一种电催化还原二氧化碳电极的制备方法及应用与流程
[0001]
本发明属于能源电化学领域,涉及一种电催化还原co2电极的制备方法及其应用。
背景技术:
[0002]
随着经济的增长,化石燃料的大量燃烧导致了co2的过度排放,进而也引发了海平面上升、冰川融化和全球变暖等环境问题。把co2转化成燃料,不仅可以降低大气中的co2浓度,也可缓解能源短缺的严峻形势。co2的电催化还原可利用风能、太阳能和潮汐能等可再生能源提供电能,在较温和的反应条件下,把co2转化成高附加值的燃料,且反应速率易控制,有望解决co2的转化利用和间歇性能源的储存问题。
[0003]
co2电催化还原反应的产物选择性较低,产物包括co、hcooh、ch3oh、ch4、c2h4、c2h5oh和副产物h2等。其中,多碳产物具有较高的能量密度和经济价值,近年来引起了研究者的广泛关注。
[0004]
目前,co2电催化还原生成多碳产物的研究,主要围绕铜、铜的氧化物和铜基分子催化剂等展开。jeon等制备出具有棱镜形貌的纳米铜,铜表面高密度的缺陷位可促进c2产物的生成。在-1.0v(vs.rhe)时,c2h4的电流密度和法拉第效率(fe)分别可达28.6ma/cm2和27.8%,但cu纳米颗粒易氧化团聚导致形貌和缺陷位数量的改变(acs catal.,2018,8,531-535)。ren等以铜片为基底,通过控制电沉积的时间和电流密度在其表面形成不同厚度的cu2o薄膜。当电解电位为-0.99v(vs.rhe)时,厚度为1.7-3.6μm的薄膜c2产物的选择性(fe
c2h4
:34-39%;fe
c2h5oh
:9-16%)最高。但操作较复杂,实现对薄膜厚度和组分的精确控制难度较大(acs catal.,2015,5,2814-2821)。weng等报道了采用2,6-二甲氧基苯甲醛和吡咯合成的5,10,15,20-四(2,6-二羟苯基)铜卟啉,在-0.976v(vs.rhe)时,co2电催化还原产物中ch4和c2h4的法拉第效率分别为27%和17%,两者的电流密度之和为21ma/cm2。但合成工艺中使用较多有毒有害的化学试剂,耗时长(j.am.chem.soc.,2016,138,8076-8079)。naonari等采用cubr、三苯基膦、4,4-联吡啶、丙酮、乙腈合成铜配位聚合物纳米催化剂,在-1.34v(vs.rhe)时,电流密度为40ma/cm2,c2h4和c2h5oh的法拉第效率分别为19.7%和33.5%。但反应的过电位高,电流密度低(acs catal.,2020,10,10412-10419)。
[0005]
综上所述,已报道的可利用co2电催化还原为多碳产物的催化剂还需以下改进:(1)简化制备工艺,降低成本;(2)降低反应的过电位;(3)提高c2产物的选择性。
技术实现要素:
[0006]
本发明的目的是提供一种电催化还原co2电极的制备方法及应用,该方法操作简单、易于控制、条件温和、环境友好。
[0007]
本发明的技术方案:
[0008]
本发明一方面提供了一种电极的制备方法,所述方法包括如下步骤:
[0009]
(1)将碳材料和铜基大环化合物超声分散于溶剂中。在20~60℃下搅拌3~10h,旋蒸除去溶剂,烘干,之后在h2、he、n2、ar、nh3中的一种或两种以上混合气氛中,200~900℃下
热处理1~5h,得到铜基大环化合物/碳电催化剂;
[0010]
(2)在20~60℃下,把铜基底材料置于乙醇、异丙醇、丙酮、稀盐酸中的一种或两种以上的混合溶剂中浸泡10~40min,然后用去离子水清洗后真空干燥,裁切备用;
[0011]
(3)取所述铜基大环化合物/碳电催化剂,依次加入水、醇和nafion后超声分散0.1~1h,配成催化剂浆液,涂于步骤(2)处理后的铜基底上,得到载有电催化剂的铜电极;
[0012]
(4)把所述载有电催化剂的铜电极在h2、he、n2、ar、nh3中的一种或两种以上混合气氛中,在500~1000℃下热处理1~5h,得到所述电极。
[0013]
上述步骤(1)和步骤(2)不分先后顺序。
[0014]
优选地,所述的碳材料在溶液中的浓度为0.1~10mg/ml;所述的铜基大环化合物的质量与碳材料的质量比大于0.1。
[0015]
优选地,所述的碳材料包括碳黑、碳纳米管、活性碳、碳纤维、石墨烯中的一种或两种以上的混合物;所述的铜基大环化合物包括铜酞菁和铜卟啉及其衍生物和类似物,为5,10,15,20-四(4-羧基苯基)铜卟啉、原卟啉氯化铜、meso-四(4-苯磺酸基)卟啉铜、四甲氧基苯基卟啉铜、铜卟啉、铜肽菁、四磺酸基四钠盐铜酞菁、meso-四(4-苯磺酸基)酞菁铜、邻菲罗啉铜中的一种或两种以上的混合物。
[0016]
优选地,所述的铜基底材料包括泡沫铜和打磨光亮的铜片。
[0017]
优选地,所述的步骤(1)中的溶剂为水、甲醇、乙醇、乙二醇、丙醇、异丙醇、二氯甲烷、n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dmac)中的一种或两种以上的混合物;所述的步骤(3)中的醇为甲醇、乙醇、丙醇、异丙醇中的一种或两种以上的混合物。
[0018]
优选地,所述的浆液中电催化剂浓度为0.1~5mg/ml;所述的铜电极上电催化剂的载量为0.1~5mg/cm2。
[0019]
优选地,所述的把催化剂浆液涂于铜基底的方法包括刷涂和喷涂。
[0020]
另一方面,本发明提供了一种电极,所述电极通过上述的方法制备。
[0021]
再一方面,本发明将上述电极用于电催化还原二氧化碳反应。
[0022]
优选地,以所述电极为工作电极,阴极;以石墨或铂电极作为对电极,阳极;以hg/hgo电极或饱和甘汞电极作为参比电极;在25℃下,采用h型电解池进行性能测试,两室中间以nafion膜隔开,两室电解质溶液体积均为100ml;阳极侧通入he、n2、ar中的一种或两种以上的混合气氛,阴极侧通入原料气;在测试前,两侧均通气至溶液饱和。
[0023]
优选地,所述的电解质溶液为0.1~1.0m的khco3、kcl、k2so4、ki、kcs、kbr、kf、kclo4、koh中的一种或两种以上的混合物。
[0024]
优选地,所述的原料气为纯co2或co2与h2、o2、n2、nh3、ar、he中的一种或几种的混合气氛;原料气中co2的体积分数为50~100%。
[0025]
优选地,所述的阴极和阳极两侧通入气体的气速相同,均为20~50ml/min。
[0026]
有益效果
[0027]
1)采用铜片和泡沫铜等作为基底材料,摒弃了溅射、电镀、化学气相沉积等复杂的工作电极制备工艺。制备催化剂的原料较廉价,反应条件温和,方法简单易行,环境友好,易于规模化生产。通过控制热处理温度、气氛、时长等条件,提高了催化剂的稳定性和导电性。
[0028]
2)制备的工作电极的co2电催化还原活性和在较低电位下的c2产物的选择性高。
附图说明
[0029]
图1本发明实施例1所得铜基大环化合物/碳电催化剂(a)和载体碳黑(b)的透射电镜照片;
[0030]
图2本发明实施例1所得铜基大环化合物/碳工作电极的co2还原反应极化曲线;
[0031]
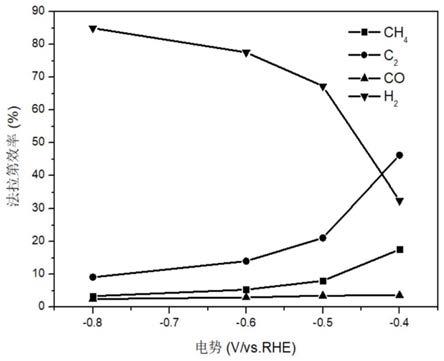
图3本发明实施例1所得铜基大环化合物/碳工作电极在-0.4v(vs.rhe)下的co2还原产物的法拉第效率;
[0032]
图4本发明实施例1在不同处理方法下制得的工作电极的co2还原反应极化曲线对比;
[0033]
图5本发明实施例1在不同处理方法下制得的工作电极在-0.4v(vs.rhe)下的co2还原产物的法拉第效率的对比。
具体实施方式
[0034]
下面结合附图和实施例对本发明作进一步的说明,以下实施例仅仅是为了更加清楚地阐述本发明,但本发明要求保护的范围并不局限于以下实施例表述的范围。
[0035]
实施例1
[0036]
铜基大环化合物/碳电极的制备:
[0037]
(1)将8mg碳黑和5mg原卟啉氯化铜分散于10ml丙醇溶液中,20℃下超声0.5h,在60℃下搅拌3h后,旋蒸除去溶剂,65℃下烘干,最终得到黑色粉末。在he气氛下,300℃热处理2h,得到铜基大环化合物/碳电催化剂;
[0038]
(2)把铜片用砂纸打磨光亮,并在60℃下的异丙醇中浸泡20min,用去离子水清洗后真空干燥,裁切备用;
[0039]
(3)称取5mg上述制得的铜基大环化合物/碳电催化剂,分别加入0.5ml水、5ml乙醇和40μl nafion溶液,混合后超声分散0.5h,配成催化剂浆液,刷涂于上述处理后的铜片上,得到载有电催化剂的铜电极;
[0040]
(4)把上述载有电催化剂的铜电极放在n2中,600℃热处理2h,得到所述电极。
[0041]
铜基大环化合物/碳电极在电催化还原二氧化碳中的应用:
[0042]
在25℃下,co2电催化还原采用h型电解池进行测试。电解质采用0.5m k2so4溶液,两室溶液体积均为100ml,中间以nafion膜隔开。阳极侧通入高纯的n2,阴极侧通入原料气co2,气速均为50ml/min。在测试前,两侧均通气至溶液饱和。用玻碳工作电极夹固定上述铜基大环化合物/碳电极作为阴极,以石墨电极为对电极,以饱和甘汞电极为参比电极。
[0043]
如图1,电镜分析表明铜基大环化合物已均匀地担载在碳黑上,未见明显颗粒团聚。
[0044]
如图2,测定铜基大环化合物/碳工作电极的co2还原性能。测试条件:以10mv/s的扫速,在0.1~-0.8v(vs.rhe)的电压范围内分别在n2和co2中进行电位扫描测试。极化曲线显示实施例1所得铜基大环化合物/碳工作电极具有较高的co2还原催化活性。
[0045]
如图3,铜基大环化合物/碳工作电极在不同电位下co2还原反应中产物选择性的比较。测试条件:在-0.4~-0.8v(vs.rhe)的电压范围内分别进行恒电位电解,进行co2电催化还原的计时电流曲线扫描,检测产物的组成及浓度,并分别计算不同电位下四类产物的法拉第效率。其中,c2产物包括c2h4和少量的c2h6(在电压考察范围内,c2h6的法拉第效率小
于3%)。在-0.4v(vs.rhe)时c2产物的法拉第效率总计达46.2%。
[0046]
对比例1-4
[0047]
对比例1:将铜片按照实施例1中步骤(2)处理,代替实施例1的工作电极;
[0048]
对比例2:将铜片按照实施例1中步骤(2)和步骤(4)进行处理(铜片未载催化剂),代替实施例1的工作电极;
[0049]
对比例3:将铜片按照实施例1中步骤(2)处理,处理后的铜片按照实施例1中步骤(1)的方法仅负载8mg碳黑,然后按照实施例1中步骤(3)和(4)进行处理,代替实施例1的工作电极;
[0050]
对比例4:实施例1不同之处在于,步骤(1)未进行热处理,代替实施例1的工作电极;
[0051]
如图4,采用不同处理方法制得的工作电极的co2还原性能比较。
[0052]
图中copper、h-copper、h-c、h-a、h-a-h-t分别代表光亮铜片(对比例1)、铜片热处理(对比例2)、铜片载碳黑后热处理(对比例3)、铜片载未经热处理的电催化剂后再经热处理(对比例4)及按实施例1所述制成的工作电极。经对比发现,实施例1所得铜基大环化合物/碳工作电极的co2还原的催化活性最高。
[0053]
如图5,在不同处理方法下制得的工作电极在-0.4v(vs.rhe)下的co2还原产物的法拉第效率的对比。与其他电极相比,按实例1所述制成的电极的烃类产物的选择性尤其是c2产物大幅提高,副产物h2的法拉第效率显著降低,仅有32.5%。
[0054]
实施例2
[0055]
将10mg碳纳米管和6mg铜卟啉分散于25ml dmac溶液中,25℃下超声0.5h,在45℃下搅拌7h后,旋蒸除去溶剂,65℃下烘干,最终得到黑色粉末。在h2/ar气氛下,500℃热处理1h,得到铜基大环化合物/碳电催化剂。
[0056]
把泡沫铜置于30℃下的乙醇中浸泡30min,用去离子水清洗后真空干燥,裁切备用。称取5mg上述制得的铜基大环化合物/碳电催化剂,分别加入0.5ml水、2ml丙醇和20μl nafion溶液混合后超声分散0.5h,配成催化剂浆液,刷涂于上述处理后的泡沫铜上,得到载有电催化剂的铜电极。之后把载有电催化剂的铜电极放在he中,700℃热处理3h,得到铜基大环化合物/碳电极。
[0057]
在25℃下,co2电催化还原采用h型电解池进行测试。电解质采用0.1m khco3溶液,两室溶液体积均为100ml,中间以nafion膜隔开。阳极侧通入高纯的ar,阴极侧通入混合原料气co2/ar(体积比:8:2),气速均为30ml/min。在测试前,两侧均通气至溶液饱和。用玻碳工作电极夹固定上述铜基大环化合物/碳电极作为阴极,以铂电极为对电极,以饱和甘汞电极为参比电极。检测在不同电位下电解产物的组成及浓度,并计算其法拉第效率。实施例2所得电催化剂的形貌和工作电极的性能与实施例1相似,具有较好的分散性,无颗粒团聚现象。在-0.5v(vs.rhe)下c2产物(包括c2h4和少量的c2h6,其中c2h6的法拉第效率小于3%)的选择性高达38.0%。
[0058]
实施例3
[0059]
将8mg石墨烯和5mg 5,10,15,20-四(对甲氧基苯基)卟啉铜分散于15ml乙二醇溶液中,30℃下超声0.2h,在50℃下搅拌6h后,旋蒸除去溶剂,65℃下烘干,最终得到黑色粉末。在nh3气氛中,600℃热处理1h,得到铜基大环化合物/碳电催化剂。
[0060]
把铜片用砂纸打磨光亮,并在50℃下的丙酮中浸泡40min,用去离子水清洗后真空干燥,裁切备用。称取5mg上述制得的铜基大环化合物/碳电催化剂,分别加入0.1ml水、1ml乙醇和10μl nafion溶液,混合后超声分散0.2h,配成催化剂浆液,喷涂于上述处理后的铜片上,得到载有电催化剂的铜电极。之后把载有电催化剂的铜电极放在n2中,800℃热处理3h,得到铜基大环化合物/碳电极。
[0061]
在25℃下,co2电催化还原采用h型电解池进行测试。电解质采用0.5m koh溶液,两室溶液体积均为100ml,中间以nafion膜隔开。阳极侧通入高纯的he,阴极侧通入混合原料气co2/n2/o2(体积比:40:7:3),气速均为20ml/min。在测试前,两侧均通气至溶液饱和。用玻碳工作电极夹固定上述铜基大环化合物/碳电极作为阴极,以石墨电极为对电极,以hg/hgo电极为参比电极。检测在不同电位下电解产物的组成及浓度,并计算其法拉第效率。实施例3所得电催化剂的形貌和工作电极的性能与实施例1相似,具有较好的分散性,无颗粒团聚现象。在-0.4v(vs.rhe)下c2产物(包括c2h4和少量的c2h6,其中c2h6的法拉第效率小于3%)的选择性高达40.0%。
[0062]
实施例4
[0063]
将10mg碳纳米管和5mg铜卟啉分散于25ml dmac溶液中,25℃下超声0.5h,在45℃下反应7h后,旋蒸除去溶剂,65℃下烘干,最终得到黑色粉末。在h2/ar气氛中,500℃热处理1h,得到铜基大环化合物/碳电催化剂。
[0064]
把泡沫铜置于30℃下的乙醇中浸泡20min,用去离子水清洗后真空干燥,裁切备用。称取5mg上述制得的铜基大环化合物/碳电催化剂,分别加入0.5ml水、2ml甲醇和20μl nafion溶液,混合后超声分散0.5h,配成催化剂浆液,刷涂于上述处理后的泡沫铜上,得到载有电催化剂的铜电极。之后把载有电催化剂的铜电极放在he中,700℃热处理3h,得到铜基大环化合物/碳电极。
[0065]
在25℃下,co2电催化还原采用h型电解池进行测试。电解质采用0.1m khco3溶液,两室溶液体积均为100ml,中间以nafion膜隔开。阳极侧通入高纯的ar,阴极侧通入混合原料气co2/ar(体积比:9:1),气速均为40ml/min。在测试前,两侧均通气至溶液饱和。用玻碳工作电极夹固定上述铜基大环化合物/碳电极作为阴极,以铂电极为对电极,以饱和甘汞电极为参比电极。检测在不同电位下电解产物的组成及浓度,并计算其法拉第效率。实施例4所得电催化剂的形貌和工作电极的性能与实施例1相似,具有较好的分散性,无颗粒团聚现象。在-0.5v(vs.rhe)下c2产物(包括c2h4和少量的c2h6,其中c2h6的法拉第效率小于3%)的选择性高达35.0%。
[0066]
实施例5
[0067]
将6mg石墨烯和4mg 5,10,15,20-四(对甲氧基苯基)卟啉铜分散于15ml乙二醇溶液中,30℃下超声0.2h,在50℃下搅拌6h后,旋蒸除去溶剂,65℃下烘干,最终得到黑色粉末。在nh3气氛中,700℃热处理1h,得到铜基大环化合物/碳电催化剂。
[0068]
把泡沫铜置于50℃下的丙酮中浸泡30min,用去离子水清洗后真空干燥,裁切备用。称取5mg上述制得的铜基大环化合物/碳电催化剂,分别加入0.1ml水、1ml异丙醇和10μl nafion溶液,混合后超声分散0.2h,配成催化剂浆液,喷涂于上述处理后的泡沫铜上,得到载有电催化剂的铜电极。之后把载有电催化剂的铜电极放在n2中,600℃热处理3h,得到铜基大环化合物/碳电极。
0.4v(vs.rhe)下c2产物(包括c2h4和少量的c2h6,其中c2h6的法拉第效率小于3%)的选择性高达39.9%。
[0078]
实施例8
[0079]
将5mg碳黑和5mg四磺酸基四钠盐铜酞菁分散于10ml丙醇溶液中,20℃下超声0.5h,在60℃下搅拌3h后,旋蒸除去溶剂,65℃下烘干,最终得到黑色粉末。在he气氛下,500℃热处理2h,得到铜基大环化合物/碳电催化剂。
[0080]
把泡沫铜置于60℃下的异丙醇中浸泡20min,用去离子水清洗后真空干燥,裁切备用。称取5mg上述制得的铜基大环化合物/碳电催化剂,分别加入0.5ml水、5ml甲醇和30μl nafion溶液,混合后超声分散0.5h,配成催化剂浆液,刷涂于上述处理后的泡沫铜上,得到载有电催化剂的铜电极。之后把载有电催化剂的铜电极放在n2中,750℃热处理2h,得到铜基大环化合物/碳电极。
[0081]
在25℃下,co2电催化还原采用h型电解池进行测试。电解质采用0.5m k2so4溶液,两室溶液体积均为100ml,中间以nafion膜隔开。阳极侧通入高纯的n2,阴极侧通入原料气co2,气速均为40ml/min。在测试前,两侧均通气至溶液饱和。用玻碳工作电极夹固定上述铜基大环化合物/碳电极作为阴极,以石墨电极为对电极,以饱和甘汞电极为参比电极。检测反应生成产物的组分及浓度,并计算其法拉第效率。实施例8所得电催化剂的形貌和工作电极的性能与实施例1相似,具有较好的分散性,无颗粒团聚现象。在-0.4v(vs.rhe)下c2产物(包括c2h4和少量的c2h6,其中c2h6的法拉第效率小于3%)的选择性高达37.4%。
[0082]
对于任何熟悉本领域的技术人员而言,在不脱离本发明技术方案范围情况下,都可利用上述揭示的技术内容对本发明技术方案作出许多可能的变动和修饰,或修改为等同变化的等效实施例。因此,凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所做的任何简单修改、等同变化及修饰,均应仍属于本发明技术方案保护的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1