面重构发生。因此,钴salen在金表面上的电聚合不仅在金表面提供稳定、不溶的薄膜以稳定钴salen/金组合,还提供了稳定的金表面重构。
电聚合后,在1M KOH电解质中测试改性电极,以评估ORR上聚合的效果。参见图4B。通过比较图4B与图4A(显示Au电极上自由钴的CV),可见当钴salen在Au表面上电聚合后,所述催化剂经过初始阶段(首个循环的可逆性相对较差),在与Au接触时比溶液中的钴salen具有更佳的催化稳定性。
扫描电子显微镜用于进一步观察Au溅射气体扩散电极(GDE)上钴salen薄膜的电聚合。使用与多晶金电极相同的条件,产生非均质薄膜。元素映射表明改性的范围良好,且薄膜的元素组成符合预期。
示例6
噻吩改性钴Salen
方案1:噻吩改性钴salen
噻吩改性钴salen通过两个步骤的程序制备。首先,由5-溴水杨醛和噻吩-2-硼酸通过铃木反应方法合成噻吩改性水杨醛。然后,通过将第一步产物和乙二胺进行缩合产生目标化学品。
噻吩改性钴salen在多晶金电极上按上述示例4中钴salen形成薄膜的方法进行电聚合。观察表面,聚合比原始钴salen电聚合膜更为容易。图5展示了噻吩改性钴,噻吩改性金电极在碱性电解质(1M氧气饱和的KOH溶液)中的CV。
示例7
过氧化氢的产生
图6展示了RRDE实验的结果,该实验使用RRDE电极(包括沉积在圆盘电极表面上的催化剂),以测定目前所公开的催化剂的电子转移数量。图7A展示了使用碳催化剂(炭黑)、Pt/C催化剂(即,炭黑上的Pt)以及目前所公开的催化剂(Au/钴salen)的催化剂翠花活性的RDE研究数据。图7B展示了图7A中催化剂(即C、Au/钴salen和Pt/C)的铁弗曲线,说明了催化剂的不同动力学性能。
所述Au/钴salen催化剂与碳相比具有较高的动力和初始电位,可促进氧气还原,同时在广泛的电位范围内仍保持对双电子氧气还原超过90%的选择性。参见图6和7A。如图6所示,所述催化剂(即,Au/钴salen,重量比为1:10)具有良好的催化活性,并在广泛的电位范围内保持双电子还原。在图7A中,所述Au/钴与碳相比具有明显更高的还原现场电位,且仅略低于Pt/C。此外,如图7B所示,所述Au/钴salen催化剂的动力学性能与在高性能燃料电池体系中最常使用的ORR催化剂—Pt的对比,同时电流密度差异(参见图7A)展示了了Pt的双电子ORR和四个电子ORR之间的差异。
高电流密度燃料电池的流场设计近来有明显的改进。但是,该知识对基于低性能碳ORR催化剂构造燃料电池无用。基于目前所公开的高性能双电子ORR催化剂,可采用燃料电池流场设计的现有知识构建高性能和高效的燃料电池,以产生过氧化氢。图8A展示了过氧化氢电解池的设计。图8B与所述电解池相关,且电解池结构如上所述。其使用镍网作为OER催化剂,AEM(阴离子交换膜)位于中间。本实验中使用的ORR催化剂为Au网,与钴salen电聚合。所述电池包括作为阴极的气体扩散电极,其中所述气体扩散电极包括由Au/钴配位络合物催化剂改性的表面层。尤其是,5-10nm的Au纳米颗粒通过溅射沉积在所述表面上。然后覆盖Au的基质以与示例5中所述相似的方式进行电聚合。所述阴极也可提供湿化的O2,并允许清除过氧化氢水溶液。所述阳极也可允许析氧。在电池阳极和阴极处发生的反应如下:
阳极:4OHˉ→4eˉ+2H2O+O2
阴极:O2+4H2O+2eˉ→2OHˉ+H2O2
使用高锰酸钾表征过氧化物的产生。图8B展示了所述催化剂的过氧化氢生产电流效率在广泛的电位范围内超过90%。
示例8
可逆空气电池
锌空气电池系统包括可制备的Au/Co salen催化层。参见图9A。所述电池壳包括锌电极(如,多孔锌电极)和允许湿化氧进入系统的空气电极。在锌电极和空气电极处发生的反应如下:
锌电极:Zn+4OHˉ→Zn(OH)42ˉ+2eˉ
空气电极:O2+2H2O+2eˉ→H2O2+2OHˉ
可使用其中包括对本领域内普通技术人员显而易见的任何材料的阴离子交换膜,包括但不限于文献(G.Merle et al.,Journal of Membrane Science 377(2011)1-35)中所公开的材料,用于将锌酸盐与空气电极分开,及提供碱性工作环境。在放电期间可产生过氧化氢溶液,并可在电池外进行保存。图9B中展示了与图9A中系统极化性能相近的系统。本实验中使用的电解质为1M KOH+2mM钴salen+10mM EDTA,而非图中的水溶液。在一些实施例中,考虑到不使用KOH溶液,可使用AEM。在这种情况下,所述改性钴salen用于催化层,水和少量过氧化物稳定剂作为电解质使用。假设放电结束时,电池外的过氧化物的浓度可达到的50%到80%的重量百分比,这种锌空气电池计算得到的能量密度为397Wh/kg到489Wh/kg。
示例9
纳米级金基催化剂
纳米级催化剂的使用提供了许多提高催化活性和增加选择性的可能。除表面积增加外,催化剂的催化性能也受到纳米尺寸的巨大影响。体相原子和面相原子的比率及边缘和表面的比率通常升高。由于所述催化剂工作机制通常基于催化剂与反应物之间的相互作用,颗粒边缘催化活性与颗粒表面不同。因此,当这两个比率增加到比体相大几个量级时,对于催化活性将有巨大的影响。
大于8nm的纳米级金颗粒通常与体相Au具有相近的催化活性。因此,首个实验涉及制造大小范围是8nm到25nm的纳米级Au颗粒。制造该大小的Au纳米颗粒的最简单方式是使用柠檬酸三钠作为还原剂还原溶液中的水合氯金酸。在该方法中,颗粒的大小通过反应时间和温度控制。三小时反应后,颗粒大小大在15nm到20nm之间。实际颗粒大小通过XRD实验测量,如示例1中所述。
纳米级Au颗粒在导电载体、碳或氧化钛上沉积。炭黑和Vulcan XC-72均用于该实验。由于碳载体为电极提供了大部分表面积,且为双电子ORR催化剂,不使用钴salen溶液对碳催化剂上的Au进行改性。所使用的替代方法是将钴salen溶解在油墨中,以形成电极。
所述油墨成分和制备方法如示例1中所述。同样按照之前示例1中所述将所述油墨沉积在RDE电极上。要验证钴salen和Au纳米级颗粒具有强烈的相互作用,最简单的方法是在氮气吹扫的碱性溶液中进行连续CV。在该电极浸入溶液后,即进行连续40个循环的CV,对其进行观察。该CV中显示的趋势表明浸入溶液后,钴salen和电极之间的相互作用变弱。此前溶解在该溶液中的钴salen扩散到大量的碱性溶液中。但是,据信,该反应对于Au纳米级颗粒的表面性质应具有充分的影响(从而改变所述催化剂的ORR催化活性)。
为对此进行验证,设计了一项比较实验。在碳粉上使用相同的Au,以及成分相同(除钴salen外)的另一种油墨进行制造。在相同条件下检测者两种催化剂的催化活性。
在纯1M KOH溶液中,该比较实验准确显示出与之前大颗粒Au电极结果的相同差异。由油墨制造,不含钴salen的催化剂没有可逆性迹象,也几乎没有过氧化物氧化迹象。另一方面,由油墨和钴salen制造的催化剂显示出很好的可逆性及过氧化物的氧化。但是,Au在由钴salen改性的碳催化剂上未显示出与大颗粒Au电极可逆性相近的可逆性。对此,两项可能的原因是:(1)所述碳载体实际上产生了过氧化物,但未将其重新氧化,因此所述电极的整体可逆性不佳,以及(2)特定区域纳米级Au的表面积大于大颗粒Au电极,产生的过氧化物更容易进一步还原。参见图13。
应理解,可在不偏离目前所公开的主题范围的前提下,对目前所公开的主题的各种细节进行更改。此外,之前描述仅出于说明而非限制目的。
缩略语
℃=摄氏度
μl-微升
μm=微米
AEM=阴离子交换膜
AgCI=氯化银
Ah=安培时
AO=蒽醌氧化
Au=金
C=碳
cm2=平方厘米
CV=循环伏安测量法或循环伏安法
DTPA=二乙烯三胺五乙酸
EDTA=乙二胺四乙酸
H2O2=过氧化氢
IHL=内亥姆霍兹层
kg=千克
KOH=氢氧化钾
kWh=千瓦·时
Li2O=氧化锂
Li2O2=过氧化锂
M=摩尔
mA=毫安
mg=毫克
ml=微升
mM=毫摩尔
mV/s=毫伏/秒
mAh=毫安培·时
O2=氧气
OER=析氧反应
OHL=外亥姆霍兹层
ORR=氧气还原反应
Pt=铂
RDE=旋转圆盘电极
RHE=可逆氢电极
rpm=每分钟转速
RRDE=旋转环盘电极
salen=N,N′-乙二胺双水杨醛席夫碱
(N,N'-ethylenebis(salicylimine))
SHE=标准氢电极
STEM=扫描透射电子显微镜
V=伏特
- 还没有人留言评论。精彩留言会获得点赞!