一种兼具定性与定量评价的三勒浆口服液质量检测方法与流程
本发明涉及三勒浆口服液的hplc指纹图谱检测方法,属于药物质量控制方法技术领域。
背景技术:
三勒浆处方源于藏药名方三果汤(又称哲布松汤),由诃子terminaliachebularetz、毛诃子terminaliabillericaroxb和余甘子phyllanthusemblical三种药物配伍而成,因三味药物在藏药中分别被称为蓭摩勒、诃梨勒与毗梨勒,又称为三勒浆。在传统藏医学中,三果汤对瘟疫、高原红细胞增多症具有显著疗效[1],被视为滋补强壮药;在现代医学中,发现其具有很好的免疫调节功能,对高脂血症[2]、肿瘤[3]等都具有一定的治疗效果。上世纪90年代,以余甘子和诃子为原料,添加l-精氨酸、l-天门冬氨酸、蜂蜜等辅料,开发了具有抗疲劳、免疫调节和耐缺氧功能的三勒浆口服液,尤其作为学生群体参加重大考试的保健品而深受欢迎。
三勒浆口服液富含多酚,但对其化学成分研究较少,质量控制也仅以柯里拉京作为内控指标,难以全面控制其质量。
技术实现要素:
本发明的目的在于提供三勒浆口服液的hplc指纹图谱检测方法。
该方法是采用hplc指纹图谱进行检测,其操作步骤如下:
(1)供试品溶液的制备:取三勒浆口服液,加入50%甲醇水溶液提取,制备供试品溶液;
(2)对照品溶液的制备:取没食子酸、没食子儿茶素、表儿茶素、柯里拉京和鞣花酸对照品,制备对照品溶液;
(3)对照药材溶液的制备:取诃子、余甘子药材粉末,加水回流提取后,制备对照药材溶液;
(4)对照指纹图谱的建立:
分别取对照品溶液、供试品溶液和诃子、余甘子对照药材溶液,注入高效液相色谱仪检测,得到各批次药材和参照物的色谱数据,色谱条件如下:
色谱柱以以十八烷基硅烷键合硅胶为填充剂,检测波长为260~280nm,流动相为0.1%磷酸水(a)-甲醇(b),进行梯度洗脱,洗脱条件为:
对色谱数据进行处理,建立三勒浆口服液的对照指纹图谱。
进一步地,检测波长为270nm。
进一步地,色谱柱为welchromc18色谱柱(4.6mm×250mm,5μm);柱温为25℃。
进一步地,流速为1ml/min。
进一步地,步骤(1)中制备供试品溶液的具体操作步骤如下:
精密量取三勒浆5ml至25ml容量瓶,加入50%甲醇水至刻度线,摇匀,用0.22μm微孔滤膜过滤,取滤液作为供试品溶液。
进一步地,步骤(2)中,以50%甲醇水溶液为溶剂,制备混合对照品溶液。
进一步地,步骤(3)中制备对照药材溶液的具体操作步骤如下:
称取诃子、余甘子粉末(过2号筛)各5g,分别置1000ml圆底烧瓶内,加入200ml水,加热回流提取1h,过滤,滤液定容至500ml,用0.22μm微孔滤膜过滤,取滤液,即得。
进一步地,所述对照指纹图谱如图1中的a所示。
进一步地,所述方法是定量检测没食子酸、没食子儿茶素、表儿茶素、柯里拉京和鞣花酸的方法。
本发明质量检测方法中,通过对不同流动相条件下色谱峰的分离度、分离时间、基线、峰数量以及峰形的考察,得出了本发明最佳的流动相系统以及梯度洗脱条件,在该条件下,所得的指纹图谱出峰多,基线平稳,分离度良好,方法重复性好,精密度高,能够更简便、快捷、全面、准确地监控三勒浆口服液的质量,采用本发明方法检测能够满足指纹图谱要求的,三嘞浆的质量优良,如果无法满足的的,质量不佳;本发明方法还可同时测得没食子酸、没食子儿茶素、表儿茶素、柯里拉京和鞣花酸的含量,实现了对三勒浆口服液定性和定量的双重研究。
本发明同时实现指纹图谱定性与多组分(没食子酸、柯里拉京、鞣花酸、表儿茶素与没食子儿茶素,见下表)含量测定的高效液相测定方法,可以更加系统、全面、直观地反映三勒浆的有效成分,确保其质量的稳定、可控。
表指标成分的分类与选择依据
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其他多种形式的修改、替换或变更。
以下通过实施例形式的具体实施方式,对本发明的上述内容作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
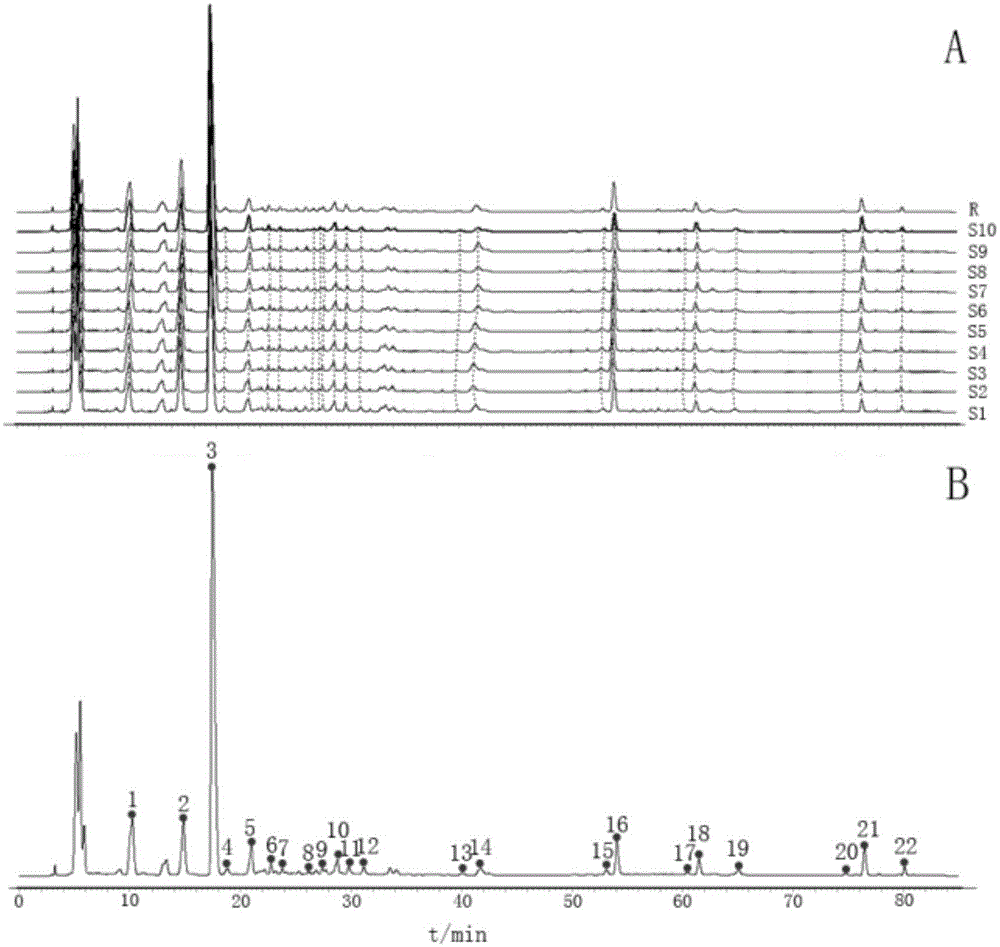
图110批三勒浆口服液hplc指纹图谱(a)和对照指纹图谱(b)
图2三勒浆hplc图。a.三勒浆样品,b.诃子对照,c.余甘子对照,d.混合对照品
具体实施方式
仪器与试药
anglent1020型高效液相色谱仪(美国agilent公司),bs110s型电子分析天平(德国sartorius公司),bp211d型电子分析天平(德国sartorius公司),milli-q超纯水仪(美国millipore公司)。
三勒浆口服液10批,四川华美制药有限公司生产,批号分别为1503003(s1)、1504002(s2)、1510002(s3)、1601001(s4)、1603001(s5)、1603004(s6)、1603005(s7)、1604002(s8)、1604005(s9)、1604008(s10)。诃子,购于四川中庸药业有限公司,产地:广西,批号z00216n01;余甘子,购于四川中庸药业有限公司,产地:四川,批号z16116n01;经成都中医药大学许润春副教授鉴定分别为使君子科植物诃子terminaliachebularetz.的干燥成熟果实,大戟科植物余甘子phyllanthusemblica的干燥成熟果实。对照品没食子酸(批号4051109,质量分数以98%计)、没食子儿茶素(批号14092403,质量分数以98%计)、表儿茶素(批号14051602,质量分数以98%计)、柯里拉京(批号prf7102406,质量分数以98%计)、鞣花酸(批号prf7101305,质量分数以98%计)均购于成都普瑞法科技开发有限公司;甲醇为色谱纯,购于fisher公司;其他试剂均为分析醇。
实施例1本发明方法的建立
(一)三勒浆口服液hplc指纹图谱的建立及方法学考察
1.1溶液的制备
1.1.1混合对照品溶液的制备
精密称取各对照品,加入50%甲醇水(指甲醇占总溶液的50%)配制成每1ml含有没食子酸846.0μg,没食子儿茶素214.5μg,表儿茶素130.75μg,柯里拉京201.0μg,鞣花酸92μg的混合溶液。
1.1.2供试品溶液的制备
精密量取三勒浆5ml至25ml容量瓶,加入50%甲醇水(指甲醇占总溶液的50%)至刻度线,摇匀,用0.22μm微孔滤膜过滤,取滤液作为供试品溶液。
1.1.3诃子、余甘子溶液的制备
称取诃子、余甘子粉末(过2号筛)各5g,分别置1000ml圆底烧瓶内,加入200ml水,加热回流提取1h,过滤,滤液定容至500ml,用0.22μm微孔滤膜过滤,取滤液,即得。
1.2指纹图谱的建立及相似度评价
色谱柱:welchromc18色谱柱(4.6mm×250mm,5μm);
检测波长:270nm;
体积流量:为1.0ml/min;
柱温:25℃;
进样量:10μl;
流动相:0.1%磷酸水-甲醇,采用梯度洗脱,洗脱程序如下:
取10批三勒浆(s1-s10),按照供试品溶液的制备方法制备供试品溶液,照以上色谱条件进行分析,将得到的色谱图导入《中药色谱指纹图谱相似度评价系统》(2012年版),以s6为参考图谱,时间窗宽度设为0.3min,根据匹配结果确定22个共有峰,并生成对照指纹图谱,计算出各批次样品的指纹图谱相似度均大于0.95。指纹图谱(s1-s10)及对照品指纹图谱见图1。
在供试品各色谱峰中,以14号峰峰面积适中,分离度及对称性均良好,故将14号峰确定为参照峰。相对峰面积和相似度结果见表1和表2。
表110批样品中共有峰的相对峰面积
表210批样品相似度评价结果
1.3方法学考察
1.3.1精密度试验
将供试品溶液(批号1604002)连续进样6次,记录色谱图,计算各主要峰相对峰面积rsd值在0.12%~1.57%,相对保留时间的rsd值在0.06%~0.95%,表明仪器精密度良好。
1.3.2稳定性试验
取同一供试品溶液(批号1604002),分别于0、3、6、9、15、18、24h在上述色谱条件下进行液相分析。计算各主要色谱峰相对峰面积rsd值在0.52%~1.72%,相对保留时间的rsd值在0.20%~1.28%,表明供试品溶液在24h内稳定。
1.3.3重复性试验
取同一批三勒浆(批号1604002),按方法平行制备6份,依次测定,计算主要色谱峰相对峰面积rsd值在0.20%~1.34%,相对保留时间的rsd值在0.11%~0.83%,表明方法重复性良好。
1.4色谱峰的指认
分别取混合对照品溶液、供试品溶液、诃子溶液和余甘子溶液,按上述色谱条件进行hplc测定,色谱图见图2。通过与诃子、余甘子和混合对照色谱峰进行比对分析,可知1、3、4、6、7、8、14、16、17、18、21和22号峰为诃子和余甘子共同拥有,9、10、11、13、15和19号峰来源于诃子,2和5号峰来源于余甘子;同时对其中5个色谱峰进行指认定性,分别为3号峰为没食子酸、8号峰为没食子儿茶素、15号峰为表儿茶素、16号峰为柯里拉京、21号峰为鞣花酸。
(二)三勒浆口服液中五个主要成分的定量测定
2.1色谱条件及样品的制备
按照“1.1.2”和“1.2”项下的方法处理。
2.2线性关系考察
精密吸取混合对照品溶液4、8、10、12、15、20l注入色谱仪进行分析,记录色谱图。以各对照品进样量x(μg)为横坐标,峰面积y为纵坐标,绘制标准曲线,求得回归方程分别为没食子酸:y=3373.3x+394.31,r=0.9999,线性范围为3.384~16.92μg;没食子儿茶素:y=266.67x-0.2481,r=1,线性范围为0.858~4.29μg;表儿茶素:y=583.34x+3.8847,r=1,线性范围为0.523~2.615μg;柯里拉京:y=1887.1x-7.0072,r=1,线性范围为0.804~4.02μg;鞣花酸:y=2446.5x+8.2644,r=0.9997,线性范围为0.368~1.84μg。
2.3仪器精密度试验
取同一批供试品溶液(批号1604002)连续进样6针,记录色谱图。结果没食子酸、没食子儿茶素、表儿茶素、柯里拉京、鞣花酸峰面积的rsd值分别为0.17%、0.92%、1.3%、0.12%和0.47%,表明仪器精密度良好。
2.4稳定性试验
取同一批供试品溶液(批号1604002),分别于0、3、6、9、15、18、24h进样,记录色谱图。结果没食子酸、没食子儿茶素、表儿茶素、柯里拉京、鞣花酸峰面积的rsd值分别为0.57%、1.77%、1.51%、0.58%和1.52%,表明供试品溶液在24h内稳定。
2.5重复性试验
取同一批样品(批号1604002),按“1.1.2”项下方法平行制备6份供试品溶液,进行测定,记录色谱图。结果没食子酸、没食子儿茶素、表儿茶素、柯里拉京、鞣花酸峰面积的rsd值分别为0.67%、0.89%、0.94%、0.72%和0.20%,表明该方法重复性良好。
2.6加样回收率试验
精密量取2.5ml已测定的三勒浆(批号1604002)6份,加入没食子酸、没食子儿茶素、表儿茶素、柯里拉京和鞣花酸对照品,按“1.1.2”项下方法制备样品,进行色谱分析,记录色谱图,计算各成分的加样回收率。结果没食子酸、没食子儿茶素、表儿茶素、柯里拉京和鞣花酸的平均加样回收率分别为101.00%、104.40%、100.36%、102.22%和100.87%,rsd分别为1.04%、1.25%、1.06%、1.17%和1.22%。
2.7样品含量的测定
取三勒浆供试品,按“1.1.2”项下方法制备,按“1.2”项下方法进行色谱分析,记录色谱图,根据上述每个对照品的标准曲线计算样品中没食子酸、没食子儿茶素、表儿茶素、柯里拉京和鞣花酸的质量浓度,结果见表3。结果表明,10批三勒浆样品中,没食子酸的含量在5.7432~7.5380mg/ml,没食子儿茶素的含量在0.4929~0.8471mg/ml,表儿茶素的含量在0.5297~0.8048mg/ml,柯里拉京的含量在0.9375~1.7565mg/ml,鞣花酸的含量在0.3527~0.5545mg/ml。
表310批三勒浆口服液测定结果
综上所述,本发明的指纹图谱出峰多,分离度好,重复性好,精密度高,为全面监控三勒浆口服液的质量提供了有效保障。
同时,利用本发明指纹图谱检测方法的提取方法和色谱条件,还可以同时测得没食子酸、没食子儿茶素、表儿茶素、柯里拉京和鞣花酸的含量,实现了对三勒浆口服液定性和定量的双重研究,从更多角度监控三勒浆口服液的质量。
- 还没有人留言评论。精彩留言会获得点赞!