一种碳纤维表面电泳沉积聚合物微纳粒子制备高性能复合材料的方法与流程
本发明属于复合材料制备技术领域,特别涉及一种碳纤维表面电泳沉积聚合物微纳粒子制备高性能复合材料的方法。
背景技术:
碳纤维(cf)增强复合材料因其性能优异而在航空航天、轨道交通、汽车等领域广泛应用。cf增强聚合物复合材料中决定其性能的关键是cf与聚合物基材间的界面粘附强度,由于cf表面化学惰性、表面自由能低、与复合材料基体的相容性差、界面黏附力低,造成复合材料的界面剪切强度、层间剪切强度等性能较差,影响复合材料的力学性能和使用寿命[pui-yanhung,kin-taklau,bronwynfox,nisharhameed,joongheelee,davidhui.surfacemodificationofcarbonfibreusinggraphenerelatedmaterialsformultifunctionalcomposites[j].compositespartb,2018,133:240-257]。
为提高cf-聚合物基体间的界面粘附性,研究者们尝试了许多cf表面改性方法,包括物理和化学改性法,如:气相沉积法和氧化法、涂层法、等离子体法、微波辐射法等,其中化学改性方法效果明显。cf表面化学改性主要涉及改变纤维表面化学组成和提高表面粗糙度。改变纤维表面组成:主要是向纤维表面引入可与基材发生相互作用或反应的活性官能基团,如羟基、羧基、环氧等基团;也可向纤维表面引入线性分子链、支化、或超支化大分子等[caifengwang,leichen,junli,yudonghuang.enhancingtheinterfacialstrengthofcarbonfiberreinforcedepoxycompositesbygreengraftingofpoly(oxypropylene)diamines[j].composites:parta,2017,99:548-557;bogao,jingzhang,lujiehuo,ruliangzhang.in-situmodificationofcarbonfiberswithhyperbranchedpolyglycerolviaanionicring-openingpolymerizationforuseinhigh-performancecomposites[j].carbon,2017,123:548-557]。提高纤维表面粗糙度:增加表面粗糙度,增加比表面积,可提高树脂浸润纤维的接触面积,同时可在碳纤维/树脂间产生机械锁链作用,从而改善界面粘合作用、提高碳纤维与基体树脂间的界面剪切强度、提高cf复合材料力学性能。
cf表面引入活性基团或分子链改变纤维表面组成时,也可增加表面粗糙度,但增加的程度有限。为有效增大纤维表面粗糙度,可将无机、有机、有机-无机复合微纳米粒子、线、棒等3d结构吸附或化学键连固定在cf表面,对纤维表面进行重构,获得表面三维结构,有效提高cf表面活性和比表面积,从而大幅提高cf/树脂界面粘合性,获得高性能复合材料。例如:采用化学接枝方法,首先对cf进行硝酸酸化等处理,使纤维表面带上-cooh、-oh等官能基团;利用硅烷偶联剂kh550-nh2处理sio2纳米粒子(nps),使粒子表面含-nh2;然后通过cf表面羧基和sio2nps表面氨基反应,在cf表面固定sio2nps,由此使复合材料界面剪切张力(ilss)和层间界面剪切张力(ifss)分别提高53.27%和40.92%,冲击强度提高34.95%(guangshunwu,lichunma,huajiang,liliu.improvingtheinterfacialstrengthofsiliconeresincompositesbychemicallygraftingsilicananoparticlesoncarbonfiber.[j].compositesscienceandtechnology,2017,153:160-167)。同样利用化学接枝方法:首先利用硅烷偶联剂kh590(-sh)处理cf,使纤维表面含-sh;利用硅烷偶联剂kh550(-c=c)处理tio2nps,使粒子表面含-c=c;然后在uv光照下通过-sh与-c=c点击反应使纤维表面接枝一层纳米粒子,所得环氧树脂基复合材料层间界面剪切强度提高78%,弯曲强度、拉伸强度分别提高32.3%和39.6%(leixiong,fengzhan,hongboliang,liangchen,daosonglan.chemicalgraftingofnano-tio2ontocarbonfiberviathiol–eneclickchemistryanditseffectontheinterfacialandmechanicalpropertiesofcarbonfiber/epoxycomposites[j].jmatersci,2018,53:2594–2603).
此外,在cf表面形成金属-有机骨架(mof)结构也是一种有效的表面结构重建改性方法。如在硝酸氧化处理cf表面原位生长出纳米多孔mof结构(uio-66-nh2),在碳纤维与树脂基体间形成一种新的界面,缓冲内/外作用力,改性纤维表面能和拉伸强度分别达到83.79mn/m和5.09gpa,分别提高102%和11.6%,复合材料的ilss提高了50.2%。(yangxb,jiangx,huangyd,etal.buildingnanoporousmetal-organicframeworks“armor”onfibersforhigh-performancecompositematerials.[j].acsappliedmaterials&interfaces,2017,9(6):5590-5599.)
将cf经浓hno3氧化处理(cf表面产生-oh和-cooh)后与六氯环三聚膦腈(hexachlorocyclotriphosphazene(hccp)和三乙胺(tea)反应形成hccp功能化cf,再与双(4-羟基苯基)砜(bis(4-hydroxyphenyl)sulfone,bps)、或4,4′-二氨基二苯醚(4,4’-oxydianiline,oda)反应、原位交联形成有机-无机杂化材料-聚膦腈细小微粒(pzsms),或使cf表面形成含过量活性氨的聚膦腈共聚物涂层-poly(ccp-co-oda),从而使单丝拉伸强度、cf与聚合物基材间的界面剪切强度(ifss)明显提高。(xiangchena,haibingxua,dongliua,chunyana,yingdanzhua.afacileone-potfabricationofpolyphosphazenemicrosphere/carbonfiberhybridreinforcementanditseffectontheinterfacialadhesionofepoxycomposites.appliedsurfacescience410(2017)530–539;xiaoqingzhang,haibingxuandxinyufan.graftingofamine-cappedcross-linkedpolyphosphazenesontocarbonfibersurfaces:anovelcouplingagentforfiberreinforcedcomposites.rscadv.,2014,4,12198-12205;haibingxu,xiaoqingzhang,dongliu,chunyan,xiangchen,davidhui,yingdanzhu.cyclomatrix-typepolyphosphazenecoating:improvinginterfacialpropertyofcarbonfiber/epoxycompositesandpreservingfibertensilestrength.compositespartb93(2016)244-251)。上述cf表面改性方法,使用强酸或强氧化剂处理cf法,常会在cf表面形成缺陷,使cf单丝拉伸强度有所降低。此外,上述改性方法大多需经历多个步骤,且各步骤反应条件严苛,如需高温和除氧环境,反应时间长(数小时)等。
技术实现要素:
本发明的目的在于克服上述现有技术的不足,提供一种碳纤维表面电泳沉积功能聚合物微球提高界面性能的方法,首先,利用多巴胺(da)处理cf:da在cf表面沉积、聚合,形成聚多巴胺(pda),以提高cf表面对聚合物粒子的粘结性;然后,通过电泳在cf表面沉积不同形貌、粒径、组成、表面含不同官能基团的聚合物微球,以提高cf与聚合物基材的界面性能。
本发明所述的碳纤维表面电泳沉积聚合物微纳粒子制备高性能复合材料的方法为:
1)cf基材表面预处理:
a)清洗:cf基材用丙酮抽提,然后超声清洗,并用乙醇和去离子水冲洗,放入烘箱干燥;
b)cf基材表面沉积聚多巴胺:将步骤a处理后的cf基材浸入浓度为1-4mg/ml的多巴胺盐酸盐的tris缓冲液中,ph值为8.5,室温静置6-12h,取出cf基材用乙醇或去离子水冲洗至溶液澄清,然后放入烘箱50-80℃干燥备用;
2)cf基材表面电泳沉积聚合物微纳粒子:
a)配制聚合物微纳粒子分散液;
b)将cf基材用导电胶固定在一铝板上,插入电泳容器,与另一铝板平行相对间隔设置,两铝板接相反电极,向烧杯中加入聚合物微纳粒子分散液,浸没cf基材进行电泳过程后,取出,室温真空干燥。
所述cf基材是丝束或织布。
所述聚合物微纳粒子为聚苯乙烯或聚苯乙烯共聚物。所述聚苯乙烯共聚物表面含有羧基、环氧基、羟基、琥珀酰亚胺基、双键基团、光敏二苯甲酮基团或-s-s-键中的至少一种,且聚合物微纳粒子表面带有负电荷。
所述聚合物微纳粒子是由含巯基、环氧基团、异氰酸酯基团中的一种或几种的单体点击聚合得到的聚合物微纳粒子,聚合物微纳粒子表面含有-sh、-nco、或环氧基团中的至少一种。
所述点击聚合时还加入了功能化合物。所述功能化合物包括荧光剂或功能药物。
所述聚合物微纳粒子的粒径为70nm-3.5μm;聚合物微纳粒子在分散介质中的浓度为0.032-0.041g/ml。
所述聚合物微纳粒子是不交联或交联的聚合物。
所述聚合物微纳粒子是实心、核-壳或空心结构;所述聚合物微纳粒子是表面光滑或粗糙的球形粒子或异形粒子。
所述电泳过程所用电压为5-20v,电泳时间为30s-120min。
所述聚合物微纳粒子分散液的分散介质是水、醇类溶剂或水与醇类溶剂的混合物。
所述醇类溶剂或水与醇类溶剂的混合物中醇与水体积比为1:1。
本发明利用电泳方法,将不同组成、形状、表面官能基团及形貌的聚合物粒子沉积吸附到聚多巴胺(pda)改性cf表面,提高cf与聚合物基材间物理机械互锁、化学键连等相互作用,从而提高界面粘附强度。tfbt测试表明:界面拉伸强度(相比未改性cf)提高了40-144%,单丝微滴脱粘法测定表明ifss提高了30-56%。
附图说明
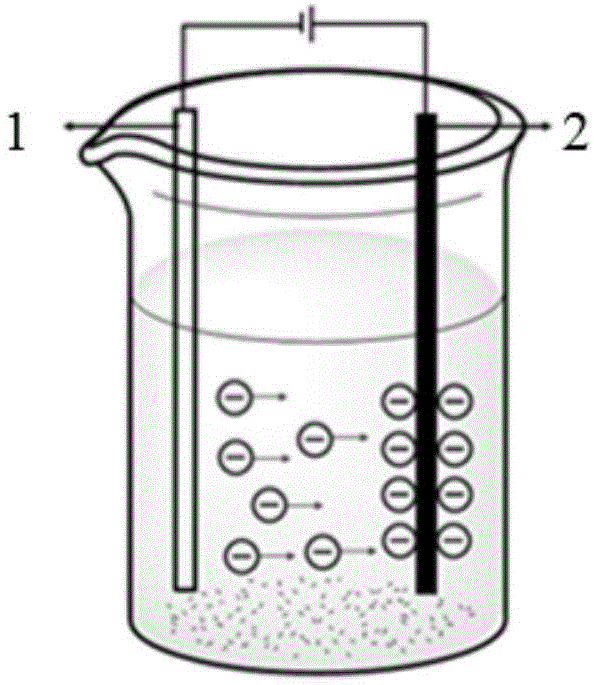
图1是电泳装置示意图,其中,1-铝板电极,2-碳纤维电极。
图2-1中,a)poly(st-co-aa)微球(70nm),b)poly(st-co-nsma)微球(98nm)的sem照片;
图2-2中,poly(st-co-gma)微球的sem、tem照片(右上),gma的mol%含量为:a-0,b-5%,c-10%,d-15%,e-20%,f-30%,g-35%,h-40%,i-50%),标尺为200nm,粒径120-230nm;
图2-3中,poly(st-co-gma)微球的sem、tem照片(右上),gma的mol%含量为:a-0,b-5%,c-10%,d-20%,e-30%,f-40%,标尺为400nm,粒径450-510nm;
图2-4中,pst微球(a),poly(st-co-dvb)微球(b,c,d)的sem、tem照片(右上),dvb含量:(b)1%,(c)5%,(d)8%,(e)10%,标尺500nm,粒径350-480nm;
图2-5中,poly(st-co-dvb-co-gma)微球的sem照片,dvb5%,gma15%,标尺为500nm,粒径550-710nm;
图2-6中,poly(st-co-dmabp)微球的sem、tem(右上、下,thf处理前后)照片,dmabp用量(a1%,b2%,c3%,d4%,e6%),sem照片标尺为500nm,tem照片标尺为200nm,粒径490-530nm;
图2-7中,poly(st-co-dsdma)微球的sem照片,dsdma5%,标尺为500nm,粒径70-610nm;
图2-8中,巯基(petmp,4-sh)-异氰酸酯(ldi+ipdi,2-nco)点击聚合物微球的sem照片,a-sh/nco=4/4,粒径1.51μm,b-sh/nco=6/6,粒径3μm;
图2-9中,巯基(petmp,4-sh)-环氧(tgmda,4-环氧)点击聚合物微球的sem照片,a-巯基/环氧=3/3,b-巯基/环氧=6/6,粒径260-610nm。
图3是碳纤维表面电泳沉积、吸附聚合物微球sem照片。
图4是cf-环氧复合材料断面sem照片,其中,a)未改性cf,b)cf-poly(st-co-gma)微球(90nm),c)cf-poly(st-co-gma)微球(270nm),d)cf-poly(st-co-gma)微球-580nm,e)cf-poly(st-co-gma)微球-920nm,标尺为3um。
图5是cf单丝-环氧微滴脱粘法测试ifss失效断面sem照片,其中,a)测试前cf单丝-环氧微滴,b)未改性cf-环氧微滴失效断面,c)cf-poly(st-co-gma)微球(90nm)、d)cf-poly(st-co-gma)微球(270nm)、e)cf-poly(st-co-gma)微球-580nm、f)cf-poly(st-co-gma)微球-920nm-环氧微滴失效断面,标尺为3um。
具体实施方式
为使本领域的技术人员更好地理解本发明的技术方案,下面结合实施例对本发明提供的一种碳纤维表面电泳沉积聚合物微纳粒子制备高性能复合材料的方法,进行详细描述。以下实施例仅用于说明本发明而非用于限制本发明的范围。
以下实施例中采用的碳纤维表面电泳沉积聚合物微纳粒子制备高性能复合材料的方法包括以下步骤:
1)cf表面预处理:
a)清洗:cf基材用丙酮抽提24h,然后超声清洗3次,并用乙醇和去离子水冲洗3次,放入烘箱50℃干燥;
b)cf基材表面沉积聚多巴胺(pda):将步骤a处理后的cf基材浸入含多巴胺盐酸盐(浓度1mg/ml)的tris缓冲液中(ph8.5),室温静置12h,取出cf基材用乙醇冲洗3次至溶液澄清,将cf放入烘箱70℃干燥备用;
2)cf表面电泳沉积聚合物微纳粒子:
a)配制聚合物微纳粒子分散液;
b)将cf基材用导电胶固定在一铝板上,插入250ml电泳烧杯,此板接电源正极或负极,另一铝板接电源负极或正极,使两板平行相对,相距3cm,装置见附图1;向烧杯中加入聚合物微纳粒子分散液,浸没碳纤维丝束或织布,在一定电压下电泳一段时间,取出,室温、真空干燥。
上述过程所用cf基材可以是丝束,每丝束含12000根cf单丝,长约40cm,也可以是cf织布,一般约为4×3cm2,实际过程可以更大或任意尺寸。
上述所用聚合物微纳粒子为聚苯乙烯(pst)及其共聚物微球,包括苯乙烯(st)与(甲基)丙烯酸及其脂类单体,如丙烯酸(aa)、甲基丙烯酸(maa)、甲基丙烯酸缩水甘油酯(gma)、甲基丙烯酸羟乙酯(hema)、甲基丙烯酸n-琥珀酰亚胺酯(nsma),以及与交联剂,如二乙烯基苯(dvb)、二甲基丙烯酸乙二醇酯(egdma)、双-甲基丙烯酸二苯甲酮酯(dmabp)、双(2-甲基丙烯)乙氧基二硫(dsdma)等共聚形成的微球。根据共聚单体的不同,上述微球表面含有羧基、环氧基、羟基、琥珀酰亚胺基或双键、光敏二苯甲酮基团或-s-s-键,微球表面带有负电荷;
上述所用聚合物微纳粒子也可以是含巯基、环氧基团、异氰酸酯基团中的一种或几种的单体点击聚合得到的聚合物微纳粒子,点击聚合制备聚合物微纳粒子所用的含巯基(-sh)单体为含3-6个-sh的单体,如:三羟甲基丙烷三(3-巯基丙酸酯),tmpmp,3-sh)、四(3-巯基丙酸)季戊四醇酯(petmp,4-sh)、六(3-巯基丙酸)二季戊四醇酯(dpmp,6-sh)等;所用含环氧单体为含2-4个环氧基团的单体,如:3,4-环氧环己基甲酸酯(erl4211,2环氧基)、三羟甲基丙烷三缩水甘油醚(tmtge,3环氧基)、三(2,3-环氧丙基)异氰尿酸酯(tgic,3环氧基)、n,n,n',n'-四环氧丙基-4,4'-二氨基二苯甲烷(tgmda,4环氧基)等;所用含异氰酸酯(-nco)单体为含2-3个-nco基团的单体,如甲苯二异氰酸酯(tdi,2-nco)、二苯基甲烷二异氰酸酯(mdi,2-nco)、二甲基联苯二异氰酸酯(4,4'-二异氰酸-3,3'-二甲基联苯,todi,2-nco)、异佛尔酮二异氰酸酯(ipdi,2-nco)、二环己基甲烷二异氰酸酯(hmdi,2-nco)、六亚甲基二异氰酸酯(hdi,2-nco)、赖氨酸二异氰酸酯(ldi,2-nco);聚六亚甲基二异氰酸酯(phdi,oligomer,3-nco)、三苯甲烷三异氰酸酯(tpmti,3-nco)、l-赖氨酸乙酯三异氰酸酯(lti,3-nco)等。上述所说点击聚合物微球表面含有-sh、或-nco、或环氧基团。
上述电泳所用聚合物微纳粒子可以是不交联或交联的,可以是球形粒子,也可以是异形粒子,如二或三部分、多凸起或凹陷的微球,微球可以是实心、核-壳或空心的,微球可具有光滑、或粗糙表面;
上述电泳所用聚合物微纳粒子的粒径为70nm~3.5μm,聚合物微纳粒子在分散介质中的浓度为0.032~0.041g/ml。
上述电泳聚合物微纳粒子的分散介质可以是水或醇类溶剂,或水与醇类溶剂的混合物,醇/水比为1/1。醇类溶剂包括甲醇或乙醇中的至少一种。
上述电泳过程所用电压为5-20v,电泳时间为30s-120min。
使用hitachis-4700型扫描电子显微镜(sem)观察cf表面粒子、cf与基材粘附状况,使用横向纤维束拉伸(transversefiberbundletensiletest,tfbttest)试验(qigc,zhangbm,yuyl.researchoncarbonfiber/epoxyinterfacialbondingcharacterizationoftransversefiberbundlecompositesfabricatedbydifferentpreparationprocesses:effectoffibervolumefraction.polymertesting2016(52)150-156)评价纤维和基材环氧树脂间的界面粘附强度(interfacialbondingstrength),也可使用单丝微滴脱粘法(mayy,yanc,xuhb,liud,shipc,zhuyd,liujl.enhancedinterfacialpropertiesofcarbonfiberreinforcedpolyamide6compositesbygraftinggrapheneoxideontofibersurface.appl.surf.sci.,2018(452)286-298)测定纤维与环氧树脂基材间的界面剪切强度(ifss)。
实施例1
将苯乙烯(st)与丙烯酸(aa)共聚微球(poly(st-co-aa,不交联),粒径70nm,表面光滑、球形、含羧基)分散在水中,形成浓度为0.032g/ml分散液;在附图1所示电泳装置中,铝板接电源负极,碳纤维(cf)丝束接电源正极,加入聚合物微球分散液,进行电泳沉积,电压为5v,电泳时间120min,然后关闭电源,取出cf丝束,在真空烘箱备中、室温干燥,待用。
实施例2
如实施例1,只是所用聚合物微球为苯乙烯(st)与甲基丙烯酸(aa)共聚微球(poly(st-co-maa),粒径81nm,表面光滑、球形、含羧基);电泳电压为10v,电泳时间30min。
实施例3
如实施例1,只是所用聚合物微球为苯乙烯(st)与甲基丙烯酸n-琥珀酰亚胺酯(nsma)共聚微球(poly(st-co-nsma),粒径98nm,表面略微粗糙、球形、含琥珀酰亚胺基团);电泳电压为15v,电泳时间10min。
实施例4
如实施例1,只是所用聚合物微球为苯乙烯(st)与甲基丙烯酸羟乙酯(hema)共聚微球(poly(st-co-hema),135nm,表面光滑、球形、含羟基基团);电泳电压为20v,电泳时间3min。
实施例5
如实施例1,只是所用聚合物微球为苯乙烯(st)与甲基丙烯酸缩水甘油酯(gma)共聚微球(poly(st-co-gma),180nm,表面多凸起、球形、含环氧基团);电泳电压为20v,电泳时间30s。
实施例6
如实施例1,只是所用聚合物微球为苯乙烯(st)与甲基丙烯酸缩水甘油酯(gma)共聚微球(poly(st-co-gma),230nm,两、三部分不规则微球、含环氧基团),微球分散在甲醇和水的混合液中,甲醇/水=1/1;电泳电压为20v,电泳时间3min。
实施例7
如实施例1,只是所用聚合物微球为苯乙烯(st)与甲基丙烯酸缩水甘油酯(gma)共聚微球(poly(st-co-gma),510nm,多凸起异形微球、含环氧基团),微球分散在甲醇和水的混合液中,甲醇/水=1/1;电泳电压为20v,电泳时间5min。
实施例8
如实施例1,只是所用聚合物微球为苯乙烯(st)与二乙烯基苯(dvb)共聚微球(poly(st-co-dvb),350nm,交联微球,表面光滑且含双键基团),微球分散在乙醇和水的混合液中,乙醇/水=1/1;电泳电压为5v,电泳时间60min。
实施例9
如实施例1,只是所用聚合物微球为苯乙烯(st)与二乙烯基苯(dvb)共聚微球(poly(st-co-dvb),400nm,交联微球,表面光滑且含双键基团,具有核-壳结构),微球分散在乙醇和水的混合液中,乙醇/水=1/1;电泳电压为10v,电泳时间15min。
实施例10
如实施例1,只是所用聚合物微球为苯乙烯(st)、二乙烯基苯(dvb)及甲基丙烯酸缩水甘油酯(gma)共聚微球(poly(st-co-dvb-co-gma),610nm,部分交联、雪人状(头部光滑、不交联,身体粗糙、交联且含环氧和双键基团),微球分散在乙醇和水的混合液中,乙醇/水=1/1;电泳电压为20v,电泳时间10min。
实施例11
如实施例1,只是所用聚合物微球为苯乙烯(st)、二乙烯基苯(dvb)及甲基丙烯酸缩水甘油酯(gma)共聚微球(poly(st-co-dvb-co-gma),680nm,交联、凹陷、含环氧和双键基团,微球分散在乙醇和水的混合液中,乙醇/水=1/1;电泳电压为20v,电泳时间10min。
实施例12
如实施例1,只是所用聚合物微球为苯乙烯(st)与二甲基丙烯酸乙二醇酯(egdma)共聚微球(poly(st-co-egdma),570nm,交联、凹陷、表面粗糙、含双键基团,微球分散在乙醇和水的混合液中,乙醇/水=1/1;电泳电压为20v,电泳时间5min。
实施例13
如实施例1,只是所用聚合物微球为苯乙烯(st)与双-甲基丙烯酸二苯甲酮酯(dmabp)共聚微球(poly(st-co-dmabp),510nm,微球呈雪人状(头部粗糙、交联、含光敏基团,身体光滑、不交联),分散在乙醇和水的混合液中,乙醇/水=1/1;电泳电压为20v,电泳时间5min。
实施例14
如实施例1,只是所用聚合物微球为苯乙烯(st)与双(2-甲基丙烯)乙氧基二硫(dsdma)共聚微球(poly(st-co-dsdma),105nm,微球交联、表面光滑,分散在水中;电泳电压为20v,电泳时间5min。
实施例15
如实施例1,只是所用聚合物微球为苯乙烯(st)与双(2-甲基丙烯)乙氧基二硫(dsdma)共聚微球(poly(st-co-dsdma),270nm,微球为核-壳结构、壳层交联、表面多凸起、含-s-s-键,分散在水中;电泳电压为20v,电泳时间5min。
实施例16
将三羟甲基丙烷三(3-巯基丙酸酯)(tmpmp)与异佛尔酮二异氰酸酯(ipdi)点击分散聚合所得微球(2μm,表面粗糙)分散在水中,形成浓度为0.041g/ml分散液;在附图1所示电泳装置中,铝板接电源负极,碳纤维(cf)丝束或织布接电源正极,加入聚合物微球分散液,进行电泳沉积,电压为20v,电泳时间5min,然后关闭电源,取出cf丝束或织布,在真空烘箱备中、室温干燥,待用。
实施例17
如实施例16,所用聚合物微球为四(3-巯基丙酸)季戊四醇酯(petmp)与异佛尔酮二异氰酸酯(ipdi)点击分散聚合所得微球,粒径1.51μm,表面粗糙、多凸起。
实施例18
如实施例16,所用聚合物微球为四(3-巯基丙酸)季戊四醇酯(petmp)与二甲基联苯二异氰酸酯(todi)点击分散聚合所得微球,粒径3.5μm,表面光滑。
实施例19
如实施例16,所用聚合物微球为三羟甲基丙烷三(3-巯基丙酸酯)(tmpmp)与甲苯二异氰酸酯(tdi)点击分散聚合所得微球,粒径1.6μm,表面粗糙。
实施例20
如实施例16,所用聚合物微球为三羟甲基丙烷三(3-巯基丙酸酯)(tmpmp)、荧光试剂7-巯基-4-甲基香豆素与二苯基甲烷二异氰酸酯(mdi)点击分散聚合所得微球,粒径1.34μm,表面粗糙。
实施例21
如实施例16,所用聚合物微球为四(3-巯基丙酸)季戊四醇酯(petmp)与二环己基甲烷二异氰酸酯(hmdi)点击分散聚合所得微球,粒径1.14μm,表面粗糙。
实施例22
如实施例16,所用聚合物微球为四(3-巯基丙酸)季戊四醇酯(petmp)与三苯甲烷三异氰酸酯(tpmti)、l-赖氨酸乙酯三异氰酸酯(lti)点击分散聚合所得微球,粒径350nm,表面粗糙、含过量巯基。
实施例23
如实施例16,所用聚合物微球为四(3-巯基丙酸)季戊四醇酯(petmp)与异佛尔酮二异氰酸酯(ipdi)、六亚甲基二异氰酸酯(hdi)点击分散聚合所得微球,粒径3.1μm,表面粗糙、含过量巯基。
实施例24
如实施例16,所用聚合物微球为四(3-巯基丙酸)季戊四醇酯(petmp)与异佛尔酮二异氰酸酯(ipdi)、聚六亚甲基二异氰酸酯(phdi)点击分散聚合所得微球,粒径3.46μm,表面粗糙、含过量巯基。
实施例25
如实施例16,所用聚合物微球为四(3-巯基丙酸)季戊四醇酯(petmp)与异佛尔酮二异氰酸酯(ipdi)、赖氨酸二异氰酸酯(ldi)、阿霉素(dox)点击分散聚合所得微球,粒径430nm,表面粗糙、含过量巯基。
实施例26
将三羟甲基丙烷三(3-巯基丙酸酯)(tmpmp)与三(2,3-环氧丙基)异氰尿酸酯(tgic)点击分散聚合所得微球(320nm,表面光滑)分散在水中,形成浓度为0.041g/ml分散液;在附图1所示电泳装置中,铝板接电源负极,碳纤维(cf)丝束或织布接电源正极,加入聚合物微球分散液,进行电泳沉积,电压为20v,电泳时间5min,然后关闭电源,取出cf丝束或织布,在真空烘箱备中、室温干燥,待用。
实施例27
如实施例26,所用聚合物微球为四(3-巯基丙酸)季戊四醇酯(petmp)与三羟甲基丙烷三缩水甘油醚(tmtge)点击分散聚合所得微球,粒径510nm,表面光滑。
实施例28
如实施例26,所用聚合物微球为四(3-巯基丙酸)季戊四醇酯(petmp)与三n,n,n',n'-四环氧丙基-4,4'-二氨基二苯甲烷(tgmda)、3,4-环氧环己基甲酸酯(erl4211)点击分散聚合所得微球,粒径610nm,表面光滑、含过量巯基。
上面结合实施例对本发明的实例作了详细说明,但是本发明并不限于上述实例,在本领域普通技术人员所具备的知识范围内,还可以在不脱离本发明宗旨的前提下作出的各种变化,也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!