一种测定普瑞巴林中残留吡啶的方法与流程
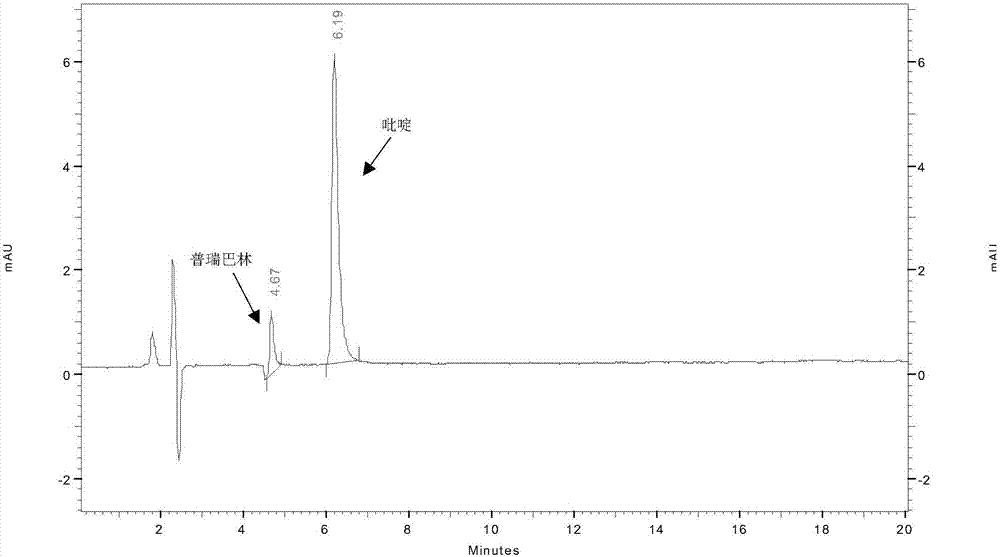
发明领域本发明涉及生物医药领域,具体的,本发明涉及一种测定普瑞巴林中残留吡啶的方法。技术背景普瑞巴林是一种新型γ-氨基丁酸(gaba)受体激动剂,用于治疗神经性疼痛,局部发作性癫痛和焦虑症。2004年12月美国食品与药品管理局((fda)批准用于治疗糖尿病性周围神经病变的神经性疼痛和带状孢疹后遗神经痛。其结构式为是白色或类白色的结晶固体,具有易溶于水、碱性的和酸性的水溶液的特性。在合成普瑞巴林时,人们常常会选用吡啶作为反应溶剂,吡啶的残留会对产品的安全性和有效性带来隐患,因此需要对吡啶残留量进行控制。吡啶的沸点为115.2℃,通常采用气相色谱顶空进样的方法进行药物中残留吡啶的测定。但是由于普瑞巴林原料药在有机溶剂中的溶解性较差,而且待测的残留溶剂吡啶为弱碱性,采用顶空气相分析时峰形较差,回收率也不好。另外采用离子色谱仪分析时,普瑞巴林离子化程度较大,会将待测溶剂吡啶峰包进去,干扰吡啶的测定。技术实现要素:本发明旨在至少在一定程度上解决相关技术中的技术问题之一。为此,本发明的一个目的在于提出了一种简便、准确、灵敏地检测普瑞巴林中残留吡啶的方法。在本发明的第一方面,本发明提出了一种测定普瑞巴林中残留吡啶的方法。根据本发明的实施例,所述方法包括:通过高效液相色谱分析方法对所述普瑞巴林中残留吡啶进行分析,以便获得色谱图;以及基于所述色谱图,确定所述普瑞巴林中残留吡啶的含量,其中,所述高效液相色谱采用以下条件:色谱柱为zorbaxeclipsexdb-c18,4.6×250mm,5微米,检测器为dad,检测波长为240nm,柱温为33℃~37℃,流动相a相为10mmol/l的磷酸氢二钾缓冲液,所述磷酸氢二钾缓冲液的ph为6.8~7.2,流动相b相为甲醇,流速为0.8ml/min~1.2ml/min,洗脱采用等度洗脱,所述等度洗脱中a相和b相的体积比为68:32~72:28,运行时间为20min。利用根据本发明实施例的检测方法,该方法具有专属性好,吡啶与相邻色谱峰分离度大于1.5;精密度好,相对标准偏差小于2.0%;准确率高,回收率在90%~110%之间,符合中国药典对原料药中残留溶剂或有关物质检测方法的要求。所述方法能准确定量8ppm至300ppm范围内普瑞巴林原料药中的吡啶。根据本发明的实施例,上述测定普瑞巴林原料药中残留吡啶的方法还可以进一步包括如下附加技术特征至少之一:根据本发明的实施例,所述磷酸氢二钾缓冲液的ph为7.0。发明人在试验中发现,磷酸氢二钾缓冲液的ph为7.0,缓冲盐的缓冲能力较强,吡啶峰对称因子为1.31,峰形较好。根据本发明的实施例,所述流速为1.0ml/min。发明人在试验中发现,流速为1.0ml/min,待测物中吡啶的保留时间6.11min,保留合适,吡啶峰与其它峰能有效分离。根据本发明的实施例,所述等度洗脱中a相和b相的体积比70:30。发明人在试验中发现,等度洗脱中a相和b相的体积比70:30,吡啶的分离度为4.57,分离度高,对称因子为1.28,峰形好,普瑞巴林中的一些杂质被检出,该条件提供足够好的吡啶检测灵敏度。根据本发明的实施例,所述柱温为35℃。发明人在试验中发现,柱温为35℃,吡啶峰对称因子1.33,峰形较好。根据本发明的实施例,所述普瑞巴林原料药是以供试品溶液的形式提供的,其中,所述供试品溶液为普瑞巴林原料药的磷酸氢二钾-甲醇溶液,并且基于每毫升所述供试品溶液,普瑞巴林原料药的含量为10mg,其中,所述磷酸氢二钾-甲醇溶液是所述a相和b相的混合溶液,所述a相和b相的体积比为7:3。发明人通过试验发现,普瑞巴林原料药在上述磷酸氢二钾-甲醇溶液中溶解能力较好,同时,普瑞巴林原料药的进样浓度在10mg/ml时,可保证足够好的吡啶检测灵敏度。根据本发明的实施例,所述供试品溶液的用量为20微升,对吡啶含量的测定更加真实、可靠、准确。根据本发明的实施例,所述基于所述色谱图,确定所述普瑞巴林原料药中吡啶的含量是通过以下公式确定的:其中,ai为供试品溶液中吡啶的峰面积;as为对照溶液中吡啶峰面积;wt为供试品的称样量,mg;ws为配制对照溶液所称取吡啶的质量,mg;dt为供试品的稀释倍数;ds为对照溶液的稀释倍数;所述对照溶液是吡啶的磷酸氢二钾-甲醇溶液,并且基于每毫升所述对照溶液,吡啶的含量为2微克。根据本发明的实施例,采用上述方式确定普瑞巴林原料药中残留吡啶的含量,准确率高,结果更加真实可靠。在本发明的第二方面,本发明提出了一种测定普瑞巴林中残留吡啶的方法。根据本发明的实施例,所述方法具体包括:(1)色谱条件色谱柱为zorbaxeclipsexdb-c18,4.6×250mm,5微米,检测器为dad,检测波长为240nm,柱温为35℃,流动相a相为10mmol/l的磷酸氢二钾缓冲溶液,所述磷酸氢二钾缓冲溶液的ph为7.0,流动相b相为甲醇,流速为1.0ml/min,洗脱梯度为等度洗脱,等度洗脱中a相和b相的体积比为70:30,运行时间为20min,(2)配制空白溶液甲醇和a相的体积比为3:7的混合溶液作为所述空白溶液,(3)配制供试品溶液精密称定供试品100mg,至10ml容量瓶中,用空白溶液溶解,并做超声处理,稀释至刻度,摇匀,即得所述供试品溶液,(4)配制对照溶液精密称定吡啶20mg,至100ml容量瓶中;用空白溶液溶解,并做超声处理,稀释至刻度,摇匀,即得对照储备液;精密移取对照储备液1.0ml至100ml容量瓶中,用空白溶液稀释至刻度,摇匀,得对照溶液,(5)将20微升供试品溶液注入色谱仪,得到色谱图,根据所述色谱图计算得到供试品溶液中吡啶的含量,其中,按以下公式计算供试品溶液中吡啶的含量,取2次测定结果的平均值作为测定结果:式中:ai为供试品溶液中吡啶的峰面积;as为连续进6针对照溶液中吡啶峰面积的平均值;wt为供试品的称样量,mg;ws为配制对照溶液所称取吡啶的质量,mg;dt为供试品的稀释倍数;ds为对照溶液的稀释倍数。所述1针对照溶液的用量为20微升。利用根据本发明实施例的检测方法,可简便、准确、灵敏地测定普瑞巴林原料药中残留吡啶的含量,从而有效控制普瑞巴林原料药的质量。附图说明图1是根据本发明实施例1的采用intersilods-3色谱柱条件下供试品加标溶液的色谱图;图2为根据本发明实施例1的采用zorbaxeclipsexdb-c18色谱柱条件下供试品加标溶液的色谱图;图3是根据本发明实施例1的吡啶的紫外吸收光谱;图4是根据本发明实施例1的普瑞巴林的紫外吸收光谱;图5是根据本发明实施例1的检测波长为240nm、245nm、250nm条件下,供试品加标溶液的色谱图;图6是根据本发明实施例1的不同ph(6.8~7.2)缓冲液条件下的吡啶对照溶液的色谱图;图7是根据本发明实施例1的流动相比例为k2hpo4:meoh=70:30条件下的供试品加标溶液的色谱图。具体实施例方式下面详细描述本发明的实施例。下面描述的实施例是示例性的,仅用于解释本发明,而不能理解为对本发明的限制。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。实施例1在本实施例中,发明人详细介绍了测定普瑞巴林中残留吡啶的方法的开发过程以及系统适用性标准选择。1.1色谱柱的选择方法开发初期,发明人在检测波长254nm,柱温35℃,流动相h2o:meoh=70:30,等度洗脱20min,进样量为20微升的色谱条件下,考察了intersilods-3(250*4.6mm,5微米)、zorbaxeclipsexdb-c18(4.6*250mm,5微米)不同品牌的色谱柱,对供试品加标溶液中普瑞巴林的保留时间、峰形以及吡啶的保留时间、峰形、峰高等影响,具体考察结果如表1所示,图1为考察intersilods-3色谱柱所得色谱图,图2为考察zorbaxeclipsexdb-c18色谱柱所得色谱图。供试品加标溶液的配制为取普瑞巴林原料药约100mg,精密称定至10ml容量瓶中,用对照溶液溶解并定容,摇匀即得,配制1份,对照溶液的配制同2.1项。表1:供试品加标溶液考察结果表1和图1结果显示,采用intersilods-3色谱,普瑞巴林保留时间为6.15min,普瑞巴林主峰前出现倒峰,吡啶的保留时间为9.72min,吡啶峰高为9110,吡啶的峰形拖尾。表1和图2结果显示,采用zorbaxeclipsexdb-c18色谱柱,普瑞巴林保留时间为4.67min,普瑞巴林主峰前出现倒峰,吡啶的保留时间6.19min,吡啶的峰高为12425,吡啶的峰形拖尾。对上述考察结果进行对比,可以得出,采用zorbaxeclipsexdb-c18色谱柱条件相比采用intersilods-3色谱柱条件的吡啶的峰高有提高,其方法的灵敏度更好,因此选择zorbaxeclipsexdb-c18色谱柱来进行后续普瑞巴林中残留吡啶的分析方法开发。1.2检测波长的确定根据图3所示,吡啶的最大紫外吸收在256nm,在235~250nm之间吸收都比较强,普瑞巴林的最大紫外吸收在196nm,如图4所示。在1.1项选定色谱柱条件下,发明人进一步对检测波长进行了考察,检测波长分别为240nm、245nm、250nm的考察结果如图5所示,考察结果显示检测波长为240nm、245nm,普瑞巴林主峰前都有倒峰,检测波长为240nm,普瑞巴林主峰的峰形正常,不出现倒峰,考虑到既要保证吡啶的响应足够也要兼顾普瑞巴林不出现倒峰,因此最终选择240nm作为检测的波长。1.3流动相、ph、流动相比例的选择与优化发明人进一步对流动相进行考察,考察结果显示流动相a相为水相,对称因子为1.68,选择流动相a相为10mmol/l磷酸氢二钾溶液缓冲盐,对称因子为1.50,与流动相a相为水相相比吡啶的峰形较好,并且吡啶在色谱柱上的保留增强,因此选择流动相a相为磷酸氢二钾溶液缓冲盐。发明人进一步对缓冲盐的ph进行考察,考察结果如图6所示,考察结果显示ph=6.8,对称因子为1.33;ph=7.0,对称因子为1.31;ph=7.2,对称因子为1.34,其缓冲盐的缓冲能力均较强,吡啶峰形均较好,其中ph=7.0,吡啶峰对称因子为1.31,峰形更好,因此优选ph=7.0。上述考察已确定色谱柱为zorbaxeclipsexdb-c18,检测波长240nm,ph=7.0的10mmol/l磷酸氢二钾-甲醇为流动相,在等度洗脱20min下,发明人进一步对流动相的比例进行考察,考察结果显示,流动相比例分别为k2hpo4:meoh=68:32、k2hpo4:meoh=70:30、k2hpo4:meoh=72:28,普瑞巴林及吡啶峰形均正常,吡啶的分离度效果均较好。其中图7显示,流动相k2hpo4:meoh=70:30,普瑞巴林中的一些杂质也被检出,吡啶的分离度为4.57,对称因子为1.28,相比灵敏度更好,因此优选该条件进行后续灵敏度的考察,具体结果如表2所示。表2:供试品加标溶液考察结果1.4流速及柱温的考察基于上述已确定条件,以及在流动相比例优选的条件下,进一步对流速进行考察,考察结果显示流速分别为0.8ml/min、1.0ml/min、1.2ml/min时,普瑞巴林及吡啶峰形均正常,吡啶的分离度效果均较好。基于流速为1.0ml/min时,待测物吡啶的保留时间6.11min,相比保留更合理,并且吡啶峰与其它峰能有效分离,吡啶峰没有被干扰,优选1.0ml/min。发明人对柱温25℃、33℃、35℃、37℃进一步考察,考察结果显示柱温为25℃,吡啶峰对称因子1.79,峰形不好,柱温分别为33℃、35℃、37℃条件下,峰形均较好,其中柱温为35℃,吡啶峰对称因子1.33,相比其它温度下峰形更好,且考虑到柱温过高容易造成色谱柱固定相的流失,因此优选35℃。1.5供试品浓度的选择理想的供试品稀释液为流动相溶液,上述优选的等度洗脱流动相比例为k2hpo4:meoh=70:30,对供试品的溶解能力较好,故发明人选择甲醇/k2hpo4缓冲液的比例为30:70的混合溶液作为供试品稀释液。关于供试品配制浓度和进样量的确定,发明人最终通过试验,确定的供试品浓度为10mg/ml,进样量为20微升,能够保证足够好的吡啶检测灵敏度。1.6系统适用性标准选择基于吡啶在zorbaxeclipsexdb-c18保留时间6.15min,对称因子为1.28及3.2项系统适用性的测试结果,选择吡啶与相邻峰的分离度≥1.5,进样6次峰面积的rsd≤10.0%,及10%对照溶液的信噪比≥10作为系统适用性标准。实施例2在本实施例中,详细介绍了发明人是如何基于实施例1所确定的色谱条件获得色谱图的,并利用色谱图如何计算得到普瑞巴林中残留吡啶的含量。2.1相关溶液的配制流动相:a相:称取约1.74g无水磷酸氢二钾,称定至1l烧杯中,加入1l超纯水溶解,混匀,用0.1mol/l的磷酸溶液或者0.1mol/l氢氧化钾溶液调节ph值为7.0,然后用0.2μm水系滤膜过滤超声即得;b相:100%甲醇;稀释液(空白溶液):a相:b相=7:3的混合液;对照储备液:取吡啶约20mg至100ml容量瓶中,精密称定,用稀释剂稀释至刻度,摇匀即得;对照溶液:移取1.0ml对照储备液至100ml容量瓶中,用稀释剂稀释至刻度,摇匀即得;灵敏度溶液:移取1.0ml对照溶液至10ml容量瓶中,用稀释剂稀释至刻度,摇匀即得;供试品溶液:取供试品约100mg,精密称定,至10ml容量瓶中,用稀释剂稀释至刻度,摇匀即得;平行配制2份。2.2色谱条件色谱柱:zorbaxeclipsexdb-c18,4.6×250mm,5微米,柱温:35℃,检测波长:240nm,流速:1.0ml/min,进样体积:20微升,流动相配制:a相(0.01mol/lph=7.0磷酸氢二钾缓冲溶液):称取约1.74g无水磷酸氢二钾,称定至1l烧杯中,加入1l超纯水溶解,混匀,用0.1mol/l的磷酸溶液或者0.1mol/l氢氧化钾溶液调节ph值为7.0,然后用0.2微米水系滤膜过滤超声即得;b相:甲醇,等度洗脱:a相:b相=70:30。2.3相关检测操作待基线平衡后,在2.2项色谱条件下,取空白溶液、灵敏度溶液、对照溶液、供试品溶液,按表3序列进样,记录各图谱。表3:进样序列溶液名称进样针数空白溶液1针灵敏度溶液1份1针对照溶液1份6针供试品溶液2份各1针系统适用性要求:灵敏度溶液中吡啶峰信噪比不小于10;第一针对照溶液中吡啶与相邻峰的分离度不小于1.5;连续进样6针吡啶峰面积的rsd不得超过10.0%。其中系统适用性满足该要求后进行供试品检测及方法学验证。2.4基于色谱图的残留吡啶的含量的计算方法按照以下公式计算供试品中吡啶的含量,以2次测定结果的平均值作为检测结果:式中:ai为供试品溶液中吡啶的峰面积;as为连续进6针对照溶液中吡啶峰面积的平均值;wt为普瑞巴林供试品的称样量,mg;ws为配制对照溶液所称取吡啶的质量,mg;dt为供试品的稀释倍数;ds为对照溶液的稀释倍数,可接受标准为吡啶含量≤200ppm。实施例3在本实施例中,发明人对本发明测定普瑞巴林中残留吡啶的方法学考察。3.1系统适用性空白溶液、对照溶液的配制方法同2.1项,各配制1份。操作:在2.2项色谱条件下,待系统平衡后,取空白溶液进样1针,灵敏度溶液进样1针,对照溶液进样6针,记录色谱图。试验结果显示连续进样6针吡啶峰面积依次为72298、74131、74240、73774、74888、74759,连续进样6针吡啶峰面积的rsd为1.27%,符合要求;灵敏度溶液中吡啶峰信噪比为32.80,符合要求;对照溶液第一针中吡啶与相邻峰的分离度为n/a,基于对照溶液中只有吡啶的峰,n/a是无限大,符合要求。因此,系统适用性符合检测要求,从而继续进行后续方法学的考察。3.2专属性空白溶液、对照储备液、对照溶液、供试品溶液的配制方法同2.1项;100%供试品加标溶液的配制:取供试品约100mg,精密称定至10ml容量瓶中,用对照溶液溶解并定容,摇匀即得,配制1份。操作:在2.2项色谱条件下,取空白溶液、供试品溶液、对照溶液、供试品加标溶液各进样1针,并记录相应色谱图,具体试验结果如表4所示。表4:专属性结果溶液名称吡啶保留时间(min)吡啶峰面积吡啶分离度空白溶液///供试品溶液///对照溶液6.1174482/供试品加标溶液6.11731397.24表4考察结果显示,空白溶液对对照溶液和供试品溶液中吡啶无干扰,符合要求;供试品溶液、对照溶液、供试品加标溶液中吡啶峰与普瑞巴林峰的分离度最小值为7.24,符合要求;供试品加标溶液中吡啶峰的保留时间与对照溶液一致,均为6.11min;供试品加标溶液中吡啶峰面积为73139,与供试品溶液相比,供试品加标溶液中吡啶的峰面积有增加,符合要求。因此方法专属性良好。3.3精密度分析重复性溶液的配制:取供试品约100mg,精密称定至10ml容量瓶中,用对照溶液溶解并定容,摇匀即得,平行配制6份,该溶液由分析人员甲配制;中间精密度溶液的配制:取供试品约100mg,精密称定至10ml容量瓶中,用对照溶液溶解并定容,摇匀即得,平行配制6份,由不同的分析人员于不同的时间在同一实验室内采用不同仪器进行如下操作。分析重复性操作:在2.2项色谱条件下,取6份分析重复性溶液,各进样1针,根据系统适用性中对照峰面积的平均值计算6份分析重复性溶液中吡啶的含量。中间精密度操作:在2.2项色谱条件下,取6份中间精密度溶液,各进样1针,根据系统适用性中对照峰面积的平均值计算6份中间精密度溶液中吡啶的含量。分析重复性试验结果显示6份分析重复性溶液中,吡啶含量的rsd值为0.52%,符合要求。中间精密度试验结果显示6份中间精密度溶液中吡啶含量的rsd值为1.24%,同时分析重复性和中间精密度的12份溶液中吡啶含量的rsd为0.92%,分析重复性和中间精密度吡啶含量平均值的绝对差值为0.60ppm,均符合要求。因此方法精密度良好。3.4线性关系空白溶液、对照储备液、对照溶液的配制方法同2.1项,各配制1份;线性储备液的配制:精密移取对照储备液25ml至100ml容量瓶中,用稀释剂稀释至刻度,摇匀即得;定量限溶液的配制:取对照溶液4ml至100ml容量瓶中,用稀释剂稀释至刻度,摇匀即得。线性溶液的配制:精密移取如表5所示的不同体积的线性储备液或对照溶液于不同体积的容量瓶中,用稀释剂稀释至刻度,摇匀,即得各浓度下的线性溶液,各配制1份。表5浓度水平10%50%100%150%线性储备液(ml)/123对照溶液(ml)1///容量瓶(ml)10505050操作:在2.2项色谱条件下,取空白溶液进样1针,取各浓度水平下线性溶液和定量限溶液,依次从低浓度到高浓度,每个浓度进样2次,记录色谱图。具体试验结果见表6,以吡啶峰面积平均值对其浓度作一元线性回归,线性回归方程为y=37784.335x-1294.439,相关系数r为1.000,符合要求;吡啶的y轴截距的绝对值与100%限度浓度对应的峰面积比值为1.74%,符合要求。本方法吡啶在0.08156μg/ml至3.0585μg/ml范围内线性良好。表6:线性结果3.5准确度空白溶液、供试品溶液的配制方法同2.1项,其中空白溶液配制1份,供试品溶液配制2份;线性储备液的配制:精密移取对照储备液25ml至100ml容量瓶中,用稀释剂稀释至刻度,摇匀即得,配制1份;准确度溶液的配制:取约500mg供试品,精密称定,置于50ml容量瓶中,平行称取9份;称好后,先向每个容量瓶中加入30~40ml稀释剂,将样品超声溶解,然后再按下表7分别移取不同体积的线性储备液于上述9个容量瓶中,每个浓度配制3份。再用稀释剂稀释至刻度,摇匀即得。表7浓度水平50%加标溶液100%加标溶液150%加标溶液线性储备液(ml)1.02.03.0容量瓶(ml)505050操作:在2.2项色谱条件下,取空白溶液进样1针,取2份供试品溶液各进1针,各浓度水平下准确度溶液各进样1针,记录色谱图。其中,按照以下公式分别计算加标前和加标后供试品溶液中吡啶含量、溶剂回收率:理论量=cs×v式中:cs为供试品加标溶液中加入的吡啶线性储备液的浓度,mg/ml;v为各供试品加标溶液中加入的吡啶线性储备液的体积,ml;cs+t为各供试品加标溶液中测得的吡啶残留量;ci为供试品溶液中测得的吡啶残留量;为供试品溶液中测得的吡啶残留量的平均值;ai为供试品溶液中吡啶峰面积;as为2针对照溶液中吡啶平均峰面积;as+t为各供试品加标溶液中吡啶的峰面积;wt为供试品溶液中供试品的称样量,mg;ws为对照溶液中吡啶的称样量,mg;ws+t为各供试品加标溶液中供试品的称样量,mg;dt为供试品溶液的稀释倍数;ds为对照溶液的稀释倍数;ds+t为各供试品加标溶液中供试品的稀释倍数。试验结果如表8所示,试验结果显示在限度浓度的50%,100%,150%下,吡啶回收率单值范围为91.20%~104.23%,在90%~110%之间,符合要求;吡啶回收率单值的rsd为4.50%,符合要求。因此该方法准确度良好。表8:准确度结果3.6耐用性考察3.6.1溶液稳定性试验对照溶液的配制方法同2.1项,配制1份;100%供试品加标溶液的配制:取供试品约100mg,精密称定至10ml容量瓶中,用对照溶液溶解并定容,摇匀即得,配制1份。操作:在2.2项色谱条件下,取对照溶液、100%供试品加标溶液在室温放置,分别于0h、7h、12h、19h、24h、48h及72h时进样1针,记录色谱图。室温下,在考察的72h内,各个时间点对照溶液中吡啶峰面积与0h峰面积比值范围为0.94~1.00(在0.90~1.10之间);100%供试品加标溶液中吡啶峰面积与0h峰面积比值范围为0.94~0.99(在0.90~1.10之间),符合要求。对照溶液和供试品溶液在室温下72小时内稳定。3.6.2色谱条件改变试验空白溶液、灵敏度溶液及对照溶液的配制方法同2.1项,空白溶液、灵敏度溶液及对照溶液各配制1份。操作:以2.2项下的色谱条件为基础条件,进行单因素改变,具体见表9。每个条件下,待基线平衡后,取空白溶液、灵敏度溶液、对照溶液各进样1针,记录色谱图。表9试验结果如表10所示,试验结果显示在改变的各色谱条件下,灵敏度溶液吡啶峰信噪比最小值为22.85;连续进样3针吡啶峰面积的rsd最大值为1.15%;对照溶液第一针中吡啶与相邻峰的分离度最小值≥1.5,均符合要求,该方法的耐用性良好。其中基于对照溶液中只有吡啶的峰,对照溶液第一针中吡啶与相邻峰的分离度是无限大。表10:耐用性结果在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1