一种利用阀切换方法同时检测植物中的无机阴离子、有机酸和三种植物素的方法与应用与流程
本发明属于分析化学领域,具体涉及一种利用阀切换方法同时检测植物中的无机阴离子、有机酸和三种植物素的方法与应用。
背景技术:
目前,检测植物中的无机阴离子、有机酸通常采用高效阴离子交换色谱法(离子色谱法),而莨菪亭、芦丁、槲皮素等植物素采用反相高效液相色谱法、分光光度法等方式进行单独测定。但在分析测试中,经常需要同时对无机阴离子、有机酸和不同的植物素进行测定。若仍然采用分开单独测定的方法,不仅会造成人力财力的浪费,而且无法满足某些对检测时效性较强的应用。目前尚没有一种有效可靠的方法能够同时测定上述物质含量,因此亟需开发一种能够同时检测植物中的无机阴离子、有机酸和多种植物素的方法。
技术实现要素:
本发明的目的在于解决现有技术中存在的问题,并提供一种利用阀切换方法同时检测植物中的有机酸、无机阴离子和三种植物素的方法与应用。具体技术方案如下:
利用阀切换方法同时检测植物中的有机酸、无机阴离子和三种植物素的方法,利用阀切换法连用高效液相色谱仪紫外检测法与高效阴离子交换色谱电导检测法对植物样品中有机酸、无机阴离子和莨菪亭、芦丁、槲皮素三种植物素进行分离检测;经前处理后的植物样品先被淋洗液带进连接在高效液相色谱仪的C18柱中,有机酸及阴离子在C18柱中不保留,通过设置合适的阀切换时间使机酸及阴离子被全部切换至离子色谱分析柱中,保留在C18柱中的三种植物素继续随着淋洗液的淋洗被冲出色谱柱,用紫外检测器检测;进入离子色谱柱的样品随着碱液淋洗液淋洗被冲出色谱柱,用电导检测器检测。
作为优选,植物样品前处理方法如下:把样品置于80℃的温度下,烘干2小时后研磨,过0.5mm的筛子,准确称取小于0.5mm的样品0.0400g,置于10mL容量瓶中,并准确加入2.00mL超纯水和8.00mL甲醇充分摇匀;设置频率为50KHz进行超声提取,提取温度为20℃,超声提取时间为1h,移入10mL聚丙烯离心管中,在2500r/min转速下离心20min,取上清液过0.45μm微孔滤膜后进样分析。
进一步的,通过阀切换实现无机阴离子、有机酸和三种植物素分离的方法如下:进样前,六通阀1保持在Load状态,样品通过手动进样装载到六通阀1中的20μL定量环中,所注入的多余样品进入废液;六通阀2保持在Inject状态;进样完成后,启动切换程序,切换六通阀1,开始进样,六通阀1处于Inject状态,六通阀2仍保持在Inject状态,淋洗液将定量环中的样品注入C18柱中,无机阴离子、有机酸在C18柱中无保留,而三种植物素在上面有保留,得以分离开来;启动切换程序1.1min后,切换六通阀2,使六通阀2处于Load状态,富集柱富集有机酸、阴离子,富集时间为2.5min;富集完成后,再次切换六通阀2,使六通阀2重新回到Inject状态,富集柱与分析柱IonPac AS11-HC相连,分离富集的有机酸、阴离子,同时C18柱分离植物素。
进一步的,离子色谱中:分析柱采用IonPac AS11-HC(4mm×250mm),保护柱采用IonPac AG11-HC(4mm×50mm),富集柱采用IonPac TAC-ULP1(5mm×23mm),色谱条件如下:流速1.0mL/min,柱温30℃;梯度洗脱程序:0-18.6min,浓度为0.8mmol/L KOH溶液;18.6-33.6min,浓度为0.8-15mmol/L KOH溶液;33.6-53.6min,浓度为15mmol/L KOH溶液;53.6-68.6min,浓度为15-50mmol/L KOH溶液;68.6-68.7min,浓度为50-0.8mmol/L KOH溶液;68.7-69.6min,浓度为0.8mmol/L KOH溶液。
高效液相色谱中:C18柱采用依利特Hypersil ODS分析柱(4.6mm×250mm,粒径5μm),流动相:甲醇(C)、水(D),流速1.0mL/min,柱温25℃,波长365nm;进样量20μL;梯度洗脱程序:0-12min,20%-40%C,80%-60%D;12-14min,40%-70%C,60%-30%D;14-22min,70%-90%C,30%-10%D;22-25min,90%-20%C,10%-80%D。
上述方法可应用于食品生产和生物医药行业中检测活性成份。
本发明的方法利用阀切换法将高效液相色谱法紫外检测法与高效阴离子交换色谱—电导检测法进行联用,可在一次检测过程中同时对植物样品中无机阴离子、有机酸和莨菪亭、芦丁、槲皮素三种植物素进行分离检测。本发明还具有重现性好、检测限低、精密度好的优点。
附图说明
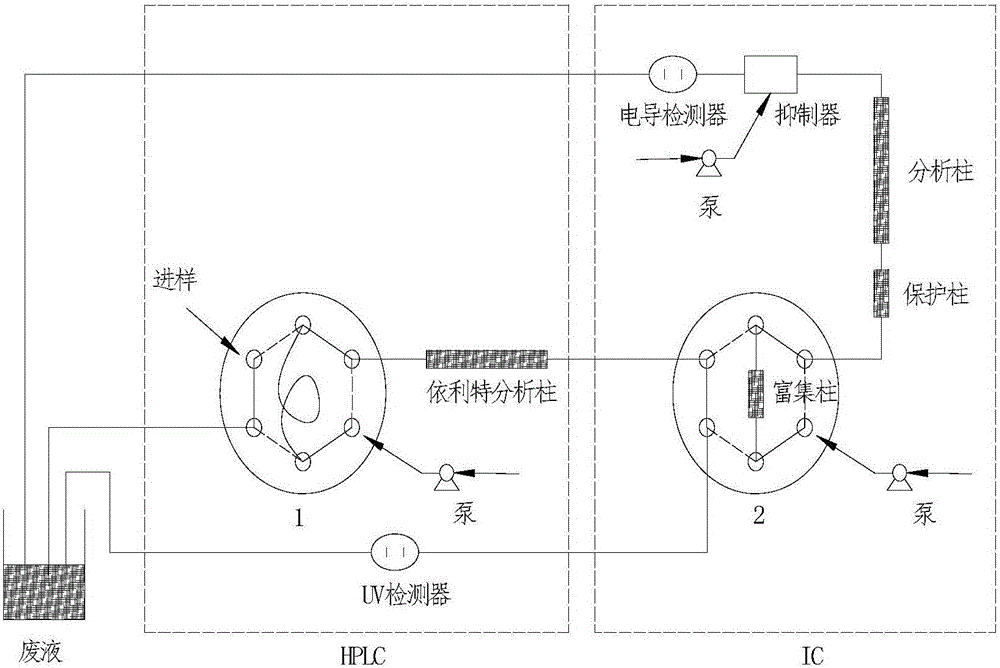
图1为阀切换装置样品装载步骤图;图中六通阀不同孔之间实线代表连通,虚线代表不连通,下同;
图2为阀切换装置依利特Hypersil ODS分析柱(C18柱)分离样品步骤图;
图3为阀切换装置富集柱富集阴离子和有机酸步骤图;
图4为阀切换装置阴离子、有机酸与植物素同时分析步骤图;
图5为50ppm有机酸标准混合溶液离子色谱图;
图6为10ppm有机酸标准混合溶液液相色谱图;
图7为第二个切换时间与阴离子、有机酸峰面积关系图;
图8为第一个切换时间与阴离子、有机酸峰面积关系图;
图9为10ppm标准混合溶液离子色谱图;其中编号分为代表:1-乳酸;2-乙酸;3-丙酸;4-甲酸;5-Cl-;6-苹果酸;7-SO42-;8-草酸;9-PO43-;10-柠檬酸。
图10为1ppm标准混合溶液液相色谱图;其中编号分别代表:1-莨菪亭;2-芦丁;3-槲皮素。
图11为甘草样品离子色谱图;其中编号分别代表:1-乳酸;2-乙酸;3-甲酸;4-Cl-;5-苹果酸;6-SO42-;7-草酸;8-PO43-;9-柠檬酸。
图12为甘草样品液相色谱图;其中编号分别代表:1-莨菪亭;2-芦丁;3-槲皮素。
具体实施方式
以下结合附图和实施案例对本发明做进一步的说明。
无机阴离子、有机酸和植物素的色谱分析,具体步骤如下:
1、仪器和试剂
本发明方法基于的阀切换装置结构如图1所示,包括泵、六通阀1、依利特Hypersil ODS分析柱(C18柱)、六通阀2、富集柱、保护柱、分析柱、抑制器、电导检测器、UV检测器。六通阀1通过定量环进行进样,并通过依利特Hypersil ODS分析柱与六通阀2相连,富集柱置于六通阀2中,六通阀2一路与UV检测器相连,另一路依次与保护柱、分析柱、抑制器、电导检测器相连。各检测元件的废液出口均与废液收集装置相连。
部分用到的仪器型号和参数具体如下:Thermo ICS 5000离子色谱仪(电导检测器,六通阀,ASRS 300(4mm)自循环电抑制器(外接水模式,采用戴安梯度泵GP50,戴安中国有限公司),KOH淋洗液自动发生器EG40,Chromeleon 6.8色谱工作站,赛默飞世尔科技中国有限公司);Agilent 1200series高效液相色谱仪(高压六通进样阀,在线脱气仪,梯度混合仪和UV-Vis检测器,安捷伦科技中国有限公司);KQ-300GVD型三频恒温数控超声波清洗器(昆山市超声仪器有限公司);BS-224S型电子分析天平(赛多利斯科学仪器(北京)有限公司);柱温箱(深圳天为机电设备有限公司)。
试剂如下:甲醇(色谱纯,美国天地TEDIA有限公司),乳酸、乙酸、丙酸、甲酸、Cl-、苹果酸、SO42-、草酸、PO43-、柠檬酸(分析纯,购自华东医药股份有限公司),莨菪亭(阿拉丁试剂)、芦丁(≥98%)、槲皮素(生化试剂)(购自华东医药股份有限公司),实验用水为超纯水(Millipore,Molsheim,France,电阻率18.2MΩ·cm)。
实验样品:甘草、白蔻和芡实样品,购自当地超市。
2、色谱条件
有机酸及阴离子的分离色谱条件及检测:赛默飞ICS5000离子色谱仪带电导检测仪分析柱IonPac AS11-HC(4mm×250mm)(括号中代表柱子型号参数,下同)+保护柱IonPac AG11-HC(4mm×50mm)和富集柱IonPac TAC-ULP1(5mm×23mm),流速1.0mL/min,柱温30℃。梯度洗脱程序:0-18.6min浓度为0.8mmol/LKOH溶液,18.6-33.6min浓度为0.8-15mmol/LKOH溶液,33.6-53.6min浓度为15mmol/LKOH溶液,53.6-68.6min浓度为15-50mmol/LKOH溶液,68.6-68.7min浓度为50-0.8mmol/LKOH溶液,68.7-69.6min浓度为0.8mmol/LKOH溶液。流动相梯度表见表1。典型的色谱图见图9。
表1离子色谱流动相梯度表
植物素的分离色谱条件及检测:安捷伦1200高液液相色谱仪带紫外检测器,依利特Hypersil ODS分析柱(4.6mm×250mm,粒径5μm),流动相:甲醇(记为C)、水(记为D),流速1.0mL/min,柱温25℃,波长365nm;进样量20μL。梯度洗脱程序:0-12min,20%-40%C,80%-60%D;12-14min,40%-70%C,60%-30%D;14-22min,70%-90%C,30%-10%D;22-25min,90%-20%C,10%-80%D。流动相梯度表见表2。典型的色谱图见图10。
表2高效液相流动相梯度表
3、标准溶液的配制
标准储备溶液:Cl-、SO42-、PO43-、乳酸、乙酸、丙酸、甲酸、苹果酸、草酸、柠檬酸由优级纯试剂配置成浓度为1000mg/L的贮备液,莨菪亭、芦丁、槲皮素由阿拉丁试剂配置成浓度为1000mg/L的贮备液,储存于4℃冰箱备用。
标准混合溶液:分别移取一定量的上述标准储备溶液至10mL的容量瓶中,用超纯水定容,配得各溶质浓度均为0.08mg/L、0.40mg/L、1.00mg/L、2.00mg/L、10.00mg/L、50.00mg/L的混合标准溶液系列备用。另外分别移取一定量的Cl-、SO42-、PO43-、乳酸、乙酸、丙酸、甲酸、苹果酸、草酸、柠檬酸标准储备溶液至10mL的容量瓶中,用超纯水定容,配得各溶质浓度均为10.00mg/L的有机酸标准混合溶液。
4、样品前处理
把样品置于80℃的温度下,烘干2小时后研磨,过0.5mm的筛子,准确称取小于0.5mm的样品0.0400g,置于10mL容量瓶中,并准确加入2.00mL超纯水和8.00mL甲醇充分摇匀;设置频率为50KHz进行超声提取,提取温度为20℃,超声提取1h后,移入10mL聚丙烯离心管中,在2500r/min转速下离心20min,取上清液过0.45μm微孔滤膜后进样分析。
5、实验步骤
进样前,六通阀1保持在Load状态,样品通过手动进样装载到六通阀1中的定量环(20μL)中,所注入的多余样品进入废液;六通阀2保持在Inject状态,这样可以避免系统中可能存在的杂质离子在富集柱上的聚集,如图1。进样完成后,启动切换程序(0min),切换六通阀1,开始进样,六通阀1处于Inject状态,六通阀2仍保持在Inject状态,淋洗液将定量环中的样品注入依利特Hypersil ODS分析柱中,有机酸和阴离子在依利特Hypersil ODS分析柱中无保留,而三种植物素在上面有保留,得以分离开来,如图2。1.1min后,切换六通阀2,使六通阀2处于Load状态,富集柱富集有机酸和阴离子,富集2.5min,如图3。富集完成后,再次切换六通阀2,使六通阀2重新回到Inject状态,富集柱与分析柱IonPac AS11-HC相连,分离富集的有机酸和阴离子,同时依利特Hypersil ODS分析柱分离植物素,如图4。
6、切换时间的选择
直接在依利特Hypersil ODS分析柱(C18柱)后面连接电导检测器(无抑制器连接)和UV-Vis检测器,观察10ppm有机酸标准混合溶液的出峰情况,如图5、6。
为了得到准确的切换时间,防止富集时间过短或过长导致富集不完全,对50ppm标准混合溶液进行一系列的测试。首先在第一个切换时间为1.4min的条件下,分别测试3.2min、3.3min、3.4min、3.5min、3.6min、3.7min时标准混合溶液的出峰情况,得到各个有机酸、阴离子的时间与峰面积关系曲线,如图7。由该曲线图可知,3.6min时,富集效果最好,因此选择3.6min为第二个切换时间。然后在第二个切换时间为3.6min的条件下,分别测试0.8min、0.9min、1.0min、1.1min、1.2min、1.3min、1.4min、1.5min、1.6min时标准混合溶液的出峰情况,得到各个有机酸、阴离子的时间与峰面积关系曲线,如图8。由该曲线图可知,1.1min时,富集效果最好,因此选择1.1min为第一个切换时间。
7、线性范围、重现性和检出限
在上文的色谱条件下,将0.08mg/L、0.40mg/L、2.00mg/L、10.00mg/L、50.00mg/L系列混合标准溶液依次进样分析,每个浓度平衡测定3次,数据通过差异性检验后,取3次所得峰面积的平均值,以峰面积(Y)为纵坐标,物质浓度(X)为横坐标建立13种物质的标准曲线。以3倍基线噪声(S/N=3)计算最低检出限,考察重现性时,以浓度为2.00mg/L的混合标准溶液在同样条件下连续进样8次(n=8),计算方法精密度。其线性方程、相关系数、线性范围、检出限及精密度见表3。
表3线性范围、重现性及检测限
8、实际样品检测及方法的回收率
对实际甘草、白蔻、芡实样品按照步骤4所述的前处理后进行检测,并加入一定浓度的标准混合溶液,进行回收率试验,结果见表4~6所示,其回收率均符合要求。实际样品离子色谱图见图11(有机酸及阴离子色谱图)高效液相色谱图见图12(植物素样品色谱图)。
表4甘草样品加标回收率
注:“-”表明未测出。
表5白蔻样品加标回收率
注:“-”表明未测出。
表6芡实样品加标回收率
注:“-”表明未测出。
- 还没有人留言评论。精彩留言会获得点赞!