本发明属于分析化学领域,具体涉及一种水溶液中短链脂肪酸的分析方法及应用。
背景技术:
短链脂肪酸属于挥发性脂肪酸,为碳原子数1~6的有机脂肪酸主要包括甲酸、乙酸、丙酸、丁酸、戊酸、己酸。短链脂肪酸包括甲酸,乙酸、丙酸、异丁酸、丁酸、异戊酸、戊酸,被后肠迅速吸收后,既储存了能量又降低了渗透压,并且短链脂肪酸对于维持大肠的正常功能和结肠上皮细胞的形态和功能具有重要作用。短链脂肪酸还可促进钠的吸收,丁酸在这方面的作用比乙酸和丙酸更强并且丁酸可增加乳酸杆菌的产量而减少大肠杆菌的数量。
油脂氧化过程生成短链脂肪酸,作为氧化二级产物生成后挥发构成油脂酸败哈喇味的一部分,以蒸馏水吸收裂解产物作为判定油脂劣化程度的指标之一。因此检测短链脂肪酸具有一定的必要性和实用性。关于短链脂肪酸的检测,国内外一般使用气相质谱联用方法,然而设备昂贵,检测费用高昂。此外也有报道气相色谱法检测短链脂肪酸,然而氢焰离子化检测器对甲酸的响应极为有限,无法实现检测目的。因此,亟待需要找到一种高效检测能够在水溶液中分析多种短链脂肪酸的分析方法。
技术实现要素:
本部分的目的在于提供一种检测水溶液中短链脂肪酸的反向高效液相色谱分析方法以及简要介绍一些较佳实施例。在本部分以及本申请的说明书摘要和发明名称中可能会做些简化或省略以避免使本部分、说明书摘要和发明名称的目的模糊,而这种简化或省略不能用于限制本发明的范围。
鉴于上述和/或现有短链脂肪酸检测中存在的问题,提出了本发明。
因此,本发明的其中一个目的是解决现有技术中的不足,提供一种保留时间短,色谱图的质量高的水溶液中短链脂肪酸的高效液相分析方法。
为解决上述技术问题,本发明提供了如下技术方案:一种水溶液中短链脂肪酸的分析方法,包括,制备标准溶液后测定标准曲线,然后将待测样品过滤膜,进高效液相色谱分析,计算各组分含量。
作为本发明所述水溶液中短链脂肪酸的分析方法的一种优选方案,其中: 所述高效液相色谱分析,其中,流速为1.0~3.0mL/min,柱温为35~45℃,进样量为10~100μL,流动相A包括磷酸二氢钾缓冲溶液,流动相B包括甲醇。
作为本发明所述水溶液中短链脂肪酸的分析方法的一种优选方案,其中:所述高效液相色谱分析,其洗脱方式为梯度洗脱,其是先以磷酸二氢钾:甲醇=80~95:5~20为起点,做7min等浓度淋洗,然后线性增大甲醇浓度,10~20min时达到磷酸二氢钾:甲醇=60~80:20~40,25~35min时以磷酸二氢钾:甲醇=60~80:20~40做等浓度淋洗,以上比例均为体积比。
作为本发明所述水溶液中短链脂肪酸的分析方法的一种优选方案,其中:所述制备标准溶液,其是分别称取甲酸、乙酸、丙酸、丁酸、戊酸、己酸用蒸馏水定容并形成不同浓度梯度,得到短链脂肪酸标准溶液。
作为本发明所述水溶液中短链脂肪酸的分析方法的一种优选方案,其中:所述高效液相色谱分析,其色谱柱选用C18柱,C18柱规格为φ4.6mm×150mm,5μm。
作为本发明所述水溶液中短链脂肪酸的分析方法的一种优选方案,其中:所述滤膜规格为0.3~0.6μm。
作为本发明所述水溶液中短链脂肪酸的分析方法的一种优选方案,其中:所述磷酸二氢钾缓冲溶液浓度为0.04~0.10mol/L,并用磷酸调节pH为2~3。
作为本发明所述水溶液中短链脂肪酸的分析方法的一种优选方案,其中:所述甲醇为色谱纯甲醇。
本发明还有一个目的是提供一种水溶液中短链脂肪酸的分析方法在油脂氧化产物样品检测方面的应用。
本发明所具有的有益效果:
(1)本发明所提供的分析方法,保留时间短,色谱图的质量高,分离效率高。
(2)本发明所提供的分析方法,相对标准差为2.30%,平均回收率96.88%,保留时间精密度为0.079~0.615%,峰面积精密度为1.12~3.50%。
(3)本发明所提供的分析方法,设备负荷小,方法简便高效。
(4)本发明所提供的分析方法可应用于油脂氧化产物检测领域,适用于样品分离纯化所得到的单一短链脂肪酸或若干短链脂肪酸的复合物。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其它的附图。其中:
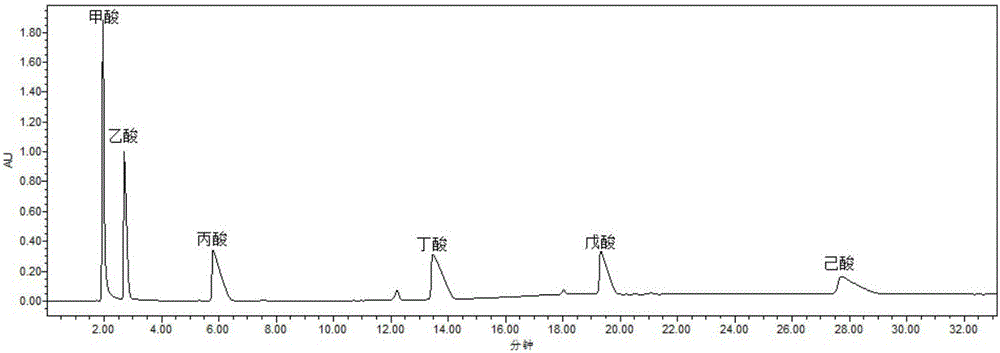
图1为本发明实施例1的混合样品的HPLC曲线。
图2为实施例4中油脂氧化过程中的短链脂肪酸变化图。
具体实施方式
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合具体实施例对本发明的具体实施方式做详细的说明。
在下面的描述中阐述了很多具体细节以便于充分理解本发明,但是本发明还可以采用其他不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似推广,因此本发明不受下面公开的具体实施例的限制。
其次,此处所称的“一个实施例”或“实施例”是指可包含于本发明至少一个实现方式中的特定特征、结构或特性。在本说明书中不同地方出现的“在一个实施例中”并非均指同一个实施例,也不是单独的或选择性的与其他实施例互相排斥的实施例。
试验仪器和色谱条件
1525型高效液相色谱仪(Waters公司,美国),SunFireTM C18色谱柱(φ4.6mm×150mm,5μm)(Waters公司,美国),实验用台式pH计(梅特勒-托利多公司,瑞士)。
用紫外分光光度仪在200nm~400nm波长内对制备的混合标准溶液进行洗脱及连续扫描,通过图像峰型、出峰情况判断每种物质的最大吸收波长,选择平均最大吸收波长215nm为本发明检测波长。
实施例1
标准曲线的测定
分别称取甲酸、乙酸、丙酸、丁酸、戊酸、己酸2g用蒸馏水定容50mL,分别量取0.5mL、1mL、2mL、4mL、8mL、16mL定容50ml,制得短链脂肪酸标准溶液。用所确定的色谱分析方法依次对标准溶液进行测定,以液相色谱出峰的绝对面积为纵坐标,标准溶液浓度为横坐标作图,得到测定水槽中各 种短链脂肪酸的线性范围和检测限。所得数据如下表:
实施例2
油脂氧化产物样品测定
采集四种油脂Rancimat氧化稳定性实验所得水溶液,经0.45μm滤膜过滤后,调整液相色谱条件如下,
色谱柱为C18柱,规格为φ4.6mm×150mm,5μm,
流速为1.0mL/min,
柱温为40℃,
检测波长为215nm,
进样量为10μL,
流动相A包括磷酸二氢钾缓冲溶液浓度为0.05mol/L,并用磷酸调节pH为2.8,流动相B包括色谱纯甲醇,
洗脱方式为梯度洗脱,其是先以磷酸二氢钾:甲醇=95:5为起点,做7min等浓度淋洗,然后线性增大甲醇浓度,16min时达到磷酸二氢钾:甲醇=60:40,然后以磷酸二氢钾:甲醇=60:40做等浓度淋洗,以上比例均为体积比。
在确定的以上液相色谱条件下测定其短链脂肪酸组成,见图1,处理结果后得到各组分的含量。
测定结果如下表:
注:ND表示为未检出。
采集样品经滤膜过滤后进样分析,在稳定操作条件下重复6次,方法的保留时间和峰面积精密度结果见表1。控制加标物体积不超过试样溶液体积的1%,加标实验结果见表2。结果表明,该法有较好的精密度和准确度。
表1方法精密度实验结果
表2加标实验结果
由此可见,本发明所提供的分析方法,相对标准差为2.30%,平均回收率96.88%,保留时间精密度为0.079~0.615%,峰面积精密度为1.12~3.50%。
实施例3(对比实施例)
油脂氧化产物样品测定
采集四种油脂Rancimat氧化稳定性实验所得水溶液,经0.45μm滤膜过滤后,调整液相色谱条件如下,
色谱柱为C18柱,规格为φ4.6mm×150mm,5μm,
流速为2.0mL/min,
柱温为22℃,
检测波长为217nm,
进样量为10ml,
流动相A包括磷酸二氢钾缓冲溶液浓度为0.05mol/L,并用磷酸调节pH为2.8,流动相B包括色谱纯甲醇,
洗脱方式为梯度洗脱,其是先以磷酸二氢钾:甲醇=90:10为起点,做15min等浓度淋洗,15min时线性调整为磷酸二氢钾:甲醇=70:30,25min时,25min时以磷酸二氢钾:甲醇=50:50做等浓度淋洗,以上比例均为体积比。
在确定的以上液相色谱条件下测定其短链脂肪酸组成,得到的谱图出现基线漂移现象,并且峰形质量差,色谱峰存在前沿与拖尾的问题。结果重复性不良,相对标准差为8.30%,精密度和准确度处于较低水平,平均回收率为89.58%。并且6次重复试验中,出现两次缓冲盐堵塞系统。设定的条件也使得机器负荷较大,色谱峰质量差与设定的条件有一定程度的关联。
发明人通过研究发现,梯度洗脱开始的时间点和前后的流动相组成,也是有选择的,尽管加大甲醇浓度缩短总体分析时间,但是又不能过快,造成AB相混合不均匀或者仪器产生气泡。甲醇含量过高,缓冲盐析出会堵塞系统,但是甲醇少了又会加长出峰时间。并且流速过快柱压太高导致仪器难以承受,随着脂肪酸碳原子数的增加,保留时间显著增加,选择合适的洗脱流速和梯度参数,对兼具分离效果与分析效率至关重要。
并且样品浓度直接影响液相出峰,浓度太小容易影响所测物质出峰,太大会导致出现平峰影响绝对面积积分。发明人进行的实验研究证实,对于在水溶液中检测分析短链脂肪酸,采用梯度洗脱能够有效地缩短液相分析时间,但梯度洗脱曲线易出现基线漂移,从图1可见,本发明提供的分析方法,色谱图质量很高。
发明人进行的实验研究证实,短链脂肪酸在95:5的流动相中分离效果佳,然而己酸的保留时间过长,达2h。进一步验证得流动相比例60:40可显著缩短戊酸和己酸的保留时间,且此比例可保证缓冲盐不会析出堵塞通道。选择丙酸出峰后开始梯度增加甲醇相,合适的梯度增加速率避免鬼峰,且最大程度缩 短了整体分析时间。
实施例4
油脂劣化评价指标方法
Rancimat氧化稳定测试仪是利用油脂氧化过程中产生的具有挥发性的二级产物改变蒸馏水电导率来测定油脂氧化酸败诱导期,并据此判断油脂氧化稳定性的分析仪器。在特定温度下向定量油脂中通入空气,油脂在热、氧环境下加速氧化产生挥发性小分子如醛、酮、挥发性酸等,小分子物质随气流进入测量池,多数酸溶解在水中,仪器依据水电导率变化判定油脂氧化程度。Rancimat氧化稳定评价实验因其操作简便、测量快速、重现性好等优点被广泛应用。
然而近年来,市场上不断出现富含不饱和脂肪酸、特殊营养成分的新型特种油品,氧化稳定性能评价过程中常常出现电导曲线异常、特定油脂测量结果重复性差等问题,而发明人通过研究发现,测量池水的电导率根本上是由溶解其中的短链脂肪酸决定。根据油脂自动氧化理论,从脂肪酸自由基反应开始,生成一级氧化产物氢过氧化物,后期氢过氧化物降解成中段碳链醛,由醛和氧气循环反应不断生成短链脂肪酸。短链脂肪酸为油脂自动氧化的二级产物,在仪器测量池中被蒸馏水吸收,导致蒸馏水电导率的改变。
大多数在Rancimat测试下产生的挥发性酸成分是甲酸和少量的乙酸、丙酸和其他酸。实验表明,甲酸的生成曲线和Rancimat电导率诱导曲线相当一致。Rancimat诱导曲线斜率跟甲酸生成速率成正比(如附图2)。油脂氢过氧化物分解遵循一级反应动力学。因此,基于油脂分解挥发脂肪酸的水溶液组成分析,可作为评价评价油脂氧化阶段的有效手段。
综上所述,本发明所提供的分析方法,保留时间短,色谱图的质量高,分离效率高;本发明所提供的分析方法,相对标准差为2.30%,平均回收率96.88%,保留时间精密度为0.079~0.615%,峰面积精密度为1.12~3.50%;本发明所提供的分析方法,设备负荷小,方法简便高效;本发明所提供的分析方法可应用于油脂氧化产物检测领域,适用于样品分离纯化所得到的单一短链脂肪酸或若干短链脂肪酸的复合物。
应说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精 神和范围,其均应涵盖在本发明的权利要求范围当中。