甲基化处理气相检测分析方法与流程
本发明涉及甲基化处理气相检测分析方法机械领域,具体涉及一种甲基化处理气相检测分析方法。
背景技术:
目前,现有灭草松(3一异丙基一(1H)一苯并一2,1,3一噻二嗪-4-酮一2,2一二氧化物)和2,4-滴(2,4-二氯苯氧基乙酸),化学结构式如下:
两者均为使用广泛的除草剂,毒性程度为低毒或中等。在GB5749-2006《生活饮用水卫生标准》规定生活饮用水中灭草松的限值为0.3mg/L,2,4-滴的限值为0.03mg/L。两者在水中残留的检测方法有国家标准和文献报道的有气相色谱、气相色谱-质谱联用、高效液相色谱、液相色谱-串联质谱、离子色谱法等方法。
国家生活饮用水检测标准GB/T5750.9-2006《生活饮用水标准检验方法农药指标》中对灭草松和2,4-滴的检测采用气相色谱法(配ECD检测器)进行检测,样品是需要衍生后方能分析的。
对于2,4-滴和灭草松直接检测的方法有离子色谱法、高效液相色谱、液相色谱-串联质谱这几种方法,但是存在以下问题:离子色谱分析时间较长,2,4-滴出峰时间12.917min、灭草松出峰时间42.947min,另外离子色谱检出限较高,2,4-滴为10μg/L、灭草松为30μg/L(崔勇,张冠英,李青.离子色谱法同时分析水中酸性除草剂灭草松和2,4-滴[J].中国卫生检验杂志,2010,20(2):295-296)。高效液相色谱法2,4-滴和灭草松的检测限可以做到3μg/L(陈锋,蔡少杰,李庆云,刘丽仪.高效液相色谱测定水中酸性除草剂灭草松和2,4-滴)[J].分析实验室,2008,27(Z1):286-288)。虽然离子色谱和高效液相色谱法的检测限均低于国家生活饮用水的执行标准GB5749-2006《生活饮用水卫生标准》的限值要求,然而比国家生活饮用水的检测标准GB/T5750.9-2006《生活饮用水标准检验方法农药指标》中的方法检出限却要高很多(在该检测标准中灭草松的检出限为0.2μg/L、2,4-滴的检出限为0.05μg/L),所以这两种方法虽然不需要进行衍生就能分析,但并不是非常的理想。
液相色谱-串联质谱在直接进样,选择多反应监测模式的条件下灭草松检出限为0.4μg/L、2,4-滴检出限为0.6μg/L(骆春迎,余辉菊.液相色谱-串联质谱测定生活饮用水中的灭草松和2,4-滴[J].中国卫生检验杂志,2014,24(21):3059-3061),考虑到该方法检出限是直接进样,水样没有经过浓缩得到的,因此推测如果水样经固相或液液萃取浓缩200倍再上仪器分析的话,检出限会低得多,应该可以满足国家生活饮用水检测标准GB/T5750.9-2006的检出限要求。尽管HPLC-MS/MS分析灭草松和2,4-滴非常方便,但目前国内还是难以大范围推广,原因之一是HPLC-MS/MS设备价格昂贵;二是环境和水质国家检测标准要使用HPLC-MS/MS来检测的项目非常少,因此大都数环境和水质的检测机构和单位并没有配置该设备;三是HPLC-MS/MS人员仪器操作培训花费的时间要比其它设备要多。
综合目前国内情况,对于生活饮用水中灭草松和2,4-滴检测使用最多的还是气相色谱(配ECD检测器)和气相色谱-气质联用两种方法,对这两种方法来说,前期的衍生化是必不可少的。衍生方法其中一种的是碘甲烷法,通过甲基化反应,使得灭草松和2,4-滴分别衍生成为甲基灭草松和2,4-滴甲酯:
这也是国家标准GB/T5750.9-2006《生活饮用水标准检验方法农药指标》规定的分析灭草松和2,4-滴的前处理方法。其它衍生方法还有重氮甲烷法(赵守成,董振霖,卫锋等.甲基化处理气相色谱-质谱法同时检测大米中12种酸性除草剂[J].质谱学报,2005,26(4):206-210),原理也是将两者甲基化后进行分析,其优点是衍生速度较快,副产物少,色谱图也很干净。但是因为重氮甲烷试剂有效期非常短,不能长久保存,难以买到商品化的试剂,此外由于重氮甲烷试剂属于高毒物质,制备过程也比较危险,所以使用该方法来做灭草松和2,4-滴的检测机构和单位是较少的。GB/T5750.9-2006《生活饮用水标准检验方法农药指标》规定的碘甲烷法是目前国内生活饮用水中灭草松和2,4-滴检测使用最普遍的方法,它的前处理大致过程是先通过乙酸乙酯萃取水中灭草松和2,4-滴,氮吹浓缩近干后加入碘甲烷-二氯甲烷溶和四丁基硫酸氢铵-氢氧化钠溶液,一并转移到烧杯里面水浴超声衍生,水浴温度需要控制在10℃—20℃,超声反应时间为50分钟,完成后再转移到分液漏斗将二氯甲烷层分离出来,再次氮吹近干,最后加正己烷定容后上气相色谱(ECD检测器)上分析。总体来说,碘甲烷法试剂易得,反应条件也不算苛刻,但是操作步骤是比较繁琐的(如氮吹就需要进行两次,液体转移次数也较多),此外超声温度控制也是有难度的(随着超声时间延长,超声仪里面的水在超声作用下温度是在逐步升高的,这在夏季尤其难控制)。
技术实现要素:
本发明针对上述问题,提出了一种甲基化处理气相检测分析方法,解决了现有甲基化处理气相检测分析方法中使用重氮甲烷法存在重氮甲烷试剂有效期非常短,不能长久保存,有高毒性;使用碘甲烷法存在操作繁琐,超声条件难以控制的缺陷。
本发明采取的技术方案如下:
一种甲基化处理气相检测分析方法,用于分析灭草松或2,4-滴;包括以下步骤:
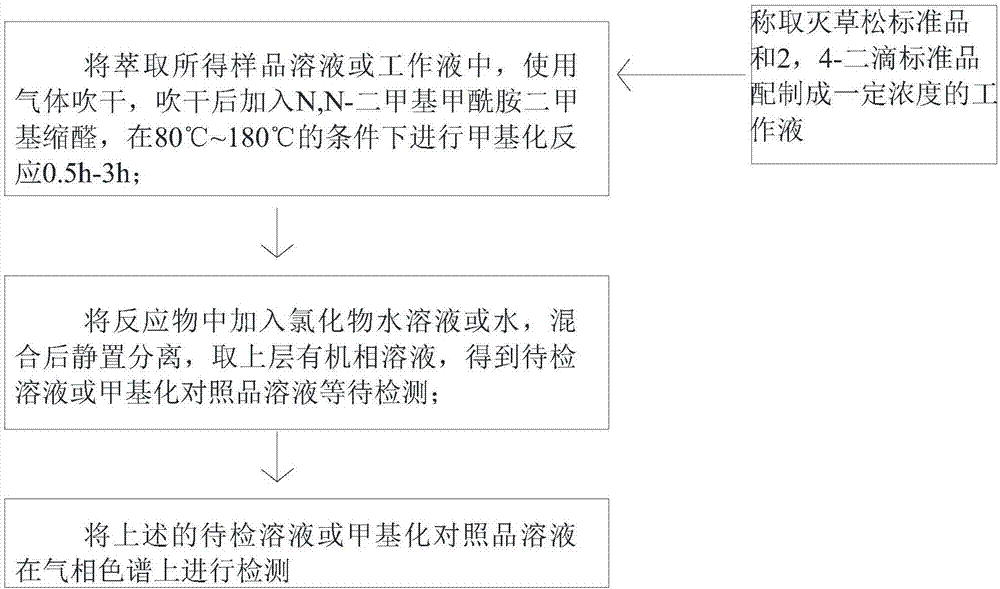
1)甲基化反应:将萃取得到的样品溶液移入容器中,使用气体吹干,吹干后加入N,N-二甲基甲酰胺二甲基缩醛,在80℃~180℃的条件下进行甲基化反应0.5h-3h;
2)萃取待检溶液:将反应物中加入氯化物水溶液或水,混合后静置分离,取上层有机相溶液,等待检测;
3)气相检测:将上述的待检溶液在气相色谱上进行检测。
本发明采用N,N-二甲基甲酰胺二甲基缩醛,将灭草松和2,4-滴甲基化,然后用气相色谱(ECD检测器又称电子俘获检测器)进行检测,该方法衍生速度快,灵敏度高,操作简单。
可选的,所述步骤1)和步骤2)中的混合采用涡旋振荡器,振荡混合时间为1min-5min。
可选的,所述步骤1)中,在甲基化反应前还加入保护溶剂。
可选的,所述保护溶剂为异辛烷。采用异辛烷为保护溶剂,不但有利于衍生反应进行,而且异辛烷会对衍生产物有保护作用,使得衍生物免遭高温破坏。
可选的,所述步骤1中,灭草松的甲基化反应更优的反应温度为160℃-170℃。在该反应温度,甲基化反应具有更优的收率。
可选的,所述步骤1)中,灭草松或2,4-滴的甲基化反应更优的反应时间为0.8h-1h。在该反应时间,甲基化反应具有更优的收率。
可选的,所述步骤1)中加入N,N-二甲基甲酰胺二甲基缩醛为200μL-500μL。
本发明的有益效果是:本发明采用N,N-二甲基甲酰胺二甲基缩醛,将灭草松和2,4-滴甲基化,然后用气相色谱(ECD检测器)进行检测,该方法衍生速度快,灵敏度高,操作简单。
附图说明:
图1是本发明甲基化处理气相检测分析方法的操作流程示意图;
图2是生活饮用水中灭草松标准曲线以及相关系数;
图3是生活饮用水中2,4滴标准曲线以及相关系数;
图4是灭草松和2,4-滴稳定性实验收率和衍生物的稳定性折线图;
图5是灭草松和2,4-滴水样加标回收率色谱图。
具体实施方式:
下面结合各附图,对本发明做详细描述。
N,N-二甲基甲酰胺二甲基缩醛(CAS号4637-24-5),简称DMFDMA,无色透明液体,沸点104℃-108℃,分子结构下:
在本发明中常用的气相检测仪器可以选用:安捷伦6890N气相色谱仪,配μECD检测器(称微电子捕获器);在实际操作中还用到了可控温烘箱、氮吹仪、漩涡振荡器。
采用的气相分析条件包括:DB-35毛细管柱,30m×0.25mm×0.25μm,进样方式为不分流进样。
用到的标准品为:灭草松标准品,德国“Dr.Ehrenstorfer”公司生产,纯度99.0%,生产批号30814;2,4-滴标准品,德国“Dr.Ehrenstorfer”公司生产,纯度99.0%,生产批号40616;甲基灭草松标准溶液,美国“O2Si smart solutions”公司生产,溶剂正己烷,浓度1012μg/mL,生产批号269733;2,4-滴甲酯标准溶液,溶剂甲醇,美国“O2Si smart solutions”公司生产,浓度99.9μg/mL,生产批号253560。
N,N-二甲基甲酰胺二甲基缩醛采用“上海百灵威化学技术有限公司”提供,纯度≥95.0%。
色谱纯乙酸乙酯可采用“上海安谱实验科技有限公司”生产,色谱纯异辛烷可采用“天津科密欧化学试剂有限公司”生产,色谱纯丙酮可采用“默克”公司生产,硝酸为优级纯,国药集团生产。
水样的处理:按照国家标准GB/T5750.9-2006《生活饮用水标准检验方法农药指标》,先用250mL水样加入1.1mL硝酸,使得水样pH<1,样品置于4℃冰箱保存。
本发明分析方法衍生物检测线性分析:
(1)工作液配制:用万分之一天平称取灭草松标准品10.0mg,2,4-滴标准品10.0mg一并到25mL棕色容量瓶内,先加10mL丙酮超声溶解,再加丙酮定容至刻度,配制成2,4-滴、灭草松的浓度各为400μg/mL母液;用1mL的胖度移液管从母液中移去1.0mL到10mL容量瓶内,加丙酮定容至刻度,配成40μg/mL的工作液。
(2)衍生过程:用移液枪移取2,4-滴、灭草松40μg/mL工作液5μL、10μL、20μL、40μL、80μL、160μL各到衍生试管中,氮气吹干,各加入400μL的N,N-二甲基甲酰胺二甲基缩醛和1.0mL异辛烷,加盖密封,漩涡1min,放入烘箱内180℃衍生30min。完毕后取出恢复室温,加入饱和氯化钠水溶液1.5mL,加盖再次漩涡1min,漩涡后静置5min,待异辛烷和水相彻底分层后,用移液枪移取上层异辛烷0.8mL装入2mL安捷伦进样小瓶内,共制得浓度为0.2μg/mL、0.4μg/mL、0.8μg/mL、1.6μg/mL、3.2μg/mL、6.4μg/mL六个浓度的标液,以萃取200mL水样来计,工作曲线质量浓度相当于水中浓度的1.0μg/L、2.0μg/L、4.0μg/L、8.0μg/L、16.0μg/L、32μg/L。灭草松的回归方程Y=2996.906X-1205.646,相关系数r为0.999;2,4-滴回归方程Y=2830.253X-1512.424,相关系数r为0.999(见附图2、3)。进一步的,样品经过GC-MS定性衍生产物为甲基灭草松和2,4-滴甲酯。
本发明分析方法衍生物收率和衍生物的稳定性:
实验过程为:
一组待处理的水样灭草松和2,4-滴均为2μg/L;另一组待处理的水样灭草松和2,4-滴均为16μg/L。
1)水样预处理:先量取水样200mL置于500mL的分液漏斗中,加乙酸乙酯50mL萃取水样,共三次,一共150mL合并于200mL烧杯中,水浴45℃进行氮吹,当乙酸乙酯还剩4-5mL时候转入衍生管,烧杯再加乙酸乙酯2mL润洗,一起合并到衍生管内,继续水浴氮吹干。
2)甲基化反应:取有机相内的样品溶液移入容器中,取有机相内的样品溶液移入容器中,氮气吹干,吹干后加入400μL的N,N-二甲基甲酰胺二甲基缩醛和1.0mL异辛烷,加盖密封,漩涡5min,放入烘箱内160℃甲基化反应0.5h。
3)萃取待检溶液:毕后取出恢复室温,加入饱和氯化钠水溶液1.5mL,加盖再次漩涡1min,漩涡后静置5min,待异辛烷和水相彻底分层后,用移液枪移取上层异辛烷0.8mL装入2mL安捷伦进样小瓶内;
4)对照品溶液的配制:配制等浓度的甲基灭草松和2,4-滴甲酯溶液作为衍生收率和稳定性的对照品溶液。
5)气相检测:样品衍生完成后立即进样,得到的该组数据为0小时数据,然后将样品放置于3-5℃的冰箱内,24小时、48小时、72小时后再次进样分析(进样前样品恢复至室温),记录的数据并编制表1、表2和附图4。样品处理和数据处理同本发明分析方法衍生物检测线性分析,说明:
表1 2,4-滴衍生物收率和稳定性实验
表2灭草松衍生物收率和稳定性实验
从实验的数据和折线图(见附图4)来看,灭草松和2,4-滴的甲基化产物尽管在72小时内含量略有降低,但不明显(最大下降幅度不超过6%)。因此,可以认为样品衍生后只要保存在3-5℃环境中,并处以2mL密封小瓶包装下,至少可以存放72小时。取6个平行样品衍生峰的峰面积平均值,并计算RSD值,规定当RSD值大于15%时,数据不采用,该组实验重新进行,同时做等浓度甲基灭草松和2,4-滴甲酯对照品溶液,通过单点比较法,计算衍生物的收率。
实施例一:本发明公开了一种甲基化处理气相检测分析方法,
1)水样预处理:先量取水样200mL置于500mL的分液漏斗中,加乙酸乙酯50mL萃取水样,共三次,一共150mL合并于200mL烧杯中,水浴45℃进行氮吹,当乙酸乙酯还剩4-5mL时候转入衍生管,烧杯再加乙酸乙酯2mL润洗,一起合并到衍生管内,继续水浴氮吹干。
2)甲基化反应:取有机相内(也就是萃取得到的)的样品溶液移入衍生试管中,氮气吹干,吹干后加入200μL的N,N-二甲基甲酰胺二甲基缩醛和1.0mL异辛烷,加盖密封,漩涡1min,放入烘箱内80℃甲基化反应0.5h。
3)萃取待检溶液:毕后取出恢复室温,加入饱和氯化钠水溶液1.5mL,加盖再次漩涡1min,漩涡后静置5min,待异辛烷和水相彻底分层后,用移液枪移取上层异辛烷0.8mL装入2mL安捷伦进样小瓶内。
4)对照品溶液的配制及衍生:根据上述的对照品溶液标准曲线的工作液配制方法进行配置,并按照其衍生过程进行操作。
5)气相检测:将上述的待检溶液和甲基化的对照品溶液在气相色谱上进行检测。分析后得灭草松和2,4-滴回收率为:2μg/L样品的2,4-滴平均回收率为81.6%,灭草松平均为82.7%;16μg/L样品2,4-滴平均回收率为79.9%,灭草松平均为80.3%;精密度:2μg/L样品2,4-滴为7.7%,灭草松7.2%;16μg/L样品2,4-滴为9.5%,灭草松8.9%。
本发明的水样预处理按照国家标准GB/T5750.9-2006《生活饮用水标准检验方法农药指标》进行操作;本发明采取氯化钠饱和水溶液,减少了目标产物溶于水而产生的损失。
实施例二:
1)水样预处理:先量取水样200mL置于500mL的分液漏斗中,加乙酸乙酯50mL萃取水样,共三次,一共150mL合并于200mL烧杯中,水浴45℃进行氮吹,当乙酸乙酯还剩4-5mL时候转入衍生管,烧杯再加乙酸乙酯2mL润洗,一起合并到衍生管内,继续水浴氮吹干。
2)甲基化反应:取有机相内的样品溶液移入容器中,取有机相内的样品溶液移入容器中,氮气吹干,吹干后加入500μL的N,N-二甲基甲酰胺二甲基缩醛和1.0mL异辛烷,加盖密封,漩涡5min,放入烘箱内180℃甲基化反应3h。
3)萃取待检溶液:毕后取出恢复室温,加入饱和氯化钠水溶液1.5mL,加盖再次漩涡1min,漩涡后静置5min,待异辛烷和水相彻底分层后,用移液枪移取上层异辛烷0.8mL装入2mL安捷伦进样小瓶内。
4)对照品溶液的配制及衍生:根据上述的对照品溶液标准曲线的工作液配制方法进行配置,并按照其衍生过程进行操作。
5)气相检测:将上述的待检溶液和对照品溶液在气相色谱上进行检测。分析后得灭草松和2,4-滴回收率为:2μg/L样品的2,4-滴平均回收率为86.4%,灭草松平均为93.7%;16μg/L样品2,4-滴平均回收率为85.2%,灭草松平均为93.3%;精密度:2μg/L样品2,4-滴为6.8%,灭草松6.3%;16μg/L样品2,4-滴为8.8%,灭草松8.2%。
实施例三:
1)水样预处理:先量取水样200mL置于500mL的分液漏斗中,加乙酸乙酯50mL萃取水样,共三次,一共150mL合并于200mL烧杯中,水浴45℃进行氮吹,当乙酸乙酯还剩4-5mL时候转入衍生管,烧杯再加乙酸乙酯2mL润洗,一起合并到衍生管内,继续水浴氮吹干。
2)甲基化反应:取有机相内的样品溶液移入容器中,取有机相内的样品溶液移入容器中,氮气吹干,吹干后加入400μL的N,N-二甲基甲酰胺二甲基缩醛和1.0mL异辛烷,加盖密封,漩涡5min,放入烘箱内160℃甲基化反应1h。
3)萃取待检溶液:毕后取出恢复室温,加入饱和氯化钠水溶液1.5mL,加盖再次漩涡1min,漩涡后静置5min,待异辛烷和水相彻底分层后,用移液枪移取上层异辛烷0.8mL装入2mL安捷伦进样小瓶内;
4)对照品溶液的配制及衍生:根据上述的对照品溶液标准曲线的工作液配制方法进行配置,并按照其衍生过程进行操作。
5)气相检测:将上述的待检溶液和对照品溶液在气相色谱上进行检测。分析后得灭草松和2,4-滴回收率为:2μg/L样品的2,4-滴平均回收率为92.6%,灭草松平均为97.7%;16μg/L样品2,4-滴平均回收率为92.9%,灭草松平均为98.9%;精密度:2μg/L样品2,4-滴为2.0%,灭草松1.8%;16μg/L样品2,4-滴为4.5%,灭草松4.2%。
实施例四:
1)水样预处理:先量取水样200mL置于500mL的分液漏斗中,加乙酸乙酯50mL萃取水样,共三次,一共150mL合并于200mL烧杯中,水浴45℃进行氮吹,当乙酸乙酯还剩4-5mL时候转入衍生管,烧杯再加乙酸乙酯2mL润洗,一起合并到衍生管内,继续水浴氮吹干。
2)甲基化反应:取有机相内的样品溶液移入容器中,取有机相内的样品溶液移入容器中,氮气吹干,吹干后加入400μL的N,N-二甲基甲酰胺二甲基缩醛和1.0mL异辛烷,加盖密封,漩涡5min,放入烘箱内170℃甲基化反应0.8h。
3)萃取待检溶液:毕后取出恢复室温,加入饱和氯化钠水溶液1.5mL,加盖再次漩涡1min,漩涡后静置10min,待异辛烷和水相彻底分层后,用移液枪移取上层异辛烷0.8mL装入2mL安捷伦进样小瓶内;
4)对照品溶液的配制及衍生:根据上述的对照品溶液标准曲线的工作液配制方法进行配置,并按照其衍生过程进行操作。
5)气相检测:将上述的待检溶液和对照品溶液在气相色谱上进行检测。分析后得灭草松和2,4-滴回收率为:2μg/L样品的2,4-滴平均回收率为93.7%,灭草松平均为97.9%;16μg/L样品2,4-滴平均回收率为93.2%,灭草松平均为98.6%;精密度:2μg/L样品2,4-滴为1.7%,灭草松1.5%;16μg/L样品2,4-滴为4.1%,灭草松3.8%,
本发明的检出限:以3倍噪声比计,本方法检出限2,4-滴为0.04μg/L,灭草松为0.03μg/L。可以比较看出,本发明检出限优于其他的检测方法(离子色谱检出限:2,4-滴为10μg/L、灭草松为30μg/L。高效液相色谱法2,4-滴和灭草松的检测限为3μg/L);也低于GB/T5750.9-2006《生活饮用水标准检验方法农药指标》标准中灭草松的检出限0.2μg/L、2,4-滴的检出限0.05μg/L。本发明采用N,N-二甲基甲酰胺二甲基缩醛,将灭草松和2,4-滴甲基化,然后用气相色谱(ECD检测器)进行检测,该方法衍生速度快,灵敏度高,操作简单。
对比例:1)水样预处理:先量取水样200mL置于500mL的分液漏斗中,加乙酸乙酯50mL萃取水样,共三次,一共150mL合并于200mL烧杯中,水浴45℃进行氮吹,当乙酸乙酯还剩4-5mL时候转入衍生管,烧杯再加乙酸乙酯2mL润洗,一起合并到衍生管内,继续水浴氮吹干。
2)甲基化反应:取有机相内的样品溶液移入容器中,取有机相内的样品溶液移入容器中,氮气吹干,吹干后加入100μL的N,N-二甲基甲酰胺二甲基缩醛和1.0mL异辛烷,加盖密封,漩涡1min,放入烘箱内78℃甲基化反应0.2h。
3)萃取待检溶液:毕后取出恢复室温,加入饱和氯化钠水溶液1.5mL,加盖再次漩涡1min,漩涡后静置5min,待异辛烷和水相彻底分层后,用移液枪移取上层异辛烷0.8mL装入2mL安捷伦进样小瓶内;
4)对照品溶液的配制及衍生:根据上述的对照品溶液标准曲线的工作液配制方法进行配置,并按照其衍生过程进行操作。
5)气相检测:将上述的待检溶液和对照品溶液在气相色谱上进行检测。分析后得灭草松和2,4-滴回收率为:2μg/L样品的2,4-滴平均回收率为70.6%,灭草松平均为68.7%;16μg/L样品2,4-滴平均回收率为72.3%,灭草松平均为68.2%;精密度:2μg/L样品2,4-滴为14.4%,灭草松15.2%;16μg/L样品2,4-滴为18.5%,灭草松19.9%。
回收率实验和精密度实验:用纯净水制备含灭草松和2,4-滴为2μg/L和16μg/L的两种加标水样,两种浓度都平行制备6个样品,分别进行萃取和衍生;分析后得到灭草松和2,4-滴水样加标回收率色谱图(见图5)。
本发明选择异辛烷作为衍生反应溶剂,具有以下优点:1)是异辛烷试剂性质稳定,即使在高温高压下自身也不会参与反应;2)是纯度高的异辛烷试剂易得,一般色谱纯级异辛烷纯度至少可以达到≥99.5%,较高的纯度可以尽可能减少由溶剂带入的杂质参与衍生反应,使得衍生反应比较专一,色谱图干扰峰少;3)是异辛烷沸点比较适中,沸点99.3℃,比正己烷(沸点68℃)要高,虽然跟衍生温度相比,异辛烷沸点还是不够高,但如果选择沸点很高的溶剂也会带来后续分析问题,比如高沸点溶剂和目标物的色谱分离难度增大等;4)是异辛烷可以和N,N-二甲基甲酰胺二甲基缩醛互溶,但只要加入水两者就可以得到分离,前期衍生和后续处理就很方便。采用异辛烷为保护溶剂,不但有利于衍生反应进行,而且异辛烷会对衍生产物有保护作用,使得衍生物免遭高温破坏。
综上所述,通过实施例一、二、三、四和对比例的数据进行比较,可以看出,甲基化反应的反应温度为160℃-170℃,反应时间为0.8h-1h,具有更高的灭草松和2,4-滴回收率和更优的精密度(实施例三、实施例四中精密度均低于5%)。超出预定的反应温度和反应时间,灭草松和2,4-滴的回收率大大低于实施例的收率;其精密度大大差于实施例的数据。而且N,N-二甲基甲酰胺二甲基缩醛加入量为400μL-500μL时;具有更高的灭草松和2,4-滴回收率和最佳的精密度。
以上所述仅为本发明的优选实施例,并非因此即限制本发明的专利保护范围,凡是运用本发明说明书及附图内容所作的等效结构变换,直接或间接运用在其他相关的技术领域,均同理包括在本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!