由薄多孔金属板组成的电化学装置的制作方法
相关专利申请的交叉引用本申请要求2018年6月6日提交的美国临时申请no.62/681,398的权益,该临时申请通过引用整体并入本文。本发明的实施例一般针对电化学系统及其操作,特别是针对由薄多孔金属板组成的电化学系统。
背景技术:
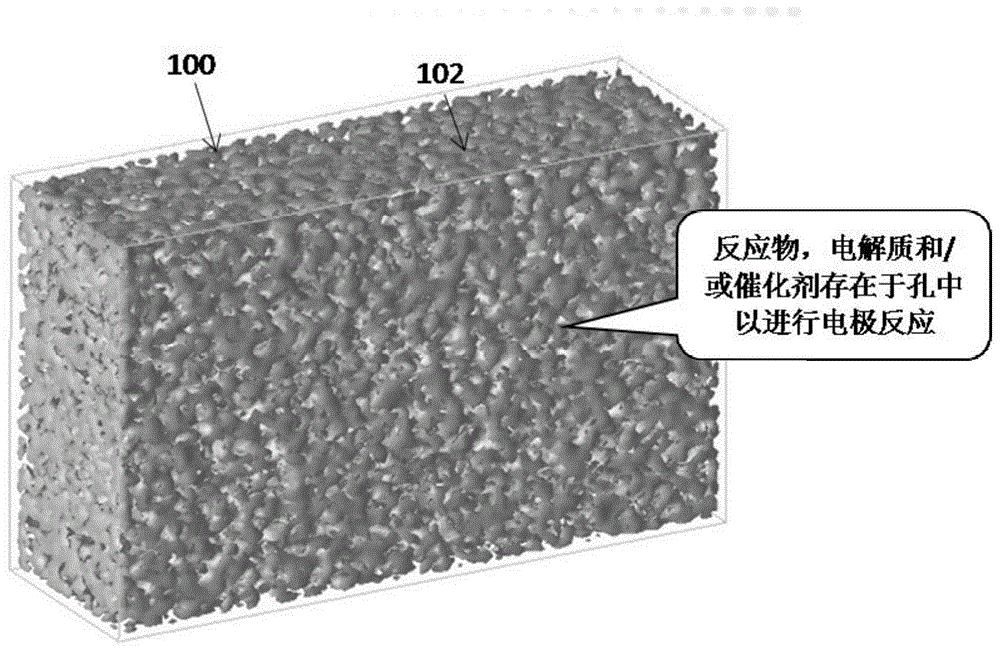
:膜分离器、正电电极和负电电极是电化学转换装置的三个基本部件,如用于电解水、氨水的电化学生产、燃料电池和电池。带负电荷的电极为电极中发生的电化学反应提供电子。在正电电极上,电子从电化学反应中被移除。例如,在电解水时会发生以下化学反应:在带负电荷的电极上:2e-+2h2o→2oh-+h2在带正电荷的电极上:2oh-→2e-+0.5o2+h2o总的反应是:h2o→h2+0.5o2在氢氧化物电解液中操作氢燃料电池时,电极上发生以下反应:在带负电荷的电极上:2e-+h2o+0.5o2→2oh-在带正电荷的电极上:h2+2oh-→2e-+2h2o总的反应是:h2+0.5o2→h2o电化学转换在许多重要的工业过程中被使用,并且对于从可再生能源或间歇性能源(如风能、太阳能和水力)中储存电能变得越来越重要。氢和氧是两种广泛使用的气体,在电解池中电解水可以产生。反之,在燃料电池中氢与氧的燃烧可以产生电能。充电电池广泛应用于电子设备和电动汽车,涉及可逆电化学反应。电化学反应在一些新兴的过程中起了作用,例如用电能从水和氮生产氨。小型电化学装置可用于根据需要生产氨,如提供氨用于选择性催化还原车辆排气中的一氧化氮。大型电化学装置可用于利用风能或太阳能生产氨,用于化肥或化学储能介质。这些电化学装置的一个常见需求是一个电极,它可以提供很高的比表面积积来让电子与反应物发生反应。大量的研究和开发工作致力于发现新的反应化学,改进电解质材料,改进催化剂和配方。然而,对电极主干结构的改进受到现有材料的局限性。这里描述的实施例填补了这个空白。除了电极的支撑结构外,下面描述的各种实施例为分离器提供了一种新的材料结构。分离器作为电化学转换装置的组成部分,具有下列一项或多项功能:i)使带正电和负电的电极彼此保持绝缘以避免短路,ii)允许电解质通过,而且iii)使两个电极的反应产物分离。在实施例中,分离器是一种膜绝缘体,在允许离子通过时不导电。例如,隔膜允许碱性电解液中的oh-离子通过,质子电解液中的h3o+离子通过,锂电池中的li+离子通过。为了具有较高的功率和能量密度,膜分离器最好尽可能薄。然而,膜分离器必须足够厚,以防止两个电极短路。因为反应是周期性发生的,膜分离器必须有足够强度以保持结构完整性。因此,为了提高电化学装置的功率和/或能量密度,需要薄的、耐用的、高渗透性的薄膜分离器。人们对更高效的电极和膜分离器的渴望,体现在利用氮气和氢气或氮气和水生产氨的电化学转化过程的发展。今天,氨主要是通过高温(350-520℃)和高压(100-300bar)下的氮和氢的热催化反应产生的。这样的生产过程,通常被称为哈伯-博世(haber-bosch)工艺过程,是资本和能源密集型的,广泛地用于高规模生产。另一方面,这种反应过程在低温下是热力学上有利的。δh=-92.4kj·mol-1美国专利号7,314,544b2(2006)公开了一种用氮和氢气从电解液中合成氨的电化学方法,该电解液由锂氢化物+锂氮化物熔盐组成。如果没有膜分离器,就会产生阳极和阴极反应的混合产物流。美国专利号8,916,123b2(2014年)披露了使用锂离子导电膜从氮化锂电解质中的氮和水生产氨,其中氮气反应氢氧化锂生成氮化锂,氮化锂再转化为氢氧化锂,同时与水反应生成氨。美国专利出版物第2016/0138176a1(2016)、美国专利出版物第20170037521(2017)和美国专利第9540737b2(2017)中披露了在低于哈伯-博世(haber-bosch)工艺的温度下用氮气和水生产氨的实际例子。这三项专利揭示了碱性电解质的使用,包括碱性氢氧化物水溶液、碱性氢氧化物和盐以及共晶熔融碱性氢氧化物。但是,没有描述膜分离器。由于氢气和氧气的混合物可能是爆炸性的,所以需要有分离器来保持从各自电极中产生的氧和氢的分离。此外,即使混合物浓度控制在爆炸极限之外,分离氢和氧的成本也很高。文献报道了用质子导电电解质(pem)膜作为空气和水的低温合成氨分离器(lan以及其他人)“环境温度和压力下空气和水直接合成氨”,科学报告,3(2013)1145。然而,这些电解质(pem)膜通常使用贵金属催化剂。陶瓷型离子导电膜已被探索研究(例如lan等人的"在中等温度下直接从湿空气中合成氨"应用催化b:《环境》(environmental)152-153(2014)212-217。总之,为了实现氮和水生产氨的实际应用,需要更高效和更高生产力的电化学装置。技术实现要素:本文描述的实施方案教导了使用高表面积多孔金属片作为电化学转换装置中的电极和/或膜分离器的骨架结构。图1示出了厚度小于约200μm的金属板100,其具有孔径为微米级和亚微米级的互联状孔结构102。其可用作图2a中带正电电极104和带负电电极的结构材料106,图2b中仅带正电的电极104或图2c图2c仅带负电的电极106。微孔金属板100是导电的并且提供大的比表面积为电子与孔内的反应物反应。互联多孔结构102使得反应物,电解质和产物能够移入和移出电极体。可以调节微孔金属板100的化学组成和/或孔结构以满足给定电极反应的特定应用需求。当微孔金属板100用于一个电极(图2b或图2c)时,另一个电极优选包括填充催化剂颗粒的糊剂或涂层,其粒径范围为几纳米至几十微米。催化剂颗粒的实例包括纳米和/或微米尺寸的过渡金属(fe,ni,co及其合金),过渡金属-二氧化铈纳米复合物,碳载过渡金属和金属氧化物,过渡金属氮化物,和碳载金属氮化物。可以将一些导电材料如炭黑添加到浆料中以增强填充颗粒层的电子传导性。可以将一些粘合剂添加到填充颗粒层中以将颗粒保持在适当位置。另一个实施例包括电极与膜分离器的集成,如图3所示,在多孔金属板上沉积了膜层。支撑膜层的微孔金属片可作为正电电极或负电电极。对于那些很难制备成自我支撑薄(<40μm)膜,脆弱的膜材料,微孔金属板可以作为一种有效的支持体。例如,陶瓷型材料由于容易断裂,很难制备成多孔的薄板使用。将这些陶瓷类材料加工成膜通常需要在如此高温(如>300℃)下进行反应和/或处理,以至于大多数有机聚合物支撑材料变得不稳定。本发明中的微孔金属板,由于金属材料的高机械强度和耐温性以及均匀的表面孔隙结构使其适合于作为这些膜的支撑体。一个实施方案涉及电化学转化装置,其包括带正电的电极,带负电的电极和膜分离器的组件。带正电的电极,带负电的电极或膜分离器中的至少一个包括至少一个导电金属基多孔片,其具有:厚度小于200μm,具有80-90%孔隙的孔隙结构尺寸小于5μm,孔隙率为20-80%。另一个实施方案涉及用于电化学生产氨的装置,其包括带正电的电极组件,所述带正电的电极包括导电的金属基多孔片,所述导电的金属基多孔片包括小于200μm的厚度,一种互联网络孔结构,其80-90%的孔的孔径小于5μm,孔隙率为20-80%,膜分离器包括厚度小于40μm的多孔陶瓷材料,和带负电的电极,包括纳米级铁催化剂,碳与纳米级铁和/或氧化铁的混合物,碳载纳米级铁,碳载纳米氧化铁,碳载纳米氮化铁,混合物碳与氮化铁或其混合物。附图说明图1是适用于根据实施例的电化学转换装置骨架结构的微孔金属板的透视图。图2a-2c是在电化学转换装置中使用图1的微孔板的实施例的示意图。图3是根据实施例支撑在微孔金属板上的膜分离器的透视图。图4a-4d是根据实施例说明微孔金属板结构特征的显微图。图5a和5b是根据实施例说明多孔金属板电极孔隙大小分布的图。图6是根据实施例在多孔金属板孔隙中加载纳米级催化剂的示意图。图7a和7b是sem显微图,分别展示了一层涂层和两层涂层在微孔金属板电极上的多孔陶瓷涂层的实施例。图8a和图8b显示了根据实施例和商业金属网格用镍片电解水的对比图。图9a和图9b是根据另一个实施例和商业金属网格对镍片进行电解的对比图。图10是根据另一个实施例和商业金属网格对镍片电解水的比较图。图11a和11b是根据实施例说明陶瓷涂层微观结构的sem显微图。图12是根据实施例经过24小时热处理后膜试样的形貌图。图13a是根据实施例说明测试单元配置的示意图。图13b是根据实施例的膜的eis曲线。图14是根据实施例用陶瓷涂层多孔镍片电极进行电解水的示意图。图15a-15d是sem显微图,说明了根据实施例在镍片孔隙内合成纳米氮化钼催化剂颗粒。图16a-16f是sem显微图,说明了根据实施例在镍片孔洞内生长的碳纳米管。图17是根据实施例连续合成纳米金属颗粒的膜反应器示意图。图18a-18c是图17所示膜反应器产生的纳米铁颗粒浆料的照片。图19a-19c是根据传统技术制造的纳米氧化铁粒子的一个实施例制作的纳米尺寸铁粒子的tem显微图。图20是根据实施例从水和氮中生产氨的电极组件的示意图。图21是根据实施例在多孔镍片电极上演示不同电压和温度下氨产率的示意图。图22a是根据一个实施方案在50,000x放大倍数下的n-ni催化多孔片材的微米和纳米结构的照片。图22b是图22a所示多孔片材在500x放大倍数的照片,包括用于原子组成分析的采样点。图23a是根据一个实施方案的10,000x放大倍数的fe/c(2m)催化剂的微米和纳米结构的照片图23b是图23a所示多孔片材在500x放大倍数的照片,包括用于原子组成分析的采样点。图24a是根据一个实施方案的50,000x放大倍数的n-fe/c(1m)催化剂的微米和纳米结构的照片。图24b是图24a所示多孔片材在500x放大倍数的照片,包括用于原子组成分析的采样点。图25a是根据一个实施方案的50,000x放大倍数的n-fe催化剂的微米和纳米结构的照片。图25b是图25a所示多孔片材在500x放大倍数的照片,包括用于原子组成分析的采样点。具体实施方式图4中示出了具有20cm×20cm×50μm多孔镍片的各种实施方案的微孔金属片电极的形态和结构特征。薄平板100提供光滑且均匀的表面(图4a),类似于金属箔。在扫描电子显微镜(sem)下显示其均匀的微结构(图4b和4c)。该表面包括在金属颗粒112之间形成的微米和亚微米级的孔110.该表面在较高孔隙率下孔结构变得加通透。三维孔互联结构102在横截面图中明显可见(图4d),孔隙在整个厚度上相互连接。优选地,金属板100是导电体,其从整个板100的电流连接点分配电子或者将电子从整个板100收集到电流连接点。金属材料本身可以用作电化学反应催化剂。或者催化剂可以沉积在孔110内部的电子传导金属颗粒112的表面上。多相反应区域增强因子定义为多孔金属片电极104,106中的电子/反应物反应区域与其几何表面积的比率。多相反应区域增强因子与孔径和孔隙率之间的关系可用下式描述:其中f=增强因子,sar=电子/化学反应区面积,sag=几何表面积,δe=薄板厚度,ε=孔隙度,dp=孔径。因此,对于致密金属箔基电极,增强因子为1。使用50μm厚(δe),50%孔隙率(ε)和0.5μm平均孔径(dp)的多孔金属板100,增强因子为200。孔隙率对增强因子的影响程度是有限的。低孔隙率(0.35)与高孔隙率板(0.70)的差异仅为2倍。对于恒定的孔隙率,孔径的影响是显着的。0.1μm孔隙片的增强系数可以是1μm孔隙片材的10倍,10μm孔隙片材的100倍。片材厚度对增强因子的影响是有限的,因为反应物进入电极内部和产品离开电极的传输阻力随着片材厚度成比例地增加。电极的体积生产率是电化学器件的一个重要的性能参数,与体积比表面(sav)有关,如下所述:比表面积随着孔径的减小而呈一级倒数的增加.上述讨论突出了在多孔电极中减小电化学反应区孔径的好处。多孔板100的孔隙率可以如下所述测量为几何孔隙率和吸附孔隙率。其中εg=几何孔隙度,ρg=几何密度,ρm=材料密度,εl=吸附孔隙度,vl=液体吸附量,vg=几何体积。基于多孔金属板相对于其材料密度的重量和体积简单地测量几何孔隙率。液体吸附孔隙率可以通过给定体积的多孔金属板100可以吸收的液体量来测量。测量中使用的液体流体应该是完全可润湿的。因此,如果所有孔110在多孔金属板100中是互连的,则几何孔隙率应等于液体吸附孔隙率。如果吸附孔隙率小于几何孔隙率,则多孔金属板100中的一些孔隙不能被液体接近。作为有效的电极结构,多孔金属板100应具有与几何孔隙率相同或非常接近的液体吸附孔隙率。对于图4中所示的多孔金属板结构,液体吸附孔隙率与几何孔隙率相同。可以在显微镜下物理检查多孔金属板100的孔径。对于微孔金属板100,通过使用现在已建立的分析程序可更好地表征孔径。多孔金属板100的外部孔径可以通过水银孔隙率测定技术表征,如图5a所示。对于不同厚度的两个多孔金属板100,在孔径分布曲线中示出了单个窄峰,其表示均匀的表面孔结构。对于这两种薄板100,2μm以上的孔数小于1%。毛细管流动是表征多孔金属板100的孔径分布的另一种常用方法。与水银孔隙率测定法不同,毛细管流动技术基于气体流动路径-即沿着片材厚度的通气流量来测量孔径。图5b显示了两个多孔金属板100的孔径分布。与图5a相比,显示了在板内部基本上存在较小的孔,孔径在0.1至0.8μm的范围内,即亚微米级。纳米催化剂120,稳定剂122和/或促进剂124可以装载在多孔金属板100的孔110内,以催化所需的反应并抑制不希望的反应,如图6所示。电解质118中的反应物114流入孔110并且反应产物116流出孔110.纳米催化剂的尺寸优选小于多孔金属板100的孔径,优选小数倍,使得孔隙开口保持在一定的比例用于反应物和产物的快速运输。优选地,催化剂与金属颗粒112紧密接触。可以使用几种方法将纳米催化剂加载到支撑孔110内,包括浸渍和化学气相沉积。采用浸渍技术,首先以溶液形式制备催化剂前体。该溶液用于填充多孔金属板100的孔102,110。然后,通过干燥除去液体载体,例如通过加热或抽真空或两者。可以通过在受控气体环境下加热来活化干燥的样品。例如,过渡金属的盐(fe,co,ni,mn等)可以溶解在水溶液中。干燥后,通过在氧化气体环境中加热样品获得纳米氧化物催化剂,如果干燥的样品在还原性气体环境中加热,则获得纳米金属催化剂。可通过浸渍添加氧化铝,氧化硅或氧化锆氧化物以促进和稳定纳米过渡金属(氧化物)催化剂的催化活性。利用化学气相沉积方法,在含有催化剂前体的气体环境中加热多孔金属板100,使得催化剂前体扩散到孔102,110中并在孔102,110内生长催化剂。可渗透离子但不导电的涂层108可沉积在微孔金属片100的一个表面上,将膜分离器108与一个或两个电极组合成一体。在一个实施方案中,膜厚度优选为涂覆的微孔金属表面上的孔口尺寸的约2至10倍,使得金属颗粒表面被连续涂层108完全覆盖。需要完全覆盖金属颗粒112来消除可能的短路。涂层108中的孔径可以由涂覆的材料的性质决定,并且可以比微孔金属板100的开口小一个数量级。适用于膜分离器108的材料包括但是不限于金属氧化物,陶瓷,玻璃和金属氧化物+聚合物混合相或复合相。膜分离器108可包括相同材料组成和/或结构的一个涂层(图7a),或不同材料和/或结构的多个层(图7b)。膜厚度可以通过在显微镜下物理检查横截面来确定,如图7a和7b所示。涂层厚度也可以通过面积加载密度来评估,其可以基于实验测量值由以下等式计算:其中wc,s=面积载荷密度(g/cm2),wc=涂覆在支撑板上的材料重量(g),sac=被涂覆的支撑板的面积(cm2),δc,s=涂层厚度(cm),ρc,p=涂层材料的填充密度(g/cc)。大多数金属氧化物的填充密度在1-3g/cc范围内。涂层厚度最好小于2x10-3cm(20μm)对应于1-6x10-3g/cc的涂层负载。可以通过水热生长在多孔金属板100上形成膜层108。分子筛的种子可以分布在支撑片表面上,然后将载种子的片浸入生长溶液中,在一定的温度和时间条件下,分子筛的晶体从种子中生长并填充晶间空隙,形成连续的膜层108。在一个实施方案中,膜层108可以通过湿化学涂层和后处理制成。粒径为200-20nm的陶瓷粉末如氧化锆可以分散在液体载体中以形成均匀的涂料浆料或溶液。将涂料溶液涂覆到金属板支撑件100上以形成陶瓷颗粒的连续填充物。然后,可以在适当的气体环境下加热涂覆的片材,使得陶瓷颗粒烧结以形成耐用的涂层108.可以选择烧结条件(气体环境,温度和时间)以获得所需的烧结程度,不会对多孔金属支撑体100造成损坏。例如,在高于500℃的温度下,如果优选使用惰性或还原性气体以防止金属板100氧化。在另一个实施方案中,通过气相沉积施加涂层。待涂覆的材料可以在气相或真空下溅射成小颗粒或碎片,碎片/颗粒沉积在多孔金属板100支撑表面上以形成连续层108。膜分离器108在电化学转换装置中的一个功能是保持相反电荷的电极104,106彼此绝缘。因此,优选耐用的绝缘材料,例如氧化锆,氧化铈,氧化铝和氧化硅。对于给定的材料,膜分离器108对短路或电穿透的抵抗力随着厚度而增加。然而,在所公开的实施例中优选地避免厚膜层108。厚膜层108的一个问题是多孔金属板支撑件100上的厚膜108易于破裂和分层。因此,厚度小于40μm是优选的。厚膜层108的另一个问题是增加了传质阻力。允许电解质118中的某些离子渗透是膜分离器的另一个功能。然而,传质阻力随膜厚度成比例地增加。作为膜分离器108,涂覆的材料可以具有固有的离子导电性,或者可以为电解质118提供来回移动的通道。例如,氧化锆材料在低温下不具有离子导电性,但多孔氧化锆膜108可通过毛细管力承载碱性氢氧化物电解质溶液。毛细管压力通过以下等式与表面张力,接触角和孔半径相关。其中δpc=毛细管压力,σ=液体的表面张力,θ=接触角,rp=孔隙半径koh溶液在37℃下具有约0.067n/m的表面张力。如果溶液在孔中完全可润湿,则对于50nm半径的孔,毛细管压力为约26巴。如果膜材料不提供离子导电性,则由对电解质溶液完全可润湿的材料制成的多孔膜层108,把孔径控制低于100nm,以牢固地把电解质溶液保持在孔隙内。实施例i.薄孔镍片电极电解水使用由moleculeworksinc.(mwi)生产的用于水电解的多孔镍合金板测试作为电极微孔金属板108的反应性。从厚度约50μm且孔隙率约40%的多孔镍片上切下两个2cm×3cm的试样片,试样用0.5mm厚的聚酯网状隔板夹在一起,以用作相应的正极和负极,然后将该组合体浸入koh溶液中。当施加到两个电极的电压超过1.3v时,气泡从两个电极出现。记录电流并与图8a和8b中的其他金属材料进行比较。这些材料的结构特征列于下表1中。对比网材料的质量面密度约为mwi镍板的1.3倍,厚度约为mwi镍板的20倍。图8a显示在25℃下用mwi的镍片获得的电流是用常规镍和不锈钢网获得的电流的约3倍。在较高温度下,其活性给进会进一步增大。在50℃时,使用mwi镍板获得的电流密度约为使用其他传统金属网获得的电流密度的5倍。使用mwi的镍片,于30%koh溶液中在50℃下进行水电解,结果与镍网和致密镍箔在图9a和9b进行比较。在1.6v(图9a),只有mwi镍片显示电流,而镍网和致密镍箔的电流太低而无法测量。在2.0v时,mwi镍板产生的电流约为镍网的三倍,是致密镍箔的四倍(图9b)。致密的镍箔具有与mwi镍片相同的几何形状,即50μm薄的平板,其材料质量面密度约为mwi镍板的1.6倍。结果清楚地表明,对于相同种类的电极材料,微孔结构102显著地改善了水电解活性。将mwi的镍板在不同电压下在100℃下的水电解与图10中的镍网和致密镍箔进行比较。在电解反应稳定在15分钟后测量电流。在相同条件下,mwi镍板的电流约为镍网和致密镍箔的两到三倍。各种条件下反应性的持续改善归因于mwi镍板中相对于金属网和箔的电子/水反应的较高表面积。表1.多孔材料作为电解水电极的信息实施例ii.在多孔金属板上覆盖多孔陶瓷膜内部制造的多孔镍片100涂覆有陶瓷材料以形成电绝缘层108,用于分离相反电荷的两个电极104,106。表2列出了厚度和孔隙率略有不同的3cm×5cm镍板100上的一组陶瓷涂层108。首先,通过真空过滤,用平均颗粒尺寸200nm含有氧化镍烧结助剂的氧化钇稳定的氧化锆(ni-ysz)涂覆多孔镍片100。50nmysz颗粒用于形成第二涂层108.将涂覆的样品干燥并在815℃下在受控气体环境中烧结。涂层的量可以通过面积加载密度(g/cm2)表征,其可以通过用涂覆区域表面积和涂层重量增加来计算。涂层厚度可以由支撑片涂覆前后的厚度差计算。通过改变用于形成第一和第二涂层108的涂层溶液的量,可以获得具有不同面积加载密度和厚度的涂层108。如果涂层孔隙率相同,则涂层厚度与面加载密度成比例地增加。具有相同或相似面加载密度的样品之间涂层厚度的变化表明这些涂层108具有不同的孔隙率。涂覆的样品做了导电性测试。表2中列出的结果表明,如果面加载密度高于约3mg/cm2和/或涂层厚度高于约10μm,则涂层108没有导电性。表2.mwi多孔镍板上的陶瓷涂层陶瓷涂层表面的微观结构如图11a和11b所示。在200nmni-ysz(加入氧化镍作为烧结促进剂的氧化钇稳定氧化锆)涂层上可以看到平均200nm大小的颗粒或晶粒(图11a)。nio作为烧结助剂加入ysz(氧化钇稳定氧化锆)晶粒中。ysz晶粒颈-颈烧烧结在一起。由于ysz粒子的随机堆积,出现了约100-200nm尺寸的空隙。这种空隙通过第二层涂层用10-50nmysz颗粒填充(图11b)。这些结果表明,多孔镍板可以被不同大小的陶瓷颗粒所覆盖,形成孔径比支撑孔小近数量级的涂层表面。陶瓷涂覆的镍片100可用作电化学转换装置的膜分离器108。通过在高压釜反应器中加热膜试样来测试涂层的稳定性。将图12中顶行的五个样品用50重量%的koh溶液浸泡并置于水蒸汽的高压釜反应器中。高压釜反应器用koh溶液填充一半,将图12底行中的三个样品浸入溶液中。将高压釜反应器在120℃下加热24小时。除50nmysz/200nmmn-ysz之外的所有膜试样都是完整的。50nmysz/200nmmn-ysz膜具有200nm尺寸的mno掺滲的ysz颗粒做为底层和50nm尺寸的ysz颗粒做为顶层。在加热后,该膜涂层破裂并从载体上剥离。具有200nmni-ysz的单层涂层和具有50nmysz/200nmni-ysz的双层涂层都是稳定的。当样品暴露于水蒸气或暴露于koh溶液时,未发现裂化或分层。结果证明了这种类型的膜材料在热水汽和电解液中的稳定性。实施例iii.koh电解液固定于陶瓷膜的离子电导率通过两次真空涂覆和随后的烧结形成用于测试的陶瓷膜108。nio促进的200nm粒径的ysz和50nm粒径的ysz分别用于第一和第二涂层。将涂覆的样品在受控的气体环境下在750°下烧结。使用电化学阻抗谱(eis)技术在环境室中测量离子电导率。测试设置如图13a所示。陶瓷涂层108被50%的koh溶液润湿,然后,裸露的多孔镍片100附着到陶瓷涂层108,其模拟在电化学转换中的膜电极组件1300。将膜电极组件1300包裹在碳纸覆盖片128中。在不同温度下在80%rh下测量的膜样品的eis图显示在图13b中。在厚度为47um,孔隙率为42%的多孔镍支撑片100上制备该膜108,涂层载量为4.3mg/cm2。在eis图中,将阻抗线外推至x轴,并且假想对应为零的实际阻抗的外推值(z'或z在z'=0)被视为膜108的电阻。所有的eis图都趋于收敛成一条线,这表明在60到100℃的温度范围内,电阻对温度的影响不大。通过减去接触电阻,得到的膜面积比电阻(asr)约为0.014欧姆·cm2。为了确认温度对电阻的影响,对另一各膜(#20180221-7)进行eis测试。为了排除陶瓷/镍片接触区域中可能的干点与测量的复杂性,在测试之前将镍支撑片/陶瓷涂层/镍覆盖片组件1300浸入koh溶液中以确保两者中陶瓷毛孔和镍孔被润湿。首先检查两个完全湿润的镍片的接触电阻。发现完全润湿的镍片具有与前两个干燥镍片相同的值(约0.026欧姆·cm2)。从测量数减去接触电阻后,在不同温度下的asr值列于下表3中。随着温度的升高,asr在60-100℃范围内略有增加。电阻值0.043-0.033欧姆·cm2略高于第一样品获得的0.014欧姆·cm2。将第二膜样品涂覆在具有47μm厚度和38%孔隙率的镍载体上,涂层载量为2.8g/cm2。使用相同的涂层材料。asr的差异归因于详细的膜结构,其可以在不同载体孔隙率和/或涂层条件下制备的样品之间变化。表3.80%rh中20180221-7膜的asr值(2.8mg/cm2涂层)温度(℃)asr值(ohm·cm2)600.043700.041800.037900.0341000.033实施例iv.陶瓷/金属板膜分离器的放大通过在21×21cm多孔镍片上制备涂层来证明陶瓷/金属片膜的放大。表4提供了以不同方式涂覆的三张膜片的数据。将片材1582分成15个3cm×5cm的涂层区域。片材1460涂覆在20cm×20cm的一个区域中。片材1854被分隔成6个5cm×10cm的涂层区域。用1m硝酸镍溶液预浸滲的200nm氧化钇稳定氧化锆(ysz),0.25m硝酸镍溶液预浸滲的的50nmysz和co和sm掺杂的50nm氧化铈粉末分別制备三种涂覆溶液。通过真空过滤将200nmni-ysz和50nmni-ysz两层涂覆在多孔镍片1582和1460上,将cosm-二氧化铈单层涂覆在多孔镍片1854上。用于涂层的溶液体积列于表4中。涂层板在连续隧道炉中在750℃的氢气中烧结。从每个片材取两个3cm×5cm的试样用以固定koh电解质。koh填充的陶瓷膜的特征用上述实施例中描述的测量技术得到的面积比电阻(asr)来表征。asr值在50和90℃下测量。在每个温度下,相对湿度(rh)从50%逐渐增加到95%。结果总结在表4中。在50℃下,asr在不同的试样和不同的rh之间可能有显著的变化。在90℃和rh≥70%时,所有膜试样的asr均小于10m·ω·cm2。表4.在21m×21cm多孔镍片片上制备的陶瓷膜及其导电性实施例v.用陶瓷膜涂覆的多孔镍片电极进行水电解使用陶瓷涂覆的多孔镍片作为电极测试水电解。通过在约30%孔隙率和50μm厚度的多孔镍支撑片上沉积200nmysz颗粒并在750℃下在受控气体环境下烧结来制备陶瓷涂层。涂层厚度约为13μm。将陶瓷/镍片与镍片片夹在一起以形成一对电极,并如上述实施例中所述进行测试。将该电极对浸入95℃的30%koh溶液中。在施加大于1.3v的电压时,在两个电极104,106处产生气体。在电解稳定后(通常在15分钟内),记录电流。陶瓷涂覆的镍片作为阴极和阳极在图14中进行比较。对于所有三对电极组件,在1.3v时没有可测量的电流,电流在1.6v时变得显著并且随着电压升高,到2.0v而迅速增加。在相同电压下,陶瓷涂覆的镍片作为阳极的电流始终高于阴极。陶瓷涂覆的镍片作为阴极的电流始终高于镍片电极。测试后,镍片上的陶瓷涂层完好无损。结果证明了陶瓷涂覆的镍片作为电极的适用性。实施例vi.将纳米催化剂颗粒结合到多孔金属板的孔中氮化钼是用于电化学反应和热催化反应的活性催化剂。通过气体/固体化学反应将纳米尺寸的氮化钼催化剂120装载在多孔镍片100的孔110内。首先,用0.2m钼酸铵四水合物溶液浸渍约50μm厚和33%孔隙率的多孔镍片100。除去镍片表面上的过量溶液并干燥湿润的镍片。然后,通过在连续流动的3%h2/n2中加热反应器内的浸渍镍板来进行原位纳米颗粒生长。将反应器以10℃/min加热至600℃并在600℃下保持1小时。为了比较的目的,将钼酸铵四水合物粉末涂抹在相同的多孔镍片100上和陶瓷板上并在相同条件下加热。通过电子扫描显微镜分析所得样品。在低放大率下,图15a显示多孔镍片100的外表面看起来与镍片相同。在高放大率下,纳米尺寸的氮化钼颗粒120在金属镍颗粒表面上的生长是明显的(图15b),粒径为约10-20nm。金属片中的微孔110还是存在的。纳米催化剂负载量为约0.9mg/cm2。相比之下,散布在多孔镍片100的外表面上的前体粉末形成几微米的催化剂晶体120(图15c)。而沉积在致密氧化铝盘子上的前体粉末形成更大的晶体(图15d)。结果证明了在多孔镍片的孔内原位合成纳米尺寸催化剂颗粒120和通过微孔金属支撑片稳定纳米催化剂颗粒120的可行性。实施例vii.多孔金属板孔内碳纳米管的生长已知碳纳米管(cnt)具有高纵横比和大的比表面积。据报道,cnt具有一些独特的电化学反应催化活性。在一个实施方案中,通过原位催化化学气相沉积将cnt加载到多孔镍片的孔中。孔隙率约为35%的多孔镍片100用0.1m过渡金属硝酸盐乙醇溶液浸渍。干燥后,将浸渍的样品装入反应器中并在连续气流中在温度分布下加热。用氮气吹扫反应器15分钟,然后将进料气体转换为氢气。在氢气流中以10℃/分钟将反应器加热至650℃。当反应器达到650℃时,进料气体转换为氮气,在用氮气吹扫反应器约4分钟后,将乙醇蒸汽引入反应器中,乙醇/氮气流中的化学气相沉积进行约10分钟,然后,关闭乙醇蒸汽的引入并冷却反应器,当样品冷却至300℃以下时,样品从反应器中卸载。镍片100从灰色金属变成黑色。片材100保持平坦并且与镍片材一样柔韧。通过sem分析揭示了孔110内的cnt1600的形成。图16显示了在用各自的fe,co和ni催化剂浸渍的三个镍片100上的cnt1600生长。装在薄片100中的cnt1600约为1mg/cm2。在fe催化片材(fe-ni片材)100的外表面上,cnt生长之前和之后的表面孔结构在低放大率下看起来相似(图16a)。从金属颗粒表面112生长的单个cnt1600在高放大倍数下是明显的(图16b)。cnt尺寸直径约为50-100nm,长度为几微米。将co催化的镍片110断裂以观察片100内的cnt生长。图16c和16d显示在孔110内广泛存在cnt1600。由于孔110内的限制,cnt管1600缠结在一起。整个片材厚度的cnt1600由ni催化的镍片显示(图16e),cnt管直径约为30-50nm(图16f)。在这些样品中还形成一些焦炭或石墨片。通过调节催化剂性质(尺寸,金属組成,负载量)和cvd条件(温度,时间,碳前体浓度等),可以针对特定应用优化cnt1600的尺寸和产率。该实施例证明了在镍片100的孔110内生长碳纳米管1600同时保持片状多孔结构102在片内完整100的可行性。其他纳米结构如石墨烯也可通过化学反应过程在孔内生长。实施例viii.纳米金属颗粒在连续膜反应器中的生长对于许多催化反应,需要廉价的过渡金属催化剂(fe,ni,co,cu等)。催化活性通常随着粒度的减小而增加。纳米过渡金属的商业来源很少。在这个实施方案中,开发了图17中所示的膜反应器方法1700,并用于通过液相反应制备小尺寸的金属颗粒1716。以fe为例,水溶液中的铁离子被硼氢化钠还原成金属铁:4fe3++3bh4-+9h2o→4fe+3h2bo3-+12h++6h2铁离子溶液可以由氯化铁或硝酸盐制成。在第一步骤1702中,通过压力脉冲发生器1706将金属(例如铁)前体提供给膜反应器1708。在第二步骤1704中,其可以与第一步骤1702同时发生,給膜反应器提供还原剂。在一个实施方案中,还原剂加在清扫流体提中。铁溶液与膜反应器1708内的还原剂接触,其中铁溶液通过膜作为纳米喷射器注入含有还原剂的清扫流体中。纳米喷射快速与还原剂反应以形成纳米尺寸的金属颗粒。纳米金属颗粒在形成后,即时从膜通道中扫出。当使用含水金属离子溶液时,使用孔径为数十至数百纳米的疏水膜在压力下产生纳米喷射。可以通过压力脉冲发生器1706脉冲注入压力以产生离散的纳米喷射。可以快速冷却反应的产物流以防止纳米颗粒的聚集,命名为步骤1710。可以通过使用磁场将纳米颗粒1716与溶液分离,因为纳米过渡金属颗粒1716通常是磁性的,命名为步骤1712.将分离的流体再循环以再次使用,命名为步骤1714。所产生的铁颗粒1716非常小,使得反应器流出物1802看起来似均匀的流体(图18a)。通过使用常规方法,例如离心和过滤,小颗粒1716难以与溶液分离。发现磁分离简单有效。如图18b所示,磁棒1804放置在容器1806的壁上。纳米颗粒1716被吸引向磁性1804杆并与溶液分离。通过除去溶液,获得浓缩的纳米鉄浆料1808(图18c),其可以用作电极催化剂。通过透射电子显微镜(tem)分析所产生的纳米鉄颗粒1716的尺寸。图19a显示约15-20nm的离散颗粒1716。可以通过使用沉淀技术制备小至5-10nm的氢氧化铁颗粒1902(图19b)。然而,一旦氢氧化铁1902煅烧成氧化铁1904,颗粒就会迅速生长。即使在适度的煅烧温度下,例如350℃,氧化铁颗粒尺寸也变为30-80nm(图19c)。如果氧化铁1902被还原成金属颗粒,则颗粒尺寸将进一步增大。膜反应器1708的连续方法可以具有高达5kg/m2/h的纳米金属生产率。方法1700提供控制粒度和组成的灵活性。单金属纳米颗粒(fe,ni,co,cu)1716可以通过供给单一金属离子溶液来制备,而金属合金颗粒可以通过供给混合离子溶液来制备。由于金属氯化物溶液彼此完全混溶,因此多金属元素的颗粒1716可以在宽范围的组成上制备。例如,feco合金可以基本上由100%fe至0%fe制成。粒径可以通过孔结构102和膜108的表面化学以及反应条件来控制。反应条件包括:进料速率,温度和停留时间。膜108确定纳米喷射的尺寸,形状,表面分布和频率。表5总结了膜反应器1708中的几次纳米鉄的合成。通过改变进料浓度,进料流速和清扫流速,可以获得反应器流出物1802中的不同生产率和固体负载量。表4.膜反应器用于合成纳米铁催化剂(2cmx10cm膜)实施例ix.从水和氮气通过电化学生产氨如前面实施例中所述的陶瓷涂覆的多孔镍片用作图20中所示的膜电极组件(mea)2000中的膜分离器108,由水蒸汽和氮气生产氨。催化剂浆料通过球磨纳米氧化铁(20-40nm)粉末和炭黑来制备,在浆料中氧化铁/碳重量比=5/1,氧化铁/去离子水重量比=1/6。在一个实施方案中,将0.5cc的糊剂施加到2.7cm×4.7cm区域中厚度约为50μm的另一多孔镍片上,将糊状物200均匀地涂布在镍片材100上,在糊状物200稍微干燥之后,将镍片材粘贴在陶瓷涂层2004上,并将两个片材100压在一起。将koh溶液浸入在多孔金属板表面上以填充镍板100的孔110.使用洁净室布擦除表面上的过量溶液。将mea2000装入测试池中,将测试池放在烤箱内,具有陶瓷涂层的多孔镍片带正电并用作阳极104,而带有催化剂膏的多孔镍片带负电荷作为阴极106.如果没有短路,则将湿的氮气流引入两,阳极104和阴极106俩侧都接近大气压下,阴极104中的潮湿氮气流提供水和氮分子作为反应物,在阴极催化剂层106中,氮和水分子与电子反应以产生氨分子和氢氧根离子,氢氧根离子扩散穿过陶瓷膜层2004并在多孔镍片阳极104中被氧化成氧和水分子。用ph为4至6,例如约4.7的稀h2so4溶液洗涤反应器流出物,使用铵离子选择的电极分析氨的存在,反应测试在恒定温度下在不同电压下进行,结果总结在图21中。在施加的每个电压下,反应进行约30分钟。似乎存在用于氨生产的最佳电压,电压应足够高以克服热力学平衡极限,如果电压超过最佳值,则副反应可能优于氨形成。温度对氨生产率的影响因副反应和反应物的吸附而变得复杂。通常,反应速率随温度而增加。然而,氮气和/或水蒸气在催化位点上的吸附可随着温度的升高而显着降低。通过使用更具活性的催化剂并优化阴极和阳极的组成和结构,可以提高氨生产率和电化学效率。本实施例证明了在大气压和低温下使用mwi多孔镍片电极通过电化学反应从水蒸气和氮气中产生氨的可行性,相对比从氮和氢气通过热催化合成氨,需要非常高的压力和温度。实施例x.用不同催化剂从水和氮电化学生产氨制备几种不同的催化剂以提高电化学电池构造中的氨生产率,如图20所示。五种催化剂的制备条件和组成列于下表6中。催化剂-n-ni薄片是通过稍微氧化多孔镍薄片制备的,该多孔镍薄片是由前面实施例中所述由moleculeworksinc.生产的。在320℃的炉中在空气中加热该薄片约2小时,然后,将片材在连续的氨气流中在350℃下处理2小时,在将反应器冷却至室温后卸载处理过的片材,得到的片材没有变形,并且保持与新片材一样平整。这种处理在多孔镍片的金属颗粒上产生纳米特征,如图22a中的sem图像的高放大率所示(50,000x)。应注意,片材保持多孔。通过能量色散谱(eds)分析片材的原子组成,用于eds分析的区域和点的结构显示在图22b中(放大500倍)。氮(n)原子存在于每个区域和采样点,平均组成列于表6中,该片含有少量的fe和co。通过x射线衍射测量晶相,对于该材料鉴定的唯一晶相是镍,表明金属氮化物以非晶态存在或者作为纳米尺寸的晶粒太小而不能通过xrd检测到。通过使用初湿浸渗技术用1m硝酸盐溶液浸渍bet表面积为1200m2/g的碳粉来制备fe/c(1m)催化剂。将湿粉末在150℃下干燥过夜,将干燥的粉末置于陶瓷舟皿内,并在管式反应器中在连续的氢气流中在400℃活化2小时,在将反应器冷却至室温并用氮气吹扫后卸载催化剂。除了使用2m硝酸铁溶液进行浸渍之外,用与fe/c(1m)相同的方法制备fe/c2m)催化剂。在图23a中,粉末催化剂在高放大倍率(10,000x)下显示出非常均匀的微观和纳米纹理,没有看到大的偏析颗粒,这表明铁很好地分散在碳基质中。eds分析表明fe是主要的金属元素,并且在图23b中的不同点的取样中证明了一些氮(n)原子的存在。fe2o3和fe被确定为该催化剂中的两个主要晶相。与fe/c(1m)不同,催化剂n-fe/c(1m)通过使用不同的活化方法制备。不是在氢气中还原,而是将fe浸渍的碳粉末在管式反应器中在连续的氨气流中在1000℃下处理2小时。即使在如此高的温度下处理之后,催化剂仍保持其均匀的微米和纳米结构,如图24a所示,放大50,000倍。在具有几百μm尺寸的单个颗粒的低放大率(500x,图24b)下,也可以看到均匀的纹理。测量催化剂中氮(n)原子的显著存在。通过xrd鉴定fe2n为唯一的晶,其相具有宽峰。xrd峰宽随晶体尺寸增加而减小,宽峰表明晶体尺寸小。用纳米氧化铁粉末(20-40mm)制备催化剂-n-fe。将粉末在管式反应器中在连续的氨气流中在1000℃下加热2小时。催化剂保持多孔,孔径为数百nm,可在50,000x放大率下看到(图25a)。大晶体用纳米颗粒装饰。低放大倍数(图25b)表示粒径在几微米到几百微米的范围内。如图25a所示,大颗粒是小得多的结晶颗粒的附聚物。显示出显着存在n原子。通过xrd确定fe2.15n是催化剂中唯一的晶相。xrd峰也宽,表明晶体尺寸小。表6.为电化学氨生产制备的五种新催化剂将表6中列出的催化剂装入电化学池中,用于由水蒸汽和氮气制备氨。电池配置与前面实施例中使用的相同,如图20所示,多孔陶瓷涂覆的镍片用作集成阳极催化剂/膜分离器。对于碳载催化剂,fe/c(1m),fe/c(2m),n-fe/c(2m),将300mg催化剂粉末放入具有10-12个氧化锆介质球的离心管中,在干燥状态下使用涡旋混合器机械研磨粉末2分钟。然后,将0.5-1.5ml脱气的去离子水加入到混合的粉末中并涡旋混合2分钟以形成均匀的催化剂糊剂。将3ml新鲜制备的50%氢氧化钾溶液加入到催化剂浆料中并涡旋混合2分钟。阳极片的陶瓷涂层表面用新鲜的koh溶液浸泡并与阴极片连接以形成单电化学电池。阴极板上的催化剂浆料涂层与陶瓷涂层接触。将阳极和阴极片均匀且牢固地压在测试池中以确保紧密接触。为了装载n-fe催化剂粉末,将300mg催化剂粉末与100mg高表面积碳粉末混合。以与前一段中所述相同的方式制备阴极片并与阳极片组装。氮化物多孔镍片-n-ni催化剂的负载与上述催化剂粉末不同。将50mg高表面积碳粉末分散在0.5ml水中,加入1-2mlkoh溶液,并混合形成碳糊。将碳糊施加到放置在洁净室布上的氮化物镍片的表面上,当碳被充分固定后,碳涂覆的氮化镍片被转移到阴极板上并压在带陶瓷膜涂层上.将测试电化学池置于烘箱内,100sccm的氩气通过水蒸发器进入测试池的阳极侧,100sccm的h2/n2(摩尔比=1/1)或n2气体通过另一个水蒸发器进入给测试池的阴极侧,化学电池温度由烘箱温度控制。在该实施例中,水蒸发器也放置在烘箱内,以确保在任何测试条件下进料气体被加湿在接近其饱和点处。阳极和阴极压力可以独立控制或保持相同。用负载有ph=2.5的h2so4溶液的吸收器洗涤阳极和阴极流出气体,分析洗涤器中的铵浓度,根据分析结果和取样条件计算氨生产率,结果总结在下表7中。表7中的测试结果均在90℃的一个恒定温度下得到,反应器压力在低压(大气压或14.7psig)下操作。对于每次测试运行,通常在不施加任何电压的情况下启动反应器以检查非电化学反应,然后,施加电压以显示电化学反应,阴极进料可以从h2/n2转换为n2以评估进料气体对反应性的影响。利用氮化物镍片阴极催化剂(n-ni),当施加1.2v时,氨生产率增加约五倍,这清楚地显示出电化学反应效应。由h2o与催化剂中n原子的副反应和/或ii)h2和n2的热催化反应,可以从没有电压的h2/n2进料气体产生氨。在没有电压的情况下从n2进料气体生产氨很可能是由h2o与催化剂中n原子的副反应引起的。在将进料气体从h2/n2切换到n2之后,在相同电压下的氨生产率保持大约相同,为3.9×10-9mol/(cm2·s)。然而,在恒定条件下,氨生产率随着运行时间而降低,290分钟后,生产率降至5.9×10-10mol/(cm2·s)。当施加1.2v电压时,碳载fe催化剂-fe/c(1m)也清楚地显示出电化学反应。氨生产率为2.0×10-9mol/(cm2·s)。使用另一种碳载fe催化剂--fe/c(2m),电化学反应的氨生产率比非电化学反应高约4倍,达到1.1×10-9mol/(cm2·s)。与催化剂-fe/c(1m)的比较表明,增加催化剂中fe负载量不一定提高氨生产率。当进料气体从h2/n2切换到n2时,氨的生产率变化不大,但是,氨生产率随着时间的推移而降低。使用碳负载的氮化fe催化剂-n-fe/c(2m)获得高氨生产率,1.2v时的电化学生产率约为初始非电化学生产率的10倍,生产率达到1.8x10-8mol/(cm2·s),进料气体从h2/n2切换到n2后,生产率降低,但当进料气体切换回n2/h2时无法恢复。通过氮化铁催化剂-n-fe显示约32倍的电化学氨生成,氨生产率在1.2v时高达5.0×10-8mol/(cm2·s),在1.5v时增加到7.6×10-8mol/(cm2·s),然而,在1.85v下,生产率降低至7.6×10-10mol/(cm2·s)。生产率降低可能是由于催化剂随时间而不是电压增加而失活,但可以说,通过增加电压不能保持高生产率。表7.表6中列出的催化剂的电化学反应测试结果该实施例证明,使用本文公开的电化学电池构造和本文公开的新催化剂可以获得高氨生产率。可以进一步优化在这些说明性实施例中呈现的催化剂,电化学电池制造和反应器设计和操作过程。在催化和催化工艺开发领域中已知的是,一旦发现新的催化化学和/或反应工艺概念,通过优化催化剂,反应器配置和工艺条件可以显着改善催化反应性能。尽管已经显示出和描述了本发明的若干实施例,但是对于本领域技术人员来说显而易见的是,在不脱离本发明的更广泛方面的情况下,可以进行许多改变和修改。因此,所附权利要求旨在涵盖所有这些变化和修改,因为它们落入本发明的真实精神和范围内。以下文献介绍了多孔金属板、电极和氨合成的制备方法,并在此引用文献:shimingchen,siglindaperathoner,claudioampelli,chalachewmebrahtu,dangshengsu和gabrielecenti“在室温和大气压下从水和氮气在碳纳米管基催化剂上电催化合成氨”angew.chem.int.ed.2017,56,2699-2703。stuartlicht,cuocochencui,baohuiwang,fangfang-fang,jasonlau,shuzhiliu通过n2和蒸汽电解在纳米级fe2o3的熔融氢氧化物悬浮液中合成氨”science,vol.345,issue6197,pp637-640(2014)。w.liu,n.l.canfield,andx.li.“薄的多孔金属板及其制造方法”美国专利9,079,136(2011年2月23日提交).p.millet,s.grigoriev“可再生氢技术生产,净化,储存,应用和安全”第2章“水电解技术”l.m.gandi'a,g.arzamendi,p.m.die'guez编辑,elsevier,2013。j.cheng,j.t.zhu,y.pan,andc.lu“硫-镍泡沫作为锂硫电池的正极材料”ecselectrochemistryletters4(2):a19-a21,2014.c.yoo,h.yoon,j.yu,j.h.joo,j.kim合成氨的装置”美国专利2016/0138176a1(2016年5月19日).jiridivisek,petermalinowski“用于碱性电解的隔膜和用于制造隔膜的方法”美国专利4636291a(1987年1月13日)。ph.vermeiren,w.adriansens,j.p.moreels,r.leysen。章节“用于碱性水电解的复合分离器”载于“氢能:理论和工程解决方案“t.o。saetre编辑,第179-184页,1997。yongjunleng,guangchen,alfonsoj.mendoza,timothyb.tighe,michaela.hickner和wang-yangwang“用碱性膜进行固态水电解”j.am.chem.soc.,134(22),pp9054-9057,2012。cecili'akristi'nkjartansdo'ttir,larsplethnielsen,“开发用于大规模碱性水电解的耐用高效电极”internationaljournalofhydrogenenergy38(2013)8221-8231。kaizeng,dongkezhang“用于制氢和应用的碱性水电解的最新进展”progressinenergyandcombustionscience36(2010)307–326。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1