测定高硅银精矿中氟和氯的方法与流程
本发明涉及矿产品分析技术领域,特别涉及测定高硅银精矿中氟和氯的方法。
背景技术:
我国每年依靠大量进口银精矿用于有色金属工业生产,银精矿由银矿石经过放射性分选、重力法选矿、或浮选等选矿过程的产物,其中银元素含量大于2500g/t,其他元素主要有硅、硫、铁、铅、铝等,二氧化硅含量一般为5~25%,硫含量一般为5~50%,其中二氧化硅含量大于25%的银精矿,通常称之为高硅银精矿。
在贸易结算方面,氯、氟等元素作为银精矿生产过程中的有害元素,已经列为进厂原料中的罚款元素而逐步受到贸易双方的重视;在选矿冶金工艺方面,氟离子、氯离子等阴离子分析是银精矿过程分析的常规项目,选冶过程对以上阴离子的监控,直接影响到后续产品的纯度;在冶炼过程中,氟和氯容易产生氟化氢和氯化氢等酸性气体,对管道设备有严重的腐蚀,影响生产设备的寿命;在国门安全把关方面,国家对涉及“安全、卫生、环保”的监测指标愈加重视,氟和氯常作为有毒有害元素的研究对象。因此,及时制定银精矿中氟、氯有害元素的检测方法,为相关机构提供检测依据显得尤为迫切。
目前测定矿石中氟的方法主要有离子选择性电极法、分光光度法,测定氯的方法主要有离子选择性电极法、电位滴定法、比浊法。以上方法只能测定氟或氯一种元素,但随着离子色谱技术的普及,离子色谱也逐渐应用到矿石领域,可以同时测定氟和氯等多种元素,缩短了检测时间,提高了检测效率。实验多采用简单的玻璃器皿自搭蒸馏装置,用硫酸溶液作为介质进行蒸馏试验,利用高沸点的酸置换低沸点酸的原理,将样品中氟和氯以氟化氢和氯化氢的形式蒸馏出来,被吸收液吸收后通过离子色谱法测定。
高硅银精矿中含硅较多,此外由于银精矿中也含有硫,二氧化硅和硫的存在导致采用常见的“直接用硫酸溶液为介质蒸馏样品”会出现两个方面的问题:在蒸馏过程中,样品中的硫会以臭鸡蛋气味的硫化氢气体溢出,对操作人员的身体健康不利;另一方面,由于高硅银精矿中二氧化硅含量较高,直接用硫酸介质蒸馏,矿样不能溶解完全,有黑渣出现,造成测定结果偏低。如采用高温水解-离子色谱法测定高硅银精矿中氟和氯需要额外购置专业的高温水解装置作为前处理设备,成本价格较高;高温水解装置需要在1000多度的高温下实验,实验中用到的样品舟及易被银精矿样品腐蚀,银精矿残渣黏附样品舟严重,样品舟不能再次使用,耗材成本高;实验过程中产生的单质硫及其他物质会残留在高温水解装置的核心部件“燃烧管”造成腐蚀,甚至断裂;实验过程中仍有臭鸡蛋气味的硫化氢气体溢出,影响操作人员身体健康。
因此有必要建立前处理成本较低、有益于操作人员健康,能够有效提取样品中氟和氯,并准确测定银精矿中氟和氯的检测方法。
技术实现要素:
有鉴于此,本发明提供一种测定高硅银精矿中氟和氯的方法。本发明采用氢氧化钠或氢氧化钾作为熔剂在高温下熔融样品,结合超声技术将坩埚壁上熔块残渣超声脱落,采用水蒸气蒸馏法蒸馏出氟和氯,以离子色谱技术为测定手段对氟和氯进行测定,实现了银精矿中中氟和氯的准确测定。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了测定高硅银精矿中氟和氯的方法,包括如下步骤:
步骤1:取待测样品经预处理与氢氧化钠或氢氧化钾混合加热后冷却,与水混合加热浸提熔融物,超声,过滤;收集滤液,洗涤滤渣后收集洗涤液与所述滤液合并获得溶液;调节ph值,蒸发至所述溶液体积五分之二的;再与2倍体积的硫酸混合制得反应液;
步骤2:取氢氧化钠溶液作为接收液放入锥形瓶,备用;
步骤3:取步骤1制得的所述反应液蒸馏,蒸馏液进入锥形瓶中与接收液混合,定容,过滤膜,离子色谱检测。
在本发明的一些具体实施方案中,步骤1中所述预处理为过150目筛,于105℃干燥1h。
在本发明的一些具体实施方案中,步骤1中所述待测样品与氢氧化钠的质量比为(0.2~0.5):(2~6)。
在本发明的一些具体实施方案中,步骤1中所述加热后冷却为:于550~650℃加热20~40min,冷却至15~35℃。
在本发明的一些具体实施方案中,步骤1中所述超声的频率为52hz,所述超声的时间为10~15min。
在本发明的一些具体实施方案中,步骤1中,以g/ml计,所述待测样品与所述硫酸的质量体积比为(0.2~0.5):(3~8)。
在本发明的一些具体实施方案中,步骤2中所述氢氧化钠溶液的摩尔浓度为0.2mol/l。
在本发明的一些具体实施方案中,步骤3中所述蒸馏的温度为160~180℃,所述蒸馏的时间为20~35min,所述蒸馏液的体积为70~120ml。
在本发明的一些具体实施方案中,步骤3中所述离子色谱检测的条件为:柱温20℃~30℃;淋洗液为3.2mmol/l碳酸钠和1.0mmol/l碳酸氢钠混合溶液,流速为0.7ml/min等度淋洗,洗脱时间为23~26min;进样量20μl。
在本发明的一些具体实施方案中,步骤3中所述离子色谱检测采用metrosepasupp5型阴离子分离柱(4mm×150mm),metrosepasupp4/5guard型保护柱(4mm×10mm)。
本发明选用氢氧化钠熔融-超声辅助对样品进行前处理后再蒸馏。高硅银精矿中二氧化硅含量较高,氢氧化钠或氢氧化钾可以与二氧化硅充分反应,更彻底的熔融样品,更好的提取样品中的氟和氯。在550~650℃高温炉中,氢氧化钠熔融样品的过程中,硫可以被氧化成高价态,防止了后期蒸馏过程中臭鸡蛋气味的硫化氢气体溢出,解决了对操作人员的身体健康不利的问题。结合了超声技术,可以将实验过程中黏附在坩埚壁上的熔块残渣超声脱落,更有效的提取氟和氯。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍。
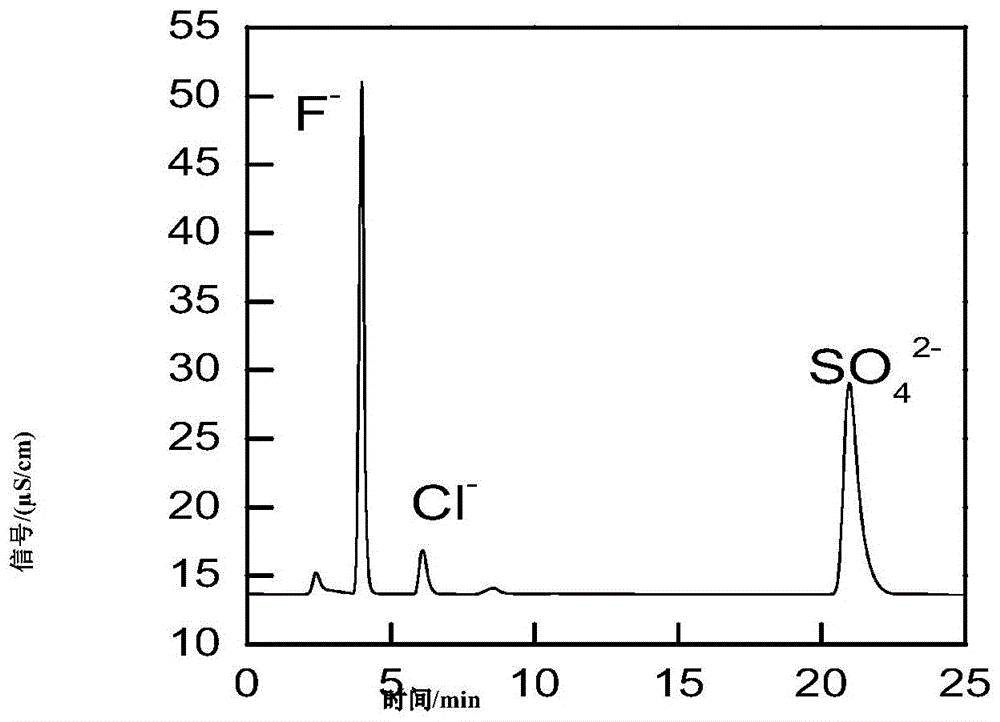
图1示样品a溶液色谱图;
图2示混合标准溶液色谱图;
图3示实验前、后的燃烧管比较图。
具体实施方式
本发明公开了一种测定高硅银精矿中氟和氯的方法,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。
本发明的技术方案为:
1、称取0.2~0.5g预干燥试样(过150目筛,在105℃干燥1h),将试样放入底部盛有1~3g氢氧化钠的镍坩埚中,加入1~3g氢氧化钠盖住试料,将坩埚放入550~650℃的马弗炉内20~40min,取出坩埚,冷却至室温。将坩埚放入预先放有60ml水的200ml烧杯中,加热浸取熔融物,至坩埚内熔块基本溶出,把烧杯放入超声仪中(超声频率为52hz)再继续超声10~15min后,坩埚壁上的熔块残渣可脱落进入到溶液中,烧杯底部可见银精矿中铅、铜、铁等元素在氢氧化钠体系下形成的沉淀。过滤溶液至另一200ml烧杯中,少量水冲洗原烧杯和滞留在滤纸上的沉淀各3次。向烧杯中加入约3~8ml硫酸调节溶液至ph=7,然后蒸发溶液至30ml,转移溶液于三口圆底烧瓶中,再加入60ml硫酸,并放入数粒玻璃珠,备用。
2、取400ml水置于水蒸气蒸馏装置中的500ml双口蒸馏瓶中,放入数粒玻璃珠,加热使水沸腾,备用。分别移取15mlnaoh溶液(0.2mol/l)于两个250ml锥形接收瓶中作为接收液,备用。
3、连接蒸馏装置进行蒸馏,控制蒸馏温度为160~180℃,当蒸馏液的体积为70~120ml时(花费时间约20~35min)取下接收瓶,将溶液转移至250ml容量瓶中,定容。容量瓶中溶液过0.22μm滤膜后注入离子色谱仪中按照仪器工作条件进行测定。
4、metrosepasupp5型阴离子分离柱(4mm×150mm),metrosepasupp4/5guard型保护柱(4mm×10mm);柱温20℃~30℃;淋洗液为3.2mmol/l碳酸钠和1.0mmol/l碳酸氢钠混合溶液,流速为0.7ml/min等度淋洗,洗脱时间为23~26min;进样量20μl。
本发明提供的测定高硅银精矿中氟和氯的方法中所用原料及试剂均可由市场购得。
仪器:
883型离子色谱仪(瑞士万通公司):配850型电导检测器,化学抑制器;milli-q型超纯水机(美国密理博公司);cpx8800h-c型超声仪(美国必能信公司);lt24/11/b180型高温炉(德国纳博热公司);0.22μm微孔纤维素过滤膜;镍坩埚;水蒸气蒸馏装置。
试剂:
氢氧化钠(优级纯,德国默克公司);硫酸(优级纯,国药集团化学试剂有限公司);氟离子标准溶液(国家钢铁材料测试中心):gsb04-2070-2007,1000mg/l;氯离子标准溶液(国家有色金属及电子材料分析测试中心):gsb04-1770-2004,1000mg/l;硫酸根离子标准溶液(国家有色金属及电子材料分析测试中心):gsb04-1773-2004,1000mg/l。
下面结合实施例,进一步阐述本发明:
实施例1:
1、称取0.2g预干燥试样c(过150目筛,在105℃干燥1h),将试样放入底部盛有1g氢氧化钠的镍坩埚中,加入1g氢氧化钠盖住试料,将坩埚放入550℃的马弗炉内20min,取出坩埚,冷却至室温。将坩埚放入预先放有60ml水的200ml烧杯中,加热浸取熔融物,至坩埚内熔块基本溶出,把烧杯放入超声仪中再继续超声10min后,坩埚壁上的熔块残渣可脱落进入到溶液中,烧杯底部可见银精矿中铅、铜、铁等元素在氢氧化钠体系下形成的沉淀。过滤溶液至另一200ml烧杯中,少量水冲洗原烧杯和滞留在滤纸上的沉淀各3次。向烧杯中加入约3ml硫酸调节溶液至ph=7,然后蒸发溶液至30ml,转移溶液于三口圆底烧瓶中,再加入60ml硫酸,并放入数粒玻璃珠,供步骤3使用。
2、取400ml水置于水蒸气蒸馏装置中的500ml双口蒸馏瓶中,放入数粒玻璃珠,加热使水沸腾,备用。分别移取15mlnaoh溶液(0.2mol/l)于两个250ml锥形接收瓶中作为接收液,供步骤3使用。
3、连接蒸馏装置进行蒸馏,控制蒸馏温度为160~180℃,当蒸馏液的体积为70ml,此时接收瓶内溶液的总体积为100ml(整个蒸馏过程约20min),取下接收瓶,将溶液转移至250ml容量瓶中,定容。容量瓶中溶液过0.22μm滤膜后注入离子色谱仪中按照仪器工作条件进行测定。
4、metrosepasupp5型阴离子分离柱(4mm×150mm),metrosepasupp4/5guard型保护柱(4mm×10mm);柱温20℃;淋洗液为3.2mmol/l碳酸钠和1.0mmol/l碳酸氢钠混合溶液,流速为0.7ml/min等度淋洗,洗脱时间为26min;进样量20μl。
表1测定结果
平行测定7个样品c,氟和氯测定值的相对标准偏差小于4%。
实施例2:
1、称取0.3g预干燥试样b(过150目筛,在105℃干燥1h),将试样放入底部盛有2g氢氧化钠的镍坩埚中,加入2g氢氧化钠盖住试料,将坩埚放入600℃的马弗炉内25min,取出坩埚,冷却至室温。将坩埚放入预先放有60ml水的200ml烧杯中,加热浸取熔融物,至坩埚内熔块基本溶出,把烧杯放入超声仪中再继续超声13min后,坩埚壁上的熔块残渣可脱落进入到溶液中,烧杯底部可见银精矿中铅、铜、铁等元素在氢氧化钠体系下形成的沉淀。过滤溶液至另一200ml烧杯中,少量水冲洗原烧杯和滞留在滤纸上的沉淀各3次。向烧杯中加入约5ml硫酸调节溶液至ph=7,然后蒸发溶液至30ml,转移溶液于三口圆底烧瓶中,再加入60ml硫酸,并放入数粒玻璃珠,供步骤3使用。
2、取400ml水置于水蒸气蒸馏装置中的500ml双口蒸馏瓶中,放入数粒玻璃珠,加热使水沸腾,备用。分别移取15mlnaoh(0.2mol/l)溶液于两个250ml锥形接收瓶中作为接收液,供步骤3使用。
3、连接蒸馏装置进行蒸馏,控制蒸馏温度为160~180℃,当蒸馏液的体积为95ml时,此时接收瓶内溶液的总体积为125ml(整个蒸馏过程约27min),取下接收瓶,将溶液转移至250ml容量瓶中,定容。容量瓶中溶液过0.22μm滤膜后注入离子色谱仪中按照仪器工作条件进行测定。
4、metrosepasupp5型阴离子分离柱(4mm×150mm),metrosepasupp4/5guard型保护柱(4mm×10mm);柱温30℃;淋洗液为3.2mmol/l碳酸钠和1.0mmol/l碳酸氢钠混合溶液,流速为0.7ml/min等度淋洗,洗脱时间为23min;进样量20μl。
表2测定结果
平行测定7个样品b,氟和氯测定值的相对标准偏差小于4%。
实施例3:
1、称取0.5g预干燥试样a(过150目筛,在105℃干燥1h),将试样放入底部盛有3g氢氧化钠的镍坩埚中,加入3g氢氧化钠盖住试料,将坩埚放入650℃的马弗炉内40min,取出坩埚,冷却至室温。将坩埚放入预先放有60ml水的200ml烧杯中,加热浸取熔融物,至坩埚内熔块基本溶出,把烧杯放入超声仪中再继续超声15min后,坩埚壁上的熔块残渣可脱落进入到溶液中,烧杯底部可见银精矿中铅、铜、铁等元素在氢氧化钠体系下形成的沉淀。过滤溶液至另一200ml烧杯中,少量水冲洗原烧杯和滞留在滤纸上的沉淀各3次。向烧杯中加入约8ml硫酸调节溶液至ph=7,然后蒸发溶液至30ml,转移溶液于三口圆底烧瓶中,再加入60ml硫酸,并放入数粒玻璃珠,供步骤3使用。
2、取400ml水置于水蒸气蒸馏装置中的500ml双口蒸馏瓶中,放入数粒玻璃珠,加热使水沸腾,备用。分别移取15mlnaoh(0.2mol/l)溶液于两个250ml锥形接收瓶中作为接收液,供步骤3使用。
3、连接蒸馏装置进行蒸馏,控制蒸馏温度为160~180℃,当蒸馏液的体积为120ml时,此时接收瓶内溶液的总体积为150ml(整个蒸馏过程约35min),取下接收瓶,将溶液转移至250ml容量瓶中,定容。容量瓶中溶液过0.22μm滤膜后注入离子色谱仪中按照仪器工作条件进行测定。
4、metrosepasupp5型阴离子分离柱(4mm×150mm),metrosepasupp4/5guard型保护柱(4mm×10mm);柱温25℃;淋洗液为3.2mmol/l碳酸钠和1.0mmol/l碳酸氢钠混合溶液,流速为0.7ml/min等度淋洗,洗脱时间为25min;进样量20μl。
表3测定结果
平行测定7个样品a,氟和氯测定值的相对标准偏差小于4%。
对比例1:碳酸钠熔融
称取0.50g(过150目筛,在105℃干燥1h)将试样a放入底部盛有3g无水碳酸钠的镍坩埚中,再加入3g无水碳酸钠,尽可能盖住试料,将坩埚放入650℃的马弗炉内40min,取出坩埚,将坩埚放入干燥器中冷却至室温。将试料熔融物置于三口圆底烧瓶中,加入60ml硫酸溶液(体积比为2∶1),并放入数粒玻璃珠,备用。
其余条件同实施例3中2~4。
对比例2:直接蒸馏
称取0.50g(过150目筛,在105℃干燥1h)将试样c直接置于三口圆底烧瓶中,加入60ml硫酸溶液(体积比为2∶1),并放入数粒玻璃珠,备用。
其余条件同实施例3中2~4。
效果例1:关于实施例3、对比例1、对比例2的对比测试结果:
表4不同前处理方法对氟和氯测定结果的影响
实施例3中氟的测定值与对比例1和对比例2相比,分别提高20.2%、14.4%;实施例3中氯的测定值与对比例1和对比例2相比,分别提高34.6%、28.3%。说明实施例3对样品中氟和氯的提取更有效。
通过运用统计学上的t检验法(双侧),可以判断两组独立数据的平均值有极显著性差异,具体分析如下:
首先比较对比例2和实施例3中“f”元素。对比例2中,f的测定次数n1=7,平均值
按照上述t的计算公式进行计算,求得t计算=23.707;再通过查ta,f值表,当p=0.99,a=0.01,f=n1+n2-2=7+7-2=12时,ta,f=3.055。则t计算>ta,f,说明两组数据存在极显著差异,实施例3对样品中氟的提取效果明显优于对比例2。
同理,对于对比例2和实施例3中“cl”元素,t计算=16.093,ta,f=3.055;则t计算>ta,f,说明两组数据存在极显著差异,实施例3对样品中氯的提取效果明显优于对比例2。
同理,对于对比例1和实施例3中“f”元素,t计算=17.346,ta,f=3.055;则t计算>ta,f,说明两组数据存在极显著差异,实施例3对样品中氟的提取效果明显优于对比例1。
同理,对于对比例1和实施例3中“cl”元素,t计算=13.809,ta,f=3.055;则t计算>ta,f,说明两组数据存在极显著差异,实施例3对样品中氯的提取效果明显优于对比例1。
对比例3:
设定样品超声时间为0min,其余条件同实施例3(因为氢氧化钠熔融样品后熔块残渣粘坩锅较严重,不能和热水充分接触,不利于残渣中氟和氯的浸出。实验中选取超声的方式,将粘附于坩埚壁的熔块残渣超声脱落下来进入水溶液中)。
对比例4:
设定样品超声时间为5min,其余条件同实施例3。
对比例5:
设定样品超声时间为10min,其余条件同实施例3。
对比例6:
设定样品超声时间为20min,其余条件同实施例3。
效果例2:关于实施例3、对比例3、对比例4、对比例5、对比例6的对比测试结果:
表5超声时间对测定结果w/%的影响
表5说明,随着超声时间的增加,氟和氯的浸出量增加,当超声时间为15min,氟和氯已经基本浸出完毕,选择超声时间为15min。
对比例7:
设定熔融温度为450℃,其余同实施例3。
对比例8:
设定熔融温度为550℃,其余同实施例3。
对比例9:
设定熔融温度为750℃,其余同实施例3。
效果例3:关于实施例3、对比例7、对比例8、对比例9的对比测试结果:
表6熔融温度对测定结果w/%的影响
表6说明,熔融温度增加有利于氟和氯的增加,当熔融温度为650℃时,氟和氯值测定值基本最大并趋于稳定。增加到750℃时,温度过高容易引起熔融物在高温炉中喷溅,影响测定。最终选择650℃。
对比例10:
设定熔融时间为10min,其余同实施例3。
对比例11:
设定熔融时间为20min,其余同实施例3。
对比例12:
设定熔融时间为60min,其余同实施例3。
效果例4:关于实施例3、对比例10、对比例11、对比例12的对比测试结果:
表7熔融时间对测定结果w/%的影响
当熔融时间为40min时,氟和氯已基本从矿物中熔出,选择熔融时间为40min。
对比例13:
设定蒸馏液的体积为45ml,其余同实施例3。
对比例14:
设定蒸馏液的体积为75ml,其余同实施例3。
对比例15:
设定蒸馏液的体积为95ml,其余同实施例3。
对比例16:
设定蒸馏液的体积为145ml,其余同实施例3。
效果例5:关于实施例3、对比例13、对比例14、对比例15、对比例16的对比测试结果:
表8接收液体积对测定结果w/%的影响
随着馏出液体积增加,接收液体积增加,当蒸馏液的体积为120ml时候,继续增加馏出液,氟和氯的值趋于稳定,故接收液体积选择为120ml。
对比例17:
设定色谱洗脱时间为10min,则10min后终止洗脱,其余同实施例3。从图1可以得知,氟离子和氯离子会从色谱柱中洗脱出来,但仍有硫酸根残留在色谱柱中,进而毒害色谱柱及影响下一个样品的分析。
效果例6:色谱分离时间的影响
银精矿样品a溶液色谱图见图1,氟离子、氯离子、硫酸根离子混合阴离子标准溶液色谱图见图2,氟离子、氯离子、硫酸根离子的保留时间分别为4.01min,6.19min,21.37min。当氟离子和氯离子流出色谱柱后,淋洗液还需继续洗脱色谱柱,将硫酸根离子从色谱柱中完全洗脱,避免硫酸根离子对分析下一个样品造成干扰,故本法将色谱洗脱时间设置为25min。
效果例7:校准曲线与检出限
配制一系列不同质量浓度的氟和氯混合标准溶液(0.5mg/l,1mg/l,5mg/l,10mg/l,20mg/l),过0.22μm微孔纤维素滤膜后,按色谱条件进行测定,以氟离子或氯离子质量浓度ρ为横坐标,对应的色谱峰峰面积a为纵坐标,绘制校准曲线,得到氟离子线性回归方程为a=0.3516ρ-1.7043,相关系数为r=0.9998;氯离子线性回归方程为a=0.2266ρ-0.5154,相关系数为r=0.9992。在实验条件下,对11份样品空白吸收液中的氟离子和氯离子测定,以结果标准偏差的3倍计算检出限,得到氟和氯的检出限分别为0.004%和0.007%。
效果例8:回收率试验
目前还未见到银精矿中含氟和氯的标准物质,也未查到高硅银精矿中氟和氯的相关方法,本文通过加标回收试验来考察方法的正确度,结果见表9。由表9可知,加标回收率为97.3%~103.5%。
表9加标回收试验结果
对比例18
通过高温水解-离子色谱法测定样品中氟和氯:称取0.5g(过150目筛,在105℃干燥1h)样品a置于高温水解装置的样品舟中,加入0.5g二氧化硅,小心混合均匀,再用约0.5g二氧化硅铺盖在上面。将试样舟放入高温水解装置中进行高温水解,水解后产生的气体经冷凝管冷凝成液体被预先放置有10ml氢氧化钠溶液(0.2mol/l)的锥形瓶接收,当锥形瓶内体积为70ml时,转移并定容到100ml容量瓶。容量瓶中溶液过0.22μm滤膜后注入离子色谱仪中按照实施例3的仪器工作条件进行测定。平行测定4个样品。
对比例19
称取0.3g(过150目筛,在105℃干燥1h)样品b,其余同对比例18。平行测定4个样品。
对比例20
称取0.2g(过150目筛,在105℃干燥1h)样品c,其余同对比例18。平行测定4个样品。
效果例9:本方法(氢氧化钠熔融-超声辅助-水蒸气蒸馏-离子色谱法)与高温水解-离子色谱法比对试验
表10结果对比
1通过运用统计学上的t检验法(双侧),可以判断两组独立数据的平均值是无显著性差异,两种测定方法的结果基本一致,具体分析如下:
首先比较对比例18和实施例3测定样品a中“f”元素。对比例18中,f的测定次数n1=4,平均值
同理。比较比较对比例18和实施例3测定样品a中“cl”元素,t计算=1.291,ta,f=1.833;则t计算<ta,f,说明两组数据不存在显著差异,两种方法测定结果基本一致。
同理。比较比较对比例19和实施例2测定样品b中“f”元素,t计算=0.877,ta,f=1.833;则t计算<ta,f,说明两组数据不存在显著差异,两种方法测定结果基本一致。
同理。比较比较对比例19和实施例2测定样品b中“cl”元素,t计算=0.887,ta,f=1.833;则t计算<ta,f,说明两组数据不存在显著差异,两种方法测定结果基本一致。
同理。比较比较对比例20和实施例1测定样品c中“f”元素,t计算=1.759,ta,f=1.833;则t计算<ta,f,说明两组数据不存在显著差异,两种方法测定结果基本一致。
同理。比较比较对比例20和实施例1测定样品c中“cl”元素,t计算=0.885,ta,f=1.833;则t计算<ta,f,说明两组数据不存在显著差异,两种方法测定结果基本一致。
2样品在高温水解实验过程中产生的单质硫及其他物质会残留在高温水解装置的核心部件“燃烧管”造成严重腐蚀,实验前和实验后的燃烧管如图3。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!