一种少腹逐瘀丸的质量检测方法与流程
本发明涉及一种中成药少腹逐瘀丸(小蜜丸)及其质量检测方法,属于中药制剂分析领域。
背景技术:
少腹逐瘀丸(小蜜丸)的主要成分和含量如下:
当归300g蒲黄300g
五灵脂(醋炒)200g赤芍200g
小茴香(盐炒)100g延胡索(醋制)100g
没药(炒)100g川芎100g
肉桂100g炮姜20g
其制备方法为:以上十味,粉碎成细粉,过筛,混匀。每100g粉末加炼蜜100~110g制成小蜜丸,即得。
中国药典收载的少腹逐瘀丸(大蜜丸),质量标准中只以显微鉴别法鉴别处方中当归、蒲黄、赤芍、肉桂、小茴香和延胡索,以薄层色谱法鉴别赤芍中芍药苷,无含量测定,质量控制方法不足以全面控制少腹逐瘀丸(大蜜丸)的产品质量。中国专利cn201610912764.7公开了一种中药复方中姜的鉴别备方法,公开了少腹逐瘀丸中炮姜的tlc鉴别方法。但现有技术中,尚没有比较全面的质量控制方法反映少腹逐瘀丸(小蜜丸)的质量状况,因而无法对少腹逐瘀丸(小蜜丸)的生产过程和产品质量进行有效地控制,不能较好的保证其药物疗效。
高效液相色谱(hplc)具有快速、灵敏、分离效能高等优点,在中药定性和定量分析中,采用hplc法分离特征成分后,用检测器进行检测分析,是全面评价中药组合物质量的有效方法。为了克服上述现有技术的不足,提高少腹逐瘀丸(小蜜丸)质量控制方法,本发明人在将少腹逐瘀丸(大蜜丸)改为小蜜丸的基础上,增加了薄层色谱法鉴别处方中当归、川芎,蒲黄,延胡索,同时采用hplc测定制剂中赤芍中芍药苷的含量,提高质量检测方法,更好的控制产品质量,该方法专属性好、耐用性强,稳定性好,能够有效保证产品质量的稳定性和可控性。
技术实现要素:
本发明的目的在于提供一种少腹逐瘀丸(小蜜丸)的质量检测方法,该方法能够较为全面、准确地分析药物活性组分,有利于产品有效成分的质量控制,保证其临床疗效。
本发明的目的通过以下技术方案实现:一种少腹逐瘀丸(小蜜丸)的质量检测方法,其特征在于,包括显微鉴别、薄层色谱定性鉴别和芍药苷的含量检测;
本发明所要解决的技术问题是为了对产品质量进行全面检测,有效控制质量质量,发明人经过大量试验,对制剂中药材及药材有效成分进行了定性和定量检测,建立了中药少腹逐瘀丸(小蜜丸)新的质量检测方法。所述少腹逐瘀丸(小蜜丸)是当归、蒲黄、五灵脂(醋炒)、赤芍、小茴香(盐炒)、延胡索(醋制)、没药(炒)、川芎、肉桂和炮姜十味中药组成,加炼蜜制成的小蜜丸。该质量检测方法是以显微鉴别法鉴别制剂中当归、蒲黄、赤芍、肉桂、小茴香和延胡索;以薄层色谱法鉴别制剂中赤芍,当归、川芎,蒲黄,延胡索;同时采用hplc测定制剂中赤芍中芍药苷的含量,从而实现对少腹逐瘀丸(小蜜丸)质量进行全面地评价和控制,为少腹逐瘀丸(小蜜丸)的真伪鉴别和内在质量检测提供全面、可靠的依据,保证了产品质量的稳定性及临床用药的安全性、有效性。
本发明技术方案以显微鉴别法鉴别制剂中当归、蒲黄、赤芍、肉桂、小茴香和延胡索;以薄层色谱鉴别少腹逐瘀丸(小蜜丸)中赤芍,当归、川芎,蒲黄,延胡索,具体包括下列步骤:
1、取本品,置显微镜下观察:薄壁细胞纺锤形,壁略厚,有极微细的斜向交错纹理(当归)。花粉粒黄色,类圆形或椭圆形,直径约30μm,外壁有网状雕纹(蒲黄)。草酸钙簇晶直径7~41μm,存在于薄壁细胞中,常排列成行或一个细胞中含有数个簇晶(赤芍)。纤维单个散在,长梭形,直径24~50μm,壁厚,木化(肉桂)。草酸钙簇晶细小,直径约5μm,一个细胞含有多个簇晶(小茴香)。糊化淀粉粒团块淡黄色(延胡索)。
2、赤芍鉴别
1)供试品溶液的制备:取本品9g,剪碎,加硅藻土10g,研匀,加乙醇50ml,超声处理20分钟,滤过,滤液蒸干,残渣用水20ml溶解,用水饱和的正丁醇振摇提取3次,每次20ml,合并正丁醇提取液,用正丁醇饱和的水洗涤3次,每次15ml,正丁醇液蒸干,残渣用乙醇20ml溶解,加活性炭2.5g,水浴加热2分钟,放冷,滤过,滤液浓缩至约1ml,作为供试品溶液;
2)对照品溶液的制备:另取芍药苷对照品,加乙醇制成每lml含2mg的溶液,作为对照品溶液;
3)点样、展开:照薄层色谱法(通则0502)试验,吸取供试品溶液5~10μl、对照品溶液2μl,分别点于同一硅胶g薄层板上,以三氯甲烷-乙酸乙酯-甲醇-甲酸(40∶5∶10∶0.2)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。
3、当归和川芎鉴别
1)供试品溶液的制备:取本品6g,剪碎,加乙醚30ml,超声处理20分钟,滤过,滤液挥干,残渣加乙酸乙酯lml使溶解,作为供试品溶液。
2)对照品溶液的制备:取当归对照药材与川芎对照药材各0.5g,同法制成对照药材溶液。
3)点样、展开:照薄层色谱法(通则0502)试验,吸取供试品溶液、对照药材溶液各5μl,分别点于同一硅胶g薄层板上,以正己烷-乙酸乙酯(4∶1)为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。
4、蒲黄鉴别
1)供试品溶液的制备:取本品10g,剪碎,加乙醚20ml,超声处理20分钟,弃去乙醚液,残渣挥干,加水30ml使溶解,用水饱和的正丁醇振摇提取2次,每次20ml,合并正丁醇液,蒸干,残渣加乙醇1ml使溶解,作为供试品溶液。
2)对照品溶液的制备:另取蒲黄对照药材1g,加水30ml,煮沸15分钟,放冷,滤过,用水饱和的正丁醇振摇提取2次,每次20ml,合并正丁醇液,蒸干,残渣加乙醇1ml使溶解,作为对照药材溶液。
3)点样、展开:照薄层色谱法(通则0502)试验,吸取供试品溶液、对照药材溶液各5μl,分别点于同一硅胶g薄层板上,以乙酸乙酯-丁酮-甲酸-水(5∶3∶1∶1)为展开剂,展开,取出,晾干,喷以5%三氯化铝乙醇溶液,105℃加热至斑点显色清晰,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。
5、延胡索鉴别
1)供试品溶液的制备:取本品30g,剪碎,加甲醇50ml,超声处理30分钟,滤过,滤液蒸干,残渣加水20ml使溶解,加浓氨试液调至碱性,用乙醚振摇提取3次,每次10ml,合并乙醚液,蒸干,残渣加甲醇lml使溶解,作为供试品溶液。
2)对照品溶液的制备:另取延胡索对照药材lg,同法制成对照药材溶液。
3)点样、展开:照薄层色谱法(通则0502)试验,吸取供试品溶液、对照药材溶液各5μl,分别点于同一用1%氢氧化钠溶液制备的硅胶g薄层板上,以甲苯-丙酮(9∶2)为展开剂,展开,取出,晾干,置碘缸中约3分钟后取出,挥尽薄层板上吸附的碘后,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。
本发明同时采用高效液相色谱法测定少腹逐瘀丸(小蜜丸)中赤芍的赤芍苷含量,包括下列步骤:
1)供试品溶液的制备:取装量差异项下少腹逐瘀丸(小蜜丸)适量,剪碎,混匀,取约1.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50ml,密塞,称定重量,待完全溶散后,超声处理(功率100w,频率40khz)30分钟,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2)对照品溶液的制备:取芍药苷对照品适量,精密称定,加甲醇制成每lml含60μg的溶液,即得;
3)hplc色谱条件:以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.1%磷酸(14∶86)为流动相;检测波长为230nm。理论板数按芍药苷峰计算应不低于3000;
4)测定方法:分别精密吸取对照品溶液与供试品溶液各10μl,注人液相色谱仪,按照步骤3)所述色谱条件,测定,即得;
本发明具有以下优点:
本发明一种少腹逐瘀丸(小蜜丸)的质量检测方法,采用显微鉴别法鉴别制剂中当归、蒲黄、赤芍、肉桂、小茴香和延胡索;以薄层色谱鉴别少腹逐瘀丸(小蜜丸)中赤芍,当归、川芎,蒲黄,延胡索,其中薄层色谱通过一个方法可一次性鉴定当归和川芎2种中药,准确性高、实用性强;采用超高效液相色谱可统一测定少腹逐瘀丸(小蜜丸)中芍药苷含量,经过方法学考察,本发明含量测定方法精密度高、稳定性好、重复性优秀,并且显著的减少了分析检测的时间,该检测方法具有较强的专属性、耐用性,其准确性、重现性、稳定性均能达到科研和生产的要求,能够有效保证产品质量的稳定性和可控性,对少腹逐瘀丸(小蜜丸)质量标准的制定具有重要的意义。
附图说明
图1为少腹逐瘀丸(小蜜丸)芍药苷紫外扫描图;
图2为少腹逐瘀丸(小蜜丸)芍药苷hplc图,从上至下依次为赤芍药材色谱图、不含赤芍的阴性色谱图、供试品色谱图、芍药苷对照品色谱图;
图3为少腹逐瘀丸(小蜜丸)芍药苷峰面积值与浓度线性关系图;
图4为少腹逐瘀丸(小蜜丸)当归显微鉴别图;
图5为少腹逐瘀丸(小蜜丸)蒲黄显微鉴别图;
图6为少腹逐瘀丸(小蜜丸)赤芍显微鉴别图;
图7为少腹逐瘀丸(小蜜丸)肉桂显微鉴别图;
图8为少腹逐瘀丸(小蜜丸)小茴香显微鉴别图;
图9为少腹逐瘀丸(小蜜丸)延胡索显微鉴别图;
图10为少腹逐瘀丸(小蜜丸)赤芍tlc色谱图,按从左至右依次为阴性供试品、芍药苷对照品、20190401批样品、20190402批样品和20190403批样品;
图11为少腹逐瘀丸(小蜜丸)当归、川芎tlc色谱图,按从左至右依次为阴性供试品、当归对照药材、川芎对照药材、20190401批样品,20190402批样品和20190403批样品;
图12为少腹逐瘀丸(小蜜丸)蒲黄tlc色谱图,按从左至右依次为阴性供试品、蒲黄对照药材、20190401批样品,20190402批样品和20190403批样品;
图13为少腹逐瘀丸(小蜜丸)延胡索tlc色谱图,按从左至右依次为阴性供试品、延胡索对照药材、20190401批样品,20190402批样品和20190403批样品。
具体实施方式
下面对本发明的实施例作详细说明,本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施例。
实施例1
赤芍苷的hplc方法学研究
1、色谱条件
仪器:ag135型电子分析天平(瑞士mettler),岛津高效液相色谱仪lc-15c型泵,spd-15c检测器,岛津shim-packvp-odsc18(250mm×4.6mm,5μm)色谱柱;乙腈-0.1%磷酸(14∶86)为流动相;检测波长为230nm;流速为1.0ml/min。
对照品:芍药苷(中国食品药品检定研究院,20mg,110736-201842,纯度为97.4%);芍药苷(中国食品药品检定研究院,20mg,110736-201716,纯度为99.3%)。
试剂:乙腈、磷酸为色谱纯,水为娃哈哈纯净水,其余试剂均为分析纯。
2、色谱条件的选择
2.1检测波长选择:精密称取芍药苷对照品适量,加甲醇制成每1ml含60µg的溶液,照紫外分光光度法(中国药典2015年版通则0401),在波长200~400nm的范围内扫描,结果波长为230nm、317nm时芍药苷有最大吸收。
2.2流动相的确定:参考药典芍药苷测定方法及参考文献,确定乙腈-0.1%磷酸(14∶86)为流动相。
3、提取方法选择:
3.1提取方法选择试验:
对照品溶液制备:取芍药苷对照品(110753-201842,纯度:97.4%)9.17mg置50ml量瓶中,用甲醇稀释至刻度,作为对照品贮备液,精密量取对照品贮备液3ml置10ml量瓶中,用甲醇稀释至刻度,即得。
供试品溶液制备:取同一份少腹逐瘀丸(小蜜丸)(批号:20190401),剪碎,混匀,分别取6份,每份约3g,精密称定,置具塞锥形瓶中,精密加人70%甲醇50ml,称定重量,待完全溶散后,其中2份超声处理(100w,40khz)30分钟,2份回流30分钟,放冷,再称定重量,用70%甲醇补足减失重量,摇匀,滤过,取续滤液,即得。
分别精密吸取对照品溶液与供试品溶液各10µl,注入液相色谱仪,测定,即得。
表1芍药苷含量测定提取方法选择结果表
结果:超声提取和回流提取对含量无影响,超声提取更为简便,故选用超声提取。
3.2提取时间选择试验:
对照品溶液制备:取“3.1提取方法选择试验”项下对照品贮备液3ml置10ml量瓶中,用甲醇稀释至刻度,即得。
供试品溶液制备:取同一份少腹逐瘀丸(小蜜丸)(批号:20190401),剪碎,混匀,分别取6份,每份约3g,精密称定,置具塞锥形瓶中,精密加人70%甲醇50ml,称定重量,待完全溶散后,其中2份超声处理(100w,40khz)20分钟,2份超声处理(100w,40khz)30分钟,2份超声处理(100w,40khz)40分钟,放冷,再称定重量,用70%甲醇补足减失重量,摇匀,滤过,取续滤液,即得。
分别精密吸取对照品溶液与供试品溶液各10µl,注入液相色谱仪,测定,即得。
表2芍药苷含量测定提取时间选择结果表
结果:超声提取20分钟、30分钟、40分钟含量一致,为保证提取充分,确定超声处理时间为30分钟。
3.3提取溶剂选择试验:
对照品溶液制备:取“3.1提取方法选择试验”项下对照品贮备液3ml置10ml量瓶中,用甲醇稀释至刻度,即得。
供试品溶液制备:取同一份少腹逐瘀丸(小蜜丸)(批号:20190401),剪碎,混匀,分别取6份,每份约3g,精密称定,置具塞锥形瓶中,两份精密加人60%甲醇50ml,两份精密加人70%甲醇50ml,两份精密加人甲醇50ml,称定重量,待完全溶散后,超声处理(100w,40khz)30分钟,放冷,再称定重量,分别用相应溶剂补足减失的重量,摇匀,滤过,取续滤液,即得。
分别精密吸取对照品溶液与供试品溶液各10µl,注入液相色谱仪,测定,即得。
表3芍药苷含量测定提取溶剂选择结果表
结果:60%甲醇提取与70%甲醇提取含量无差别,用甲醇提取样品不能充分溶散,提取不充分,故选用原标准70%甲醇提取。
3.4溶剂用量选择试验:
供试品溶液制备:取同一份少腹逐瘀丸(小蜜丸)(批号:20190401),剪碎,混匀,分别取6份,每份约3g,精密称定,置具塞锥形瓶中,两份精密加入70%甲醇40ml,两份精密加入70%甲醇50ml,两份精密加入70%甲醇75ml,称定重量,待完全溶散后,分别超声处理(100w,40khz)30分钟,放冷,再称定重量,用70%甲醇补足减失重量,摇匀,滤过,取续滤液,即得。
分别精密吸取对照品溶液与供试品溶液各10µl,注入液相色谱仪,测定,即得。
表4芍药苷含量测定溶剂用量选择结果表
结果:提取溶剂为40ml、50ml、75ml时含量变化不大,为便于试验,确定溶剂用量为50ml。
3.4供试品溶液制备:
取少腹逐瘀丸(小蜜丸),剪碎,取约1.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50ml,称定重量,待完全溶散后,超声处理(功率:100w,频率:40khz)30min,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
4、稳定性试验:
供试品溶液制备:取少腹逐瘀丸(小蜜丸)(批号:20190401)1.5g,按“3.4供试品溶液制备”项下方法处理,分别于0小时、2小时、4小时、6小时、8小时、12小时,18小时进样,结果见表5。
对照品溶液制备:取芍药苷对照品(110736-201716;纯度:99.3%)20.05mg置100ml量瓶中,用甲醇稀释至刻度,作为对照品贮备液,精密量取对照品贮备液3ml置10ml量瓶中,用甲醇稀释至刻度,即得。分别于0小时、1小时、2小时、4小时、6小时、8小时进样。
表5芍药苷含量测定稳定性试验结果表
结果表明:供试品溶液在18小时内峰面积rsd为0.52%,对照品在8小时内峰面积积分值rsd为0.75%,证明芍药苷供试品溶液和对照品溶液稳定性良好。
5、精密度试验:
对照品溶液制备:精密量取“稳定性试验”项下对照品贮备液3ml,用甲醇稀释至10ml,即得。
精密吸取芍药苷对照品溶液10μl,注入液相色谱仪,并记录色谱图(附图)。连续进样6次,计算峰面积积分值的rsd
表6芍药苷含量测定精密度试验结果表
结果表明:对照品rsd为1.56%,证明仪器精密度良好。
6、线性关系考察:取芍药苷对照品9.17mg(110736-201842;纯度97.4%)置50ml量瓶中,用甲醇稀释至刻度,作为对照品贮备液,精密量取对照品贮液2.0、3.0、4.0、5.0、6.0、7.0置10ml量瓶中,用甲醇稀释至刻度,摇匀,作为对照品溶液。分别吸取上述对照品溶液各10μl进样,注入液相色谱仪,测定。以芍药苷浓度(mg/ml)为横坐标,峰面积(a)为纵坐标,绘制标准曲线,回归方程为:a=13189906.8c-38468.2,r=0.9996。
表7芍药苷含量测定线性范围测定结果表
以上结果表明:0.0357mg/ml~0.1250mg/ml范围内,芍药苷峰面积值与浓度线性关系良好。
7、重复性试验:
对照品溶液制备:取芍药苷对照品(110736-201842;纯度97.4%)17.35mg置100ml量瓶中,用甲醇定容至刻度,作为对照品贮备液,精密量取对照品贮备液3ml置10ml量瓶中,用甲醇稀释至刻度,即得。
供试品溶液:取同一批号(批号:20190401)的少腹逐瘀丸(小蜜丸)6份,分别按“3.4供试品溶液制备”项下方法制备,测定芍药苷的含量。
表8芍药苷含量测定重复性试验结果表
结果:试验方法的重现性良好。
8、加样回收率试验:
对照品溶液制备:精密量取“重复性试验”项下对照品贮备液3ml,置10ml量瓶中,用甲醇稀释至刻度,摇匀,即得。
供试品溶液制备:取同一批(批号:20190401)已知含量的少腹逐瘀丸(小蜜丸)(含量2.86mg/g),剪碎,分别取6份,每份约1.5g,精密称定,均加入芍药苷对照品溶液(浓度168.989μg/ml)10ml,按3.4供试品溶液制备项下方法处理,测定含量,以下式计算回收率。
回收率=(实测值-样品含量)/对照品加入量×100%
表9芍药苷含量测定加样回收率试验结果表
结果:试验方法的回收率良好。
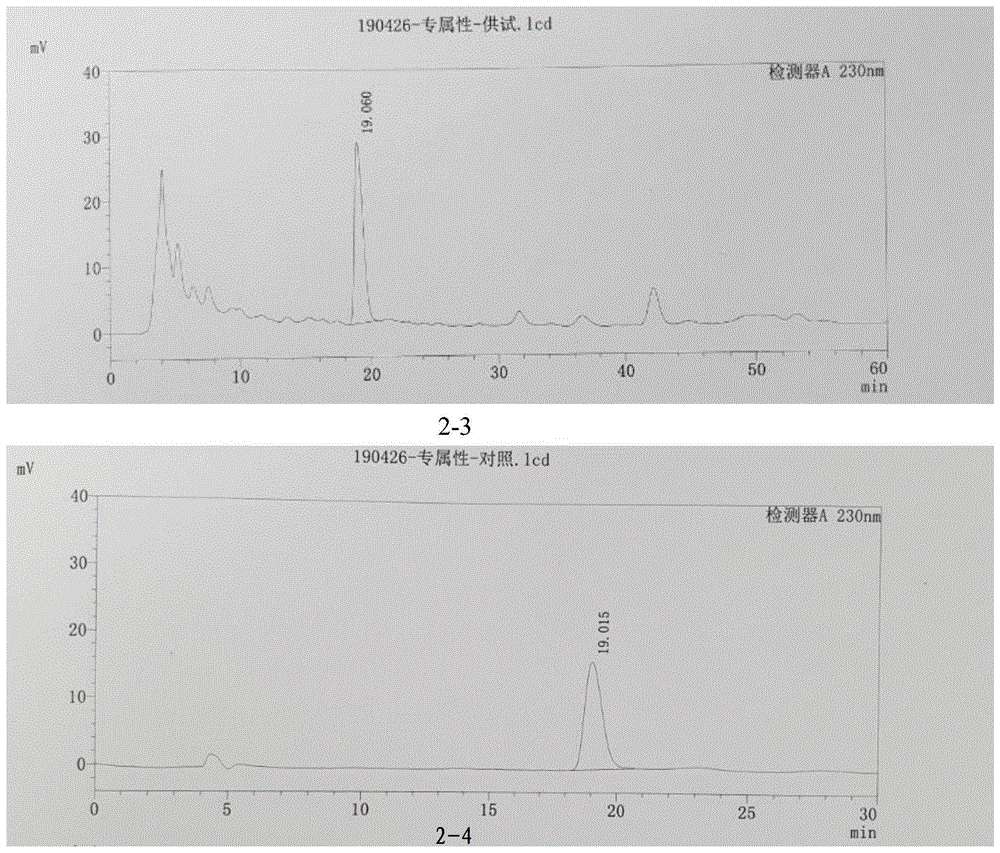
9、专属性:
供试品溶液制备:取少腹逐瘀丸(小蜜丸)(批号:20190401)1.5g,精密称定,按“3.4供试品溶液制备”处理,即得。
对照品溶液制备:取精密度项下对照品溶液,即得。
赤芍药材溶液制备:取赤芍药材约0.1g,精密称定,按“3.4供试品溶液制备”方法制备,即得。
阴性供试品溶液制备:取不含赤芍的阴性样品1.5g,精密称定,按“3.4供试品溶液制备”方法制备,即得。
分别精密吸取赤芍药材溶液、阴性供试品溶液、供试品溶液、对照品溶液各10μl,注入液相色谱仪,并记录色谱图。
结果表明:芍药苷可有效检出,且阴性供试品溶液无干扰。
10、样品含量测定:取样品按正文方法测定,芍药苷含量测定结果如下。
表10少腹逐瘀丸(小蜜丸)样品中芍药苷含量测定结果表
本品3批样品以高效液相法芍药苷的平均含量为2.84mg/g。
根据上述试验结果,考虑药材来源,以及制剂生产、贮藏等因素,故暂定本品每g含赤芍以芍药苷(c23h28o11)计,不得少于0.8mg。
实施例2少腹逐瘀丸(小蜜丸)中当归、蒲黄、赤芍、肉桂、小茴香和延胡索显微鉴别
1)薄壁细胞纺锤形,壁略厚,有极微细的斜向交错纹理(当归)。
2)花粉粒黄色,类圆形或椭圆形,直径约3μm,外壁有网状雕纹(蒲黄)。
3)草酸钙簇晶直径7~41μm,存在于薄壁细胞中,常排列成行或一个细胞中含有数个簇晶(赤芍)。
4)纤维单个散在,长梭形,直24~50μm,壁厚,木化(肉桂)。
5)草酸钙簇晶细小,直径约5μm,一个细胞含有多个簇晶(小茴香)。
6)糊化淀粉粒团块淡色(延胡索)。
结果:图4~9为少腹逐瘀丸(小蜜丸)中当归、蒲黄、赤芍、肉桂、小茴香和延胡索显微鉴别图,结果表明显微鉴别方法简便,专属性强,阴性无干扰,显微特征突出,可以作为少腹逐瘀丸(小蜜丸)中的质量检测方法。
实施例3少腹逐瘀丸(小蜜丸)中赤芍鉴别
1)供试品溶液的制备:取本品9g,剪碎,加硅藻土10g,研匀,加乙醇50ml,超声处理20分钟,滤过,滤液蒸干,残渣用水20ml溶解,用水饱和的正丁醇振摇提取3次,每次20ml,合并正丁醇提取液,用正丁醇饱和的水洗涤3次,每次15ml,正丁醇液蒸干,残渣用乙醇20ml溶解,加活性炭2.5g,水浴加热2分钟,放冷,滤过,滤液浓缩至约1ml,作为供试品溶液;
2)对照品溶液的制备:另取芍药苷对照品,加乙醇制成每lml含2mg的溶液,作为对照品溶液;
3)点样、展开:照薄层色谱法(通则0502)试验,吸取供试品溶液5~10μl、对照品溶液2μl,分别点于同一硅胶g薄层板上,以三氯甲烷-乙酸乙酯-甲醇-甲酸(40∶5∶10∶0.2)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。;
4)结果:图10为少腹逐瘀丸(小蜜丸)中赤芍的薄层色谱图,结果表明:含有赤芍的样品在与芍药苷对照品色谱对应的位置上,显相同颜色的斑点。色谱分离良好,紫花地丁的特征斑点突出,斑点清晰,方法简便,专属性强,除去赤芍的阴性对照无干扰,建立的tlc方法可以作为少腹逐瘀丸(小蜜丸)中赤芍的质量检测方法。
实施例4少腹逐瘀丸(小蜜丸)中当归和川芎鉴别
1)供试品溶液的制备:取本品6g,剪碎,加乙醚30ml,超声处理20分钟,滤过,滤液挥干,残渣加乙酸乙酯lml使溶解,作为供试品溶液。
2)对照品溶液的制备:取当归对照药材与川芎对照药材各0.5g,同法制成对照药材溶液。
3)点样、展开:照薄层色谱法(通则0502)试验,吸取供试品溶液、对照药材溶液各5μl,分别点于同一硅胶g薄层板上,以正己烷-乙酸乙酯(4∶1)为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。
4)结果:图11为少腹逐瘀丸(小蜜丸)中当归、川芎的薄层色谱图,结果表明:含有当归、川芎的样品在与当归、川芎对照药材色谱对应的位置上,显相同颜色的斑点。色谱分离良好,紫花地丁的特征斑点突出,斑点清晰,方法简便,专属性强,除去当归、川芎的阴性对照无干扰,建立的tlc方法可以作为少腹逐瘀丸(小蜜丸)中当归、川芎的质量检测方法。
实施例5少腹逐瘀丸(小蜜丸)中蒲黄鉴别
1)供试品溶液的制备:取本品10g,剪碎,加乙醚20ml,超声处理20分钟,弃去乙醚液,残渣挥干,加水30ml使溶解,用水饱和的正丁醇振摇提取2次,每次20ml,合并正丁醇液,蒸干,残渣加乙醇1ml使溶解,作为供试品溶液。
2)对照品溶液的制备:另取蒲黄对照药材1g,加水30ml,煮沸15分钟,放冷,滤过,用水饱和的正丁醇振摇提取2次,每次20ml,合并正丁醇液,蒸干,残渣加乙醇1ml使溶解,作为对照药材溶液。
3)点样、展开:照薄层色谱法(通则0502)试验,吸取供试品溶液、对照药材溶液各5μl,分别点于同一硅胶g薄层板上,以乙酸乙酯-丁酮-甲酸-水(5∶3∶1∶1)为展开剂,展开,取出,晾干,喷以5%三氯化铝乙醇溶液,105℃加热至斑点显色清晰,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。
4)结果:图12为少腹逐瘀丸(小蜜丸)中蒲黄的薄层色谱图,结果表明:含有蒲黄的样品在与蒲黄对照药材色谱对应的位置上,显相同颜色的斑点。显相同颜色的斑点斑点清晰,方法简便,专属性强,除去蒲黄的阴性对照无干扰,色谱分离良好,蒲黄的特征斑点突出,建立的tlc方法可以作为少腹逐瘀丸(小蜜丸)中蒲黄的质量检测方法。
实施例6少腹逐瘀丸(小蜜丸)中延胡索鉴别
1)供试品溶液的制备:取本品30g,剪碎,加甲醇50ml,超声处理30分钟,滤过,滤液蒸干,残渣加水20ml使溶解,加浓氨试液调至碱性,用乙醚振摇提取3次,每次10ml,合并乙醚液,蒸干,残渣加甲醇lml使溶解,作为供试品溶液。
2)对照品溶液的制备:另取延胡索对照药材lg,同法制成对照药材溶液。
3)点样、展开:照薄层色谱法(通则0502)试验,吸取供试品溶液、对照药材溶液各5μl,分别点于同一用1%氢氧化钠溶液制备的硅胶g薄层板上,以甲苯-丙酮(9∶2)为展开剂,展开,取出,晾干,置碘缸中约3分钟后取出,挥尽薄层板上吸附的碘后,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。
4)结果:图13为少腹逐瘀丸(小蜜丸)中延胡索的薄层色谱图,结果表明:含有延胡索的样品在与延胡索对照药材色谱对应的位置上,显相同颜色的斑点。色谱分离良好,延胡索的特征斑点突出,显相同颜色的荧光斑点。斑点清晰,方法简便,专属性强,除去延胡索的阴性对照无干扰,建立的tlc方法可以作为少腹逐瘀丸(小蜜丸)中延胡索的质量检测方法。
实施例7高效液相色谱法测定少腹逐瘀丸(小蜜丸)中赤芍的芍药苷
1)供试品溶液的制备:取装量差异项下少腹逐瘀丸(小蜜丸)适量,剪碎,混匀,取约1.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50ml,密塞,称定重量,待完全溶散后,超声处理(功率100w,频率40khz)30分钟,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2)对照品溶液的制备:取芍药苷对照品适量,精密称定,加甲醇制成每lml含60μg的溶液,即得;
3)hplc色谱条件:以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.1%磷酸(14∶86)为流动相;检测波长为230nm。理论板数按芍药苷峰计算应不低于3000;
4)测定方法:分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,按照步骤3)所述色谱条件,测定,即得;
以上所述仅为本发明的优选实施例,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制,应当指出,对于本领域的普通技术人员来说,在不脱离本发明原理及构思的前提下,还可以做出若干修饰和改进,或直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!