一种壳聚糖填充微型基质固相分散方法与流程
本发明属于天然药物提取领域,涉及一种提取测定青果中有效成分的方法,具体涉及一种采用壳聚糖作为吸附剂的基质固相分散技术提取检测青果中有效成分的方法。
背景技术:
青果为橄榄科植物橄榄Canarium album Raeusch.的干燥成熟果实。秋季果实成熟时采收,干燥。具有清热解毒,利咽,生津等功能。可用于咽喉肿痛,咳嗽痰黏,烦热口渴,鱼蟹中毒等。青果一般用于鲜吃、加工凉果和制药。不同品种的青果某些化学成分的含量差别很大,而对不同用途的青果的药效成分有不同的要求,因此应对青果不同品种中的有效成分进行分析测定。在分析检测之前需要对样品进行预处理,传统的预处理过程(例如超声提取,回流提取,索氏提取等)一般包括提取和净化两步,因而它存在操作复杂、耗时、有机溶剂消耗量大,污染环境等缺点。
基质固相分散(MSPD)是一种样品前处理的新技术,由于它集样品分散、提取和净化与一体,因此相比于传统的方法可简化步骤,节约时间。目前传统的基质固相分散方法的吸附剂量(1~10g)、药材的用量(1g以上)、洗脱剂的用量(2~20ml)都比较大,其所用的吸附剂一般为氧化铝,C18,硅胶等,他们普遍缺乏选择性。因此开发小型的新型的基质固相分散方法也是非常有意义的。
壳聚糖是甲壳素脱乙酰基产物,它具有游离的氨基和羟基,有良好的吸附性能,壳聚糖及其衍生物具有吸附容量大、成本低、可再生性、有生物相容性、无毒、易于化学改性等优点,因而被广泛应用于医药、纺织、水处理、生物工程等领域。由于壳聚糖具有良好的吸附性能,安全无毒,因此适合用作基质固相分散的吸附剂。
目前将壳聚糖作为吸附剂结合微型基质固相分散技术用于青果中化学成分的测定尚未见报道。本发明采用壳聚糖作为基质固相的吸附剂,并极大地减少了传统基质固相萃取技术的样品用量,并可高效提取青果的有效成分。
技术实现要素:
本发明是要解决目前提取检测青果中有效成分的方法中步骤复杂、有机溶剂 用量大、费时、选择性低等问题,采用壳聚糖为吸附剂的基质固相分散技术提取检测青果中的有效成分的方法。
壳聚糖作为吸附剂的基质固相分散技术提取检测青果中的有效成分的方法,具体是按照以下步骤完成的:
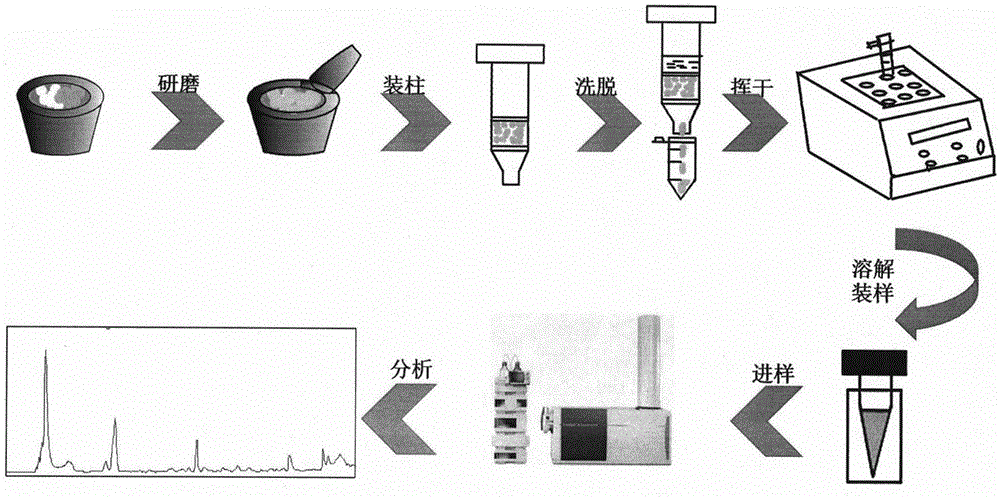
(1)称取青果粉末50mg与一定质量比的吸附剂于研钵中,研磨一定的时间,将研磨好的固体作为填料装入1ml规格的固相萃取小柱中,用0.5ml的洗脱液对小柱洗脱三次,将收集到的洗脱液挥干,再用200μl的洗脱液溶解,将离心管内的液体涡旋10s后13000rpm离心5min。取上层清液装样,用超高效液相色谱-四极杆飞行时间质谱联用仪(UHPLC-Q-TOF/MS)采样分析,得到青果有效成分的色谱图。
(2)将没食子酸对照品配制成不同浓度的对照品溶液,按富集样液的条件用超高效液相色谱-四极杆飞行时间质谱联用仪检测,获得没食子酸的谱图,以没食子酸的进样量为横坐标,以没食子酸对照品溶液的谱图中的峰面积为纵坐标制作没食子酸标准曲线,按同样方法制作羟基酪醇、没食子酸甲酯、木犀草苷、芦丁、橄榄苦苷、鞣花酸的标准曲线。根据提取液各成分色谱图中的峰面积和各成分的标准曲线,计算得到提取液中各成分的含量。
其中(1)中所述吸附剂包括:中分子壳聚糖、低分子壳聚糖、低粘度壳聚糖、高纯度壳聚糖、羧化壳聚糖,优选为中分子壳聚糖。
所述吸附剂的质量为:6.25mg、12.5mg、25mg、37.5mg、50mg、62.5mg、75mg、87.5mg,优选为25mg。
所述研磨时间为:20s、40s、60s、80s、100s,优选为60s。
所述洗脱液包括:60%甲醇、80%甲醇、100%甲醇、乙腈、乙醇、乙酸乙酯、正丁烷,优选为60%甲醇。
本发明的有益效果是:本发明采用基质固相分散技术提取青果中的有效成分,是提取和净化一步完成,具有操作简便,节约时间,有机溶剂及样品用量少等优点。其选用壳聚糖作为吸附剂具有吸附性能好,成本低,安全无毒,可再生性等特点,提高了分析方法的选择性。使其大大优于传统的提取方法。此外本发明以超高效液相色谱-四极杆飞行时间质谱联用仪(UHPLC-Q-TOF/MS)作为检测器具有快速、稳定、准确的特点。
附图说明
图1为本发明基质固相分散萃取方法的工艺流程图。
图2为考察吸附剂种类的基质固相分散萃取效果柱状图。图中,1、2、3、4、5、6、7分别代表没食子酸、羟基酪醇、没食子酸甲酯、木犀草苷、芦丁、鞣花酸、橄榄苦苷。
图3为考察药材和中分子壳聚糖之间不同质量比例下的基质固相分散萃取效果折线图。图中,1、2、3、4、5、6、7分别代表没食子酸、羟基酪醇、没食子酸甲酯、木犀草苷、芦丁、鞣花酸、橄榄苦苷。
图4为考察不同研磨时间的基质固相分散萃取效果折线图。图中,1、2、3、4、5、6、7分别代表没食子酸、羟基酪醇、没食子酸甲酯、木犀草苷、芦丁、鞣花酸、橄榄苦苷。
图5为考察洗脱剂种类的基质固相分散萃取效果柱状图。图中,1、2、3、4、5、6、7分别代表没食子酸、羟基酪醇、没食子酸甲酯、木犀草苷、芦丁、鞣花酸、橄榄苦苷。
图6为青果混合对照品的UHPLC-Q-TOF/MS色谱图。图中,1、2、3、4、5、6、7分别代表没食子酸、羟基酪醇、没食子酸甲酯、木犀草苷、芦丁、鞣花酸、橄榄苦苷。
图7为青果活性成分提取液的UHPLC-Q-TOF/MS色谱图。
具体实施方式
下面结合附图及具体实施例对本发明作进一步说明。
以下所述,仅是本发明的较佳实施例而已,但这些具体实施方式不以任何方式限制本发明的保护范围。
实施例1
一、称取50mg的青果粉末5份与25mg不同种类的壳聚糖(中分子壳聚糖、低分子壳聚糖、低粘度壳聚糖、高纯度壳聚糖、羧化壳聚糖)分别加入五组研钵中,研磨60s。
二、将研磨好的固体作为填料装入1ml规格的固相萃取小柱中,用0.5ml的甲醇对小柱洗脱三次,将收集到的洗脱液挥干,再用200μl的洗脱液溶解,将离 心管内的液体涡旋10s后13000rpm离心5min。
三、取上层清液装样,用超高效液相色谱-四极杆飞行时间质谱联用仪(UHPLC-Q-TOF/MS)采样分析。
本实验采用的仪器为安捷伦超高效液相色谱-四极杆飞行时间质谱联用仪(UHPLC-Q-TOF/MS)。仪器:安捷伦超高效液相色谱系统(Agilent 1290 Infinity LC,Agilent Technologies,Santa Clara,CA,USA)配备真空抽气泵,二元泵流动相系统,恒温自动进样器,恒温柱温箱。色谱柱Agilent Zorbax Extend-C18 column(2.1mm×50mm,1.8μm),柱温40℃。流动相组成包括甲醇(A)和0.1%甲酸水溶液(B),梯度洗脱为0-1min:5%A,1-3.0min:5-20%A,3.0-4.0min:20-30%A,4.0-5.0min:30-30%A,5.0-6.0min:30-40%A,6.0-7.0min:40-45%A,7.0-8.0min:45-50%A,8.0-9.0min:50-80%A,9.0-10min:80-100%A,10-11min:100-100%A,11-12min:100-5%A,平衡时间5min。进样体积1μL,流速0.4mL/min。
该UHPLC系统与下述质谱系统联用:6530Q-TOF/MS(Agilent Technologies,Santa Clara,CA,USA)配备dual ESI离子源,仪器参数:负离子模式,雾化气压45psig,干燥气流速12L/min,干燥气温度350℃,毛细管电压3500V,裂解电压175V,四极杆电压65V,八极杆电压750V,质谱检测范围为100-700m/z。数据解析软件为Mass Hunter software(version B 05.00 Qualitative Analysis)。
不同种类吸附剂的萃取效果柱状图如图2所示,结果表明,中分子壳聚糖的萃取效果最佳。可能由于它黏度、颗粒大小等适中能更好的与青果中的有效成分作用并能紧密地填在小柱内。故而,优选中分子壳聚糖作为吸附剂。
实施例2
一、称取9份50mg的青果粉末于9组研钵中,并称取8份不同质量的中分子壳聚糖(6.25mg,12.5mg,25mg,37.5mg,50mg,62.5mg,75mg,87.5mg)分别加入8组研钵中,剩下一组研钵不加吸附剂,研磨60s。
二、将研磨好的固体作为填料装入1ml规格的固相萃取小柱中,用0.5ml的甲醇对小柱洗脱三次,将收集到的洗脱液挥干,再用200μl的洗脱液溶解,将离心管内的液体涡旋10s后13000rpm离心5min。
三、取上层清液装样,用超高效液相色谱-四极杆飞行时间质谱联用仪(UHPLC- Q-TOF/MS)采样分析。
不同青果与中分子壳聚糖的质量比的萃取效果如图3中折线图所示。由图中可以看到随着吸附剂量的增加萃取效果增强,基本增至25mg后萃取效果趋于平缓甚至略有下降,虽然25mg的中分子壳聚糖对物质1没食子酸的萃取效果不是最佳,但综合考虑,对总体来说是最优的。原因可能是,吸附剂用量的增加可以更多的吸附青果中的活性成分,但当活性成分基本被吸附之后,过多的吸附剂会使活性成分难以解吸,从而是萃取效果有所下降,
实施例3
一、称取5份50mg的青果粉末和5份25mg的中分子壳聚糖放入5组研钵中,分别研磨不同的时间(20s,40s,60s,80s,100s)。
二、将研磨好的固体作为填料装入1ml规格的固相萃取小柱中,用0.5ml的甲醇对小柱洗脱三次,将收集到的洗脱液挥干,再用200μl的洗脱液溶解,将离心管内的液体涡旋10s后13000rpm离心5min。
三、取上层清液装样,用超高效液相色谱-四极杆飞行时间质谱联用仪(UHPLC-Q-TOF/MS)采样分析。
不同研磨时间的萃取效果折线图如图4所示。结果表明,随着研磨时间的增加萃取效果增强,增至最优值60s后,随着研磨时间的增加萃取效果反而有所下降。可能是由于充分的研磨有助于吸附剂更好的接触青果中的活性成分,萃取效果增强,但是过度的研磨会使活性成分被挤压到吸附剂中致密的孔径中,使得活性成分较难洗脱下来,从而使萃取效果下降。
实施例4
一、称取6份50mg的青果粉末和6份25mg的中分子壳聚糖放入5组研钵中,研磨60s。
二、将研磨好的固体作为填料分别装入6支1ml规格的固相萃取小柱中,分别用0.5ml不同的洗脱液(60%甲醇、80%甲醇、甲醇、乙腈、乙醇、乙酸乙酯)对小柱洗脱三次,将收集到的洗脱液挥干,再用200μl的洗脱液溶解,将离心管内的液体涡旋10s后13000rpm离心5min。
三、取上层清液装样,用超高效液相色谱-四极杆飞行时间质谱联用仪(UHPLC-Q-TOF/MS)采样分析。
不同洗脱剂的萃取效果柱状图如图5所示。结果显示,随着洗脱液极性的增强萃取效果逐渐加强,即60%的甲醇洗脱效果最佳,可能由于60%的甲醇极性与目标分子接近并能较好地溶解目标分子,且其洗脱时间相对较长,能与活性成分更好地接触、相互作用,因此萃取效果最好。
日内精密度
一、称取50mg的青果粉末和25mg的中分子壳聚糖放入研钵中,研磨60s。
二、将研磨好的固体作为填料装入1ml规格的固相萃取小柱中,用0.5ml 60%的甲醇对小柱洗脱三次,将收集到的洗脱液挥干,再用200μl的洗脱液溶解,将离心管内的液体涡旋10s后13000rpm离心5min。
三、取上层清液装样,用超高效液相色谱-四极杆飞行时间质谱联用仪(UHPLC-Q-TOF/MS)采样分析。在同一天内连续进样6次。
日间精密度
一、称取50mg的青果粉末和25mg的中分子壳聚糖放入研钵中,研磨60s。
二、将研磨好的固体作为填料装入1ml规格的固相萃取小柱中,用0.5ml60%的甲醇对小柱洗脱三次,将收集到的洗脱液挥干,再用200μl的洗脱液溶解,将离心管内的液体涡旋10s后13000rpm离心5min。
三、取上层清液装样,用超高效液相色谱-四极杆飞行时间质谱联用仪(UHPLC-Q-TOF/MS)采样分析。将该样品连续进样3天,每天2次。
日内、日间精密度实验结果汇总如下表1:
表1
1,2,3,4,5,6,7分别代表没食子酸、羟基酪醇、没食子酸甲酯、木犀草苷、芦丁、鞣花酸、橄榄苦苷。
重复性考察
一、称取50mg的青果粉末和25mg的中分子壳聚糖放入研钵中,研磨60s。
二、将研磨好的固体作为填料装入1ml规格的固相萃取小柱中,用0.5ml60%的甲醇对小柱洗脱三次,将收集到的洗脱液挥干,再用200μl的洗脱液溶解,将离心管内的液体涡旋10s后13000rpm离心5min。
三、取上层清液装样,用超高效液相色谱-四极杆飞行时间质谱联用仪(UHPLC-Q-TOF/MS)采样分析。按上述步骤重复做三组,做为重复性考察。
药材含量测定
一、称取50mg的青果粉末和25mg的中分子壳聚糖放入研钵中,研磨60s。
二、将研磨好的固体作为填料装入1ml规格的固相萃取小柱中,用0.5ml 60%的甲醇对小柱洗脱三次,将收集到的洗脱液挥干,再用200μl的洗脱液溶解,将离心管内的液体涡旋10s后13000rpm离心5min。
三、取上层清液装样,用超高效液相色谱-四极杆飞行时间质谱联用仪(UHPLC-Q-TOF/MS)采样分析。获得提取液的UHPLC-Q-TOF/MS色谱图。
图7为青果活性成分提取液的UHPLC-Q-TOF/MS色谱图。
取没食子酸、羟基酪醇、没食子酸甲酯、木犀草苷、芦丁、鞣花酸、橄榄苦苷的对照品适量,精密称定,置于棕色量瓶中,加甲醇制成每1mL含没食子酸250μg、羟基酪醇250μg、没食子酸甲酯250μg、木犀草苷250μg、芦丁250μg、鞣花酸250μg、橄榄苦苷250μg的对照品溶液。将制得的对照品溶液配置成一系列不同浓度的混合对照品溶液,按同样的条件用UHPLC-Q-TOF/MS检测,获得7种对照品的色谱图,以对照品的进样量为横坐标,对应色谱峰的峰面积为纵坐标,分别制作这7种物质的标准曲线。以青果提取液的色谱峰峰面积及标准曲线计算得到青果中没食子酸、羟基酪醇、没食子酸甲酯、木犀草苷、芦丁、鞣花酸、橄榄苦苷的含量。
没食子酸、羟基酪醇、没食子酸甲酯、木犀草苷、芦丁、鞣花酸、橄榄苦苷浓度均为100μg/mL的混合对照品溶液的色谱图如图6所示。
7种成分的标准曲线以及检测限和定量限如下表2所示:
表2.
回收率实验
一、称取2份50mg的青果粉末和2份25mg的中分子壳聚糖分别放入2组研钵中,其中一组加入20μl100μg/ml的混和对照品溶液,研磨60s。
二、将研磨好的固体作为填料装入1ml规格的固相萃取小柱中,用0.5ml 60%的甲醇对小柱洗脱三次,将收集到的洗脱液挥干,再用200μl的洗脱液溶解,将离心管内的液体涡旋10s后13000rpm离心5min。
三、取上层清液装样,用超高效液相色谱-四极杆飞行时间质谱联用仪(UHPLC-Q-TOF/MS)采样分析。
重复性、含量测定、回收率实验结果汇总如下表3:
表3
根据表中数据显示,本发明方法的重复性好,回收率高,检测准确性高。
- 还没有人留言评论。精彩留言会获得点赞!