一种醒脑静注射液或其中间体中多指标成分含量的测定方法与流程
本发明属于中药成分含量测定领域,具体涉及一种醒脑静注射液或其中间体中多指标成分含量的测定方法。
背景技术:
醒脑静注射液是由麝香、郁金、栀子、冰片四味中药制成的注射剂,具有清热泻火、凉血解毒、开窍醒脑的功效,临床上主要用于中风、脑缺血、意识障碍、急性酒精中毒等病症。醒脑静注射液的现行质量标准是2003年的国家药品标准(ws3-b-3353-98-2003),该标准仅对注射液中的麝香酮进行鉴别,对冰片进行含量测定,而对于栀子、郁金中的成分并没有进行质量控制。
多篇文献报道了对醒脑静注射液中麝香酮和冰片的含量测定方法。有文献报道了对醒脑静注射液中来源于郁金的莪术双环烯酮、莪术烯醇、莪术二酮、莪术酮、吉马酮5个倍半萜类成分的含量测定方法。公开号为cn105548385a的中国专利文献公开了一种醒脑静注射液液相指纹图谱测定方法,公开号为cn102091283a的中国专利文献公开了一种醒脑静注射液气相指纹图谱测定方法。指纹图谱测定方法虽然能以半定量的方式对注射液质量进行评价,但不能对注射液中各指标成分的含量进行准确测定。
另一方面,栀子在脑血管疾病中的治疗作用已被多篇文献报道,而对于醒脑静注射液中源于栀子的成分目前尚缺少含量测定方法,这不利于实现对该产品的全面质量控制。此外,对于醒脑静注射液中莪术呋喃二烯酮的含量测定方法,目前尚无报道。
由于栀子、郁金中的挥发性成分并不稳定,且莪术酮、莪术双环烯酮、4-亚甲基-异佛尔酮等成分的对照品非常紧缺且价格昂贵,若采用常规的多个对照品外标定量测定方法,分析成本太高。一测多评法是一种基于待测成分间的相对校正因子,以一个内参物对照品代替多个成分对照品进行定量的方法,可用于解决对照品紧缺和检测成本高的问题,是一种适合中药特点的多指标质量评价新模式。因此为了在生产中控制好醒脑静注射液中间体及其成品的质量,很有必要在现有技术基础之上研究设计出能准确且低成本地测定源于栀子和郁金的多指标成分含量的方法。
技术实现要素:
本发明的目的是为了解决上述问题,基于一测多评法,提供一种能够准确测定醒脑静注射液或其中间体中异佛尔酮、4-亚甲基-异佛尔酮、莪术双环烯酮、莪术烯醇、莪术二酮、莪术酮、莪术呋喃二烯酮、莪术醇和吉马酮9种成分含量的高效液相色谱分析方法。该方法检测灵敏度高,操作简便、实用性高、检测成本低,可以客观、全面、准确地评价醒脑静注射液或其中间体的质量,解决因对照品缺乏而无法准确测定多种指标成分含量的问题。
为了实现以上目的,本发明采取的技术方案为:
一种醒脑静注射液或其中间体中多指标成分含量的测定方法,包括以下步骤:
(1)供试品溶液的制备
直接将醒脑静注射液或其中间体作为供试品溶液进行分析;本发明醒脑静注射液中间体指栀子和郁金的水蒸气蒸馏提取液;
(2)对照品溶液的制备
分别精密称取异佛尔酮、4-亚甲基-异佛尔酮、莪术双环烯酮、莪术烯醇、莪术二酮、莪术酮、莪术呋喃二烯酮、莪术醇、吉马酮9种成分的对照品适量,分别用质量分数为30~100%的甲醇水溶液溶解并定容,制成具有已知浓度的各指标成分的单一对照品储备液;
再分别精密吸取各成分的单一对照品储备液适量,混合后加质量分数为10~50%的甲醇水溶液稀释得到适宜浓度的混合对照品溶液,备用;
(3)相对校正因子的测定
取步骤(2)制备的混合对照品溶液,注入高效液相色谱仪,得到混合对照品溶液中莪术二酮和其他各待测成分的峰面积,以莪术二酮为内参物,以相对校正因子计算公式fx/s=fx/fs=(mx×as)/(ms×ax)分别计算异佛尔酮、4-亚甲基-异佛尔酮、莪术双环烯酮、莪术烯醇、莪术酮、莪术呋喃二烯酮、莪术醇和吉马酮8种成分对莪术二酮的相对校正因子,
式中,fs为莪术二酮的校正因子,fx为除莪术二酮以外的某一待测成分的校正因子,ms是混合对照品溶液中莪术二酮的进样质量,as是混合对照品溶液中莪术二酮进样质量为ms时测得的峰面积,mx是混合对照品溶液中除莪术二酮以外的某一待测成分的进样质量,ax是混合对照品溶液中除莪术二酮以外的某一待测成分进样质量为mx时测得的峰面积;
(4)样品中多指标成分的测定
精密吸取供试品溶液,注入高效液相色谱仪,得到供试品溶液中各待测成分的峰面积,供试品溶液中莪术二酮的进样质量ms’由莪术二酮单一对照品溶液以外标定量法测定得到,供试品溶液中除莪术二酮以外的其他某一待测成分的进样质量mx’由公式mx’=fx/s×ms’×(ax’/as’)计算得到,
式中,as’和ax’分别是供试品溶液中测得的莪术二酮和除莪术二酮以外的其他某一待测成分的峰面积,fx/s是步骤(3)测得的异佛尔酮、4-亚甲基-异佛尔酮、莪术双环烯酮、莪术烯醇、莪术酮、莪术呋喃二烯酮、莪术醇和吉马酮8种成分对莪术二酮的相对校正因子;
供试品溶液中各待测成分的浓度由测定或计算得到的进样质量ms’和mx’经换算得到。
本发明醒脑静注射液中间体指栀子和郁金的水蒸气蒸馏提取液,由于栀子、郁金中的挥发性成分并不稳定,且莪术酮、莪术双环烯酮、4-亚甲基-异佛尔酮等成分的对照品非常紧缺且价格昂贵,若采用常规的多个对照品外标定量测定方法,分析成本太高,本方法基于一测多评法,能准确且低成本地测定源于栀子和郁金的多指标成分含量的方法。
所述测定方法中,高效液相色谱仪测定的条件为:色谱柱:反相c18色谱柱;流动相:a相为磷酸水溶液,b相为乙腈;洗脱程序为线性梯度洗脱;流速:0.5~0.7ml/min;柱温:35~45℃;检测波长:异佛尔酮,238nm±2nm;4-亚甲基-异佛尔酮,275nm±2nm;莪术双环烯酮,256nm±2nm;莪术烯醇,261nm±2nm;莪术二酮,220nm±2nm;莪术酮,274nm±2nm;莪术呋喃二烯酮,274nm±2nm;莪术醇,210nm±2nm;吉马酮,210nm±2nm。
作为优选,所述的流动相中,磷酸水溶液的质量分数为0.05~0.2%。
作为优选,所述的洗脱程序中,磷酸水溶液占流动相体积比为:0min,28~32%;10min,53~57%;24min,53~57%;24.1min,68~72%;35min,68~72%;40min,98~100%。
作为优选,步骤(3)中,异佛尔酮、4-亚甲基-异佛尔酮、莪术双环烯酮、莪术烯醇、莪术酮、莪术呋喃二烯酮、莪术醇、吉马酮与莪术二酮间的相对校正因子分别为0.177±0.01、0.208±0.01、0.497±0.01、0.602±0.01、1.59±0.05、0.768±0.02、0.790±0.05、0.275±0.01。
与现有技术相比,本发明具有以下有益效果:
(1)本发明提供一种能够准确测定醒脑静注射液或其中间体中9种成分含量的高效液相色谱分析方法。既能对醒脑静注射液中源于栀子的2个主要成分(异佛尔酮和4-亚甲基-异佛尔酮)进行定量测定,又能对源于郁金的7个主要成分(莪术双环烯酮、莪术烯醇、莪术二酮、莪术酮、莪术呋喃二烯酮、莪术醇、吉马酮)进行定量测定,可以更全面、准确地评价醒脑静注射液或其中间体的质量。
(2)本发明对流动相的组成、流动相流速、柱温进行了大量筛选,确定采用优选的色谱条件,可以同时保证9个指标成分在色谱分析中达到很好的分离。本发明通过紫外全波长扫描,确定9个指标成分合适的检测波长,可以灵敏地检测各指标成分。
(3)本发明基于一测多评法,利用待测成分间的相对校正因子,以莪术二酮单一对照品作为内参物代替多个成分对照品进行定量,可用于解决对照品紧缺和检测成本高的问题。本方法检测成本低、实用性高,具有很好的应用前景和使用价值。
(4)对本发明提供的分析方法进行方法学验证表明,该方法精密度、重复性、准确性、灵敏度均很好。本发明的测定结果通过外标定量法进行验证,结果表明,本发明的测定结果与外标定量法相似度极高,可以准确灵敏地测定各指标成分。
附图说明
图1为实施例1中混合对照品的高效液相色谱图;
图2为实施例1中栀子和郁金水蒸气蒸馏提取液的高效液相色谱图;
图3为实施例2中醒脑静注射液的高效液相色谱图。
具体实施方式
下面结合具体实施例进一步阐明本发明,应理解这些实施例仅用于说明本发明而不用于限制本发明的范围,在阅读了本发明之后,本领域技术人员对本发明的各种等价形式的修改均落于本申请所附权利要求所限定的范围。
实施例1
一种醒脑静注射液中间体中多指标成分含量的测定方法,该方法包括以下步骤:
(1)栀子和郁金水蒸气蒸馏提取液的制备
栀子、郁金药材分别粉碎、过筛至20~80目,各取30g粉末,加蒸馏水1500ml,进行水蒸气蒸馏至蒸馏液体积为1000ml时停止蒸馏,即得栀子和郁金水蒸气蒸馏提取液。
(2)供试品溶液的制备
直接将步骤(1)得到的栀子和郁金水蒸气蒸馏提取液(即醒脑静注射液生产过程中的中间体)作为供试品溶液,无前处理和稀释。
(3)对照品溶液的制备
精密称取21.67mg异佛尔酮、12.90mg4-亚甲基-异佛尔酮、13.73mg莪术双环烯酮、16.82mg莪术烯醇、16.25mg莪术二酮、10.32mg莪术酮、12.00mg莪术呋喃二烯酮、11.11mg莪术醇、16.11mg吉马酮9种成分的对照品,分别加质量分数为80%甲醇水溶液溶解并定容至250、50、50、50、50、5、25、50、50ml,制成单一对照品储备液;
然后分别精密吸取上述各成分的单一对照品储备液240、295、330、930、1250、120、250、200、146μl,混合后加质量分数为20%甲醇水溶液稀释定容至25ml,得到异佛尔酮、4-亚甲基-异佛尔酮、莪术双环烯酮、莪术烯醇、莪术二酮、莪术酮、莪术呋喃二烯酮、莪术醇、吉马酮浓度分别为0.834、3.04、3.62、12.5、16.2、9.91、4.80、1.78、1.88μg/ml的混合对照品溶液,于4℃保存,备用。
(4)相对校正因子的测定
以莪术二酮为内参物(以符号s表示),ms是其进样质量,as是内参物进样质量为ms时测得的峰面积,fs=ms/as是其校正因子。对于其他待测成分(以符号x表示),mx是x成分的进样质量,ax是x成分进样质量为mx时测得的峰面积,fx=mx/ax是x成分的校正因子。x成分与s成分间的相对校正因子fx/s的计算公式为fx/s=fx/fs=(mx×as)/(ms×ax)。
取步骤(3)制备得到的混合对照品溶液,进样50μl进行高效液相色谱分析,得到混合对照品溶液的高效液相色谱图(如图1所示),图1中,检测波长:0~15.5min,238nm;15.5~19min,275nm;19~20min,256nm;20~24min,261nm;24~26min,220nm;26~30.5min,274nm;30.5min后,210nm),峰1:异佛尔酮;峰2:4-亚甲基-异佛尔酮;峰3:莪术双环烯酮;峰4:莪术烯醇;峰5:莪术二酮;峰6:莪术酮;峰7:莪术呋喃二烯酮;峰8:莪术醇;峰9:吉马酮。
色谱图积分得到莪术二酮和其他各待测成分的峰面积,并以公式fx/s=(mx×as)/(ms×ax)分别计算各待测成分与莪术二酮间的相对校正因子,异佛尔酮、4-亚甲基-异佛尔酮、莪术双环烯酮、莪术烯醇、莪术酮、莪术呋喃二烯酮、莪术醇和吉马酮8种成分对于莪术二酮的相对校正因子分别为0.177、0.209、0.497、0.600、1.54、0.750、0.842、0.269。
(5)样品中多指标成分的测定
取步骤(2)的供试品溶液,进样50μl注入高效液相色谱仪进行测定,得到栀子和郁金水蒸气蒸馏提取液的高效液相色谱图(如图2所示),图2中,检测波长:0~15.5min,238nm;15.5~19min,275nm;19~20min,256nm;20~24min,261nm;24~26min,220nm;26~30.5min,274nm;30.5min后,210nm),峰1:异佛尔酮;峰2:4-亚甲基-异佛尔酮;峰3:莪术双环烯酮;峰4:莪术烯醇;峰5:莪术二酮;峰6:莪术酮;峰7:莪术呋喃二烯酮;峰8:莪术醇;峰9:吉马酮,色谱图积分得到莪术二酮和其他各待测成分的峰面积。
供试品溶液中莪术二酮的进样质量ms’由莪术二酮对照品以外标定量法测定得到,供试品溶液中其他待测成分的进样质量mx’由公式mx’=fx/s×ms’×(ax’/as’)计算得到。此公式中的as’和ax’是对供试品溶液分析测得的内参物和其他待测成分的峰面积,fx/s是步骤(3)测得的各待测成分与莪术二酮间的相对校正因子。由测定或计算得到的进样质量ms’和mx’,除以进样体积,计算得到样品中各待测成分的浓度,结果如下:异佛尔酮0.851μg/ml,4-亚甲基-异佛尔酮3.22μg/ml,莪术双环烯酮3.60μg/ml,莪术烯醇10.3μg/ml,莪术二酮13.2μg/ml,莪术酮9.89μg/ml,莪术呋喃二烯酮4.46μg/ml,莪术醇1.07μg/ml,吉马酮2.04μg/ml。
(6)步骤(4)和步骤(5)所用的高效液相色谱参数如下:
仪器:waterse2695高效液相色谱仪;
色谱柱:迪马钻石c18色谱柱(5μm,250×4.6mm);
流动相:质量分数0.1%磷酸水溶液-乙腈;
洗脱程序为线性梯度洗脱,0.05%~0.2%磷酸水溶液占流动相体积比为:0min,30%;10min,55%;24min,55%;24.1min,70%;35min,70%;40min,100%;
流速:0.6ml/min;
柱温:40℃;
检测波长:异佛尔酮,238nm;4-亚甲基-异佛尔酮,275nm;莪术双环烯酮,256nm;莪术烯醇,261nm;莪术二酮,220nm;莪术酮,274nm;莪术呋喃二烯酮,274nm;莪术醇,210nm;吉马酮,210nm。
(7)本发明方法的方法学考察结果如下:
(7.1)线性关系考察
精密吸取步骤(3)制备得到的异佛尔酮、4-亚甲基-异佛尔酮、莪术双环烯酮、莪术烯醇、莪术二酮、莪术酮、莪术呋喃二烯酮、莪术醇、吉马酮单一对照品储备液240、295、330、930、1250、120、250、200、175μl,混合后加质量分数为20%甲醇水溶液稀释定容至5ml,制成9种成分浓度分别为4.17、15.2、18.1、62.6、81.2、49.5、24.0、8.89、11.3μg/ml的混合对照品溶液。按步骤(6)中的色谱条件分别进样1、2、5、10、20、50μl进行测定,记录相应的峰面积。以各成分浓度(μg/ml)乘以进样体积(μl)得到被测成分进样质量(ng)作为自变量x,以峰面积作为因变量y,进行线性回归。9种成分的回归方程和线性范围如表1所示。结果表明,9个成分在考察的浓度范围内线性关系良好。
表1.线性关系考察结果
(7.2)精密度、重复性、稳定性考察
取步骤(2)制备得到的同一份供试品溶液,按步骤(6)中的色谱条件重复进样6次,按步骤(5)测得9个成分浓度的rsd如表2所示。结果表明,精密度良好。
由于建立的分析方法没有供试品前处理和稀释过程,所以重复性试验考察方式为取步骤(1)得到的同一份栀子-郁金蒸馏液,装到6个进样瓶中作为供试品溶液按步骤(6)中的色谱条件分别进样分析,按步骤(5)测得9个成分浓度的rsd如表2所示。结果表明,重复性良好。
取步骤(2)制备得到的供试品溶液,在常温下放置0、1、2、4、8、12小时后按步骤(6)中的色谱条件进样,按步骤(5)测得9个成分浓度的rsd如表2所示。结果表明,9个成分在常温下12小时内稳定。
表2.精密度、重复性、稳定性考察结果
(7.3)加样回收率考察
按步骤(1)和步骤(2)分别制备得到6份供试品溶液,按步骤(5)测定9种成分的浓度。分别取6份供试品溶液各0.4ml,精密加入步骤(3)得到的对照品溶液0.4ml混匀,按步骤(5)测定9种成分浓度,并计算各成分的加样回收率。测定结果如表3所示,加样回收率在95.3~105.4%范围内,表明该方法的准确度良好。
表3.加样回收率(%)考察结果
(8)本发明方法与外标法测定结果比较
以外标法对步骤(5)中的同一份供试品溶液进行测定,得到各待测成分浓度如下:异佛尔酮0.853μg/ml,4-亚甲基-异佛尔酮3.24μg/ml,莪术双环烯酮3.61μg/ml,莪术烯醇10.3μg/ml,莪术二酮13.2μg/ml,莪术酮9.75μg/ml,莪术呋喃二烯酮4.45μg/ml,莪术醇1.06μg/ml,吉马酮2.01μg/ml。步骤(5)中本发明方法测定结果与外标法基本一致,相对偏差在2%以内,表明本发明方法能准确且低成本地测定9种指标成分的含量。
实施例2
一种醒脑静注射液中多指标成分含量的测定方法,该方法包括以下步骤:
(1)供试品溶液的制备
直接将醒脑静注射液作为供试品溶液,无前处理和稀释。
(2)对照品溶液的制备
精密称莪术二酮的对照品16.25mg,加质量分数为80%甲醇水溶液溶解并定容至50ml,制成对照品储备液。然后精密吸取上述对照品储备液1250μl,加浓度为30%甲醇水溶液稀释定容至25ml,得到浓度为16.2μg/ml的莪术二酮对照品溶液,于4℃保存,备用。
(3)样品中多指标成分的测定
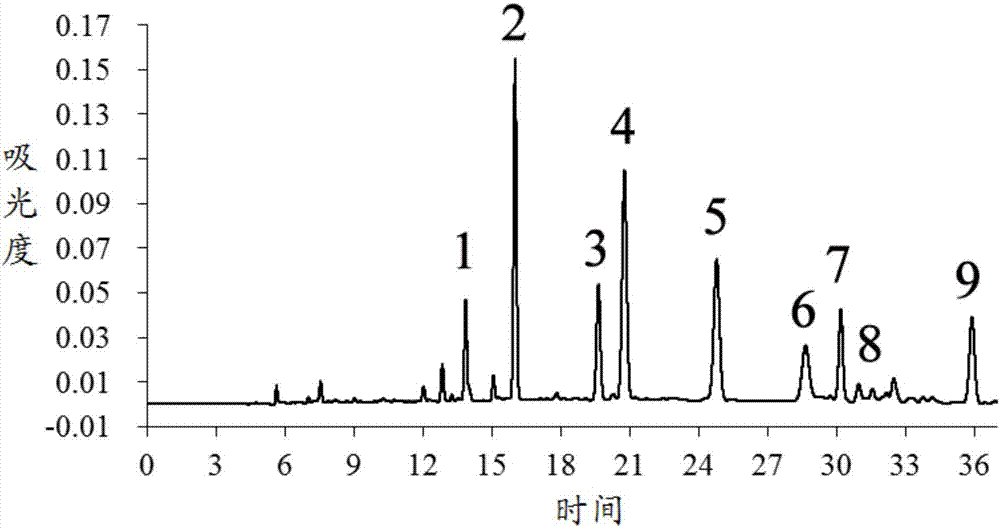
取步骤(1)和步骤(2)得到的供试品溶液和莪术二酮对照品溶液,分别进样50μl进行高效液相色谱分析,得到醒脑静注射液的高效液相色谱图(如图3所示)和莪术二酮对照品溶液的高效液相色谱图,图3中,检测波长:0~15.5min,238nm;15.5~19min,275nm;19~20min,256nm;20~24min,261nm;24~26min,220nm;26~30.5min,274nm;30.5min后,210nm),峰1:异佛尔酮;峰2:4-亚甲基-异佛尔酮;峰3:莪术双环烯酮;峰4:莪术烯醇;峰5:莪术二酮;峰6:莪术酮;峰7:莪术呋喃二烯酮;峰8:莪术醇;峰9:吉马酮,色谱图积分得到醒脑静注射液中各待测成分的峰面积及莪术二酮对照品溶液中莪术二酮的峰面积。
供试品溶液中莪术二酮的进样质量ms’由莪术二酮对照品溶液以外标定量法测定得到,供试品溶液中其他待测成分的进样质量mx’由公式mx’=fx/s×ms’×(ax’/as’)计算得到。
异佛尔酮、4-亚甲基-异佛尔酮、莪术双环烯酮、莪术烯醇、莪术酮、莪术呋喃二烯酮、莪术醇和吉马酮8种成分对于莪术二酮的相对校正因子在本发明方法的开发过程中已测定,具体测定过程可参考实施例1的步骤(4),测定结果分别为0.176、0.207、0.496、0.600、1.59、0.756、0.802、0.276,可直接使用。
由测定或计算得到的进样质量ms’和mx’,除以进样体积,计算得到样品中各待测成分的浓度,结果如下:异佛尔酮0.600μg/ml,4-亚甲基-异佛尔酮0.854μg/ml,莪术双环烯酮2.64μg/ml,莪术烯醇5.42μg/ml,莪术二酮9.77μg/ml,莪术酮5.71μg/ml,莪术呋喃二烯酮1.63μg/ml,莪术醇1.23μg/ml,吉马酮1.49μg/ml。
(4)步骤(3)所用的高效液相色谱参数如下:
仪器:安捷伦1200高效液相色谱仪;
色谱柱:akzonobelkromasilc18(5μm,250×4.6mm);
流动相:质量分数0.1%磷酸水溶液-乙腈;
洗脱程序为线性梯度洗脱,0.1%磷酸水溶液占流动相体积比为:0min,30%;10min,55%;24min,55%;24.1min,70%;35min,70%;40min,100%;
流速:0.6ml/min;
柱温:40℃;
检测波长:异佛尔酮,238nm;4-亚甲基-异佛尔酮,275nm;莪术双环烯酮,256nm;莪术烯醇,261nm;莪术二酮,220nm;莪术酮,274nm;莪术呋喃二烯酮,274nm;莪术醇,210nm;吉马酮,210nm。
(5)本发明方法与外标法测定结果比较
以外标法对步骤(3)中的同一份供试品溶液进行测定,得到各待测成分浓度如下:异佛尔酮0.601μg/ml,4-亚甲基-异佛尔酮0.858μg/ml,莪术双环烯酮2.64μg/ml,莪术烯醇5.45μg/ml,莪术二酮9.77μg/ml,莪术酮5.72μg/ml,莪术呋喃二烯酮1.66μg/ml,莪术醇1.24μg/ml,吉马酮1.47μg/ml。步骤(3)中本发明方法测定结果与外标法基本一致,相对偏差在2%以内,表明本发明方法能准确且低成本地测定9种指标成分的含量。
以上所述仅是本发明的优选实施方式,对于本技术领域的普通技术人员,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!