一种表面活性剂的微量快速检测方法与流程
本发明涉及化合物检测技术的
技术领域:
,尤其是涉及一种表面活性剂的微量快速检测方法。
背景技术:
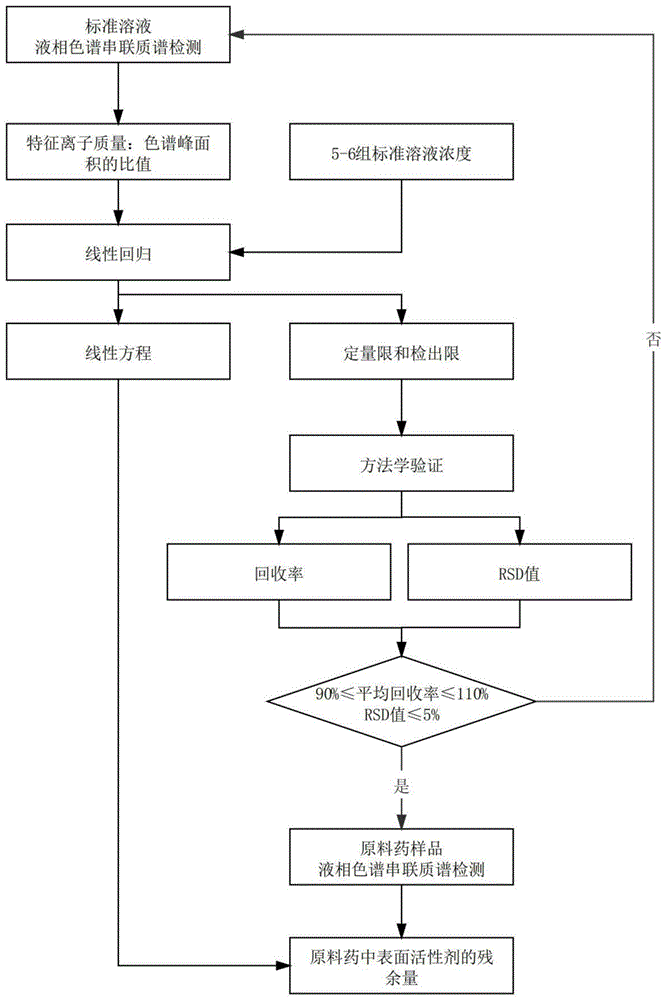
:表面活性剂是指加入少量能使其溶液体系的界面状态发生明显变化的物质,其包括阴离子型表面活性剂、阳离子型表面活性剂、两性离子型表面活性剂、非离子型表面活性剂等。其中,阴离子型表面活性剂包括肥皂类、硫酸化物和磺酸化物;阳离子型表面活性剂包括苯扎氯铵和苯扎溴铵;两性离子型表面活性剂包括卵磷脂、氨基酸型和甜菜碱;非离子型表面活性剂包括烷基葡糖苷、脂肪酸甘油酯、多元醇、聚乙烯型和聚氧乙烯-聚氧丙烯共聚物。目前,常见的表面活性剂有十二烷基硫酸钠、苯扎溴铵、3-磺丙基十四烷基二甲甜菜碱、吐温20、吐温80、曲拉通x-100、曲拉通x-114等。这些表面活性剂由于具有润湿或抗粘、乳化或破乳、起泡或消泡以及增溶、分散、洗涤、防腐、抗静电等一系列物理化学作用,除了在日常生活中作为洗涤剂外,其他应用几乎可以覆盖所有的精细化工领域。尤其是在医药领域的生物药制备过程中,常常会添加上述常见的表面活性剂用作蛋白溶解增溶剂、病毒灭活处理、发酵中消泡剂、以及制剂抗聚合稳定剂等。但是这些表面活性剂同时也对人体具有毒性,对皮肤、消化系统等存在刺激,容易引发过敏相关的不良反应。因此,现在需要一种适应这些表面活性剂残余量检测的检测方法,以便在药物质量控制中高效准确地控制表面活性剂的残留量。申请号为cn200810067212.6的中国专利公开了一种液相色谱-串联质谱联用技术测定烷基季铵盐类阳离子型表面活性剂的方法。申请号为cn200710040617.6的中国专利公开了一种磺酸类阴离子型表面活性剂的快速检测方法。申请号为cn201811634778.2的中国专利公开了一种吐温系列辅料高效液相色谱-高分辨质谱联用分析和鉴定方法。上述三篇专利分别通过液相/气相色谱串联质谱检测的方式,完成对离子型表面活性剂或非离子型表面活性剂的检测。虽然上述技术都涉及到了类似的方法,但是这些方法的具体检测步骤不一,所涉及的仪器和检测条件也各不相同。这就导致本领域技术人员在检测表面活性剂残余量时,需要更换不同的检测方法和设备材料,以达到定量检测的目的,有待改进。为此,黄雯雯,张梅,魏晓晓,etal.一种未知表面活性剂产品的成分剖析[j].中国测试,2017(8).一文中公开了利用lc-ms结合ir、gc、gc-ms等方法,对阴离子型表面活性剂和非离子型表面活性剂进行定性分析的方法。这种方法虽然在一定程度上对多种表面活性剂的检测具有通用性,但是所涉及的仪器较多,检测速度较慢,且不能进行定量分析,有待改进。技术实现要素:本发明要解决的问题是针对现有技术中所存在的上述不足而提供一种表面活性剂的微量快速检测方法,其通过进行液相色谱串联质谱检测并建立线性关系,解决了现有技术中的定量检测方法适用的表面活性剂的种类单一的问题,达到了能较快地对多种表面活性剂进行定量分析的目的。本发明的上述发明目的是通过以下技术方案得以实现的:一种表面活性剂的微量快速检测方法,包括以下步骤,s1将多组表面活性剂的标准溶液分别进行液相色谱串联质谱检测,得到所述表面活性剂的特征离子质量和色谱峰面积;s2以s1得到的特征离子质量对色谱峰面积的比值、以及所述表面活性剂的标准溶液浓度进行线性回归,确定线性关系、检出限和定量限;s3将原料药样品进行高效液相色谱串联质谱检测;s4根据s3得到的检测数据和s2的线性回归数据计算出所述表面活性剂的含量;其中,所述表面活性剂为阴离子型表面活性剂、阳离子型表面活性剂、两性离子型表面活性剂、以及非离子型表面活性剂。通过采用上述技术方案,利用质谱分析法是通过对被测表面活性剂离子的质荷比的测定来进行分析的一种分析方法,被分析的表面活性剂首先要离子化,然后利用不同离子在电场或磁场的运动行为的不同,把离子按质荷比分开而得到质谱,通过表面活性剂的质谱和相关信息,可以得到表面活性剂的定性定量结果;在此过程中,通过采用液相色谱串联质谱检测的方法,将液相色谱的分离能力和质谱的高选择性、高灵敏度结合起来,具有提供相对分子质量和结构信息,同时,能够快速微量精准检测的优点,为表面活性剂的残留检测提供了一种高效准确的分析方法,解决现有技术检测适用的表面活性剂的种类单一的问题;另外,在最初的线性回归并确定线性关系、检出限和定量限后,只需对原料药样品进行一次液相色谱串联质谱检测,即可较快地计算出表面活性剂的含量,操作简易快捷。进一步地,所述s1中,所述液相色谱串联质谱检测的具体步骤为,s1质谱检测,根据所述表面活性剂的相对分子质量,建立质谱条件,然后使用质谱仪依据质谱条件进行检测,找到响应值最大的一级离子、二级离子和对应的碰撞能量,同时保存源区参数;s2液相色谱检测,根据所述表面活性剂在色谱柱上的保留时间,建立色谱条件和液相洗脱梯度,然后使用超高效液相色谱仪依据优化后的色谱条件进行检测,计算色谱峰面积。通过采用上述技术方案,利用液相色谱串联质谱技术的优势上,分别优化液相和质谱条件,选择合适的洗脱梯度;其中,对质谱条件进行了优化,对各个表面活性剂进行一级和二级扫描,选择最优二级离子作为特征离子,最终实现对各类表面活性剂的定量分析,同时具有检测时间短、检测灵敏度高、线性关系较好的优点。具体地,所述质谱仪为三重四极杆液质联用仪。三重四极杆液质联用仪能快速地进行二级谱图扫描确认,并可以同时进行定量和定性分析,有利于提高本方法的检测速率和检测灵敏度。具体地,所述质谱条件为,离子源:电喷雾正离子扫描,esi+;监测反应:多反应监测扫描mrm;离子源温度:200-400℃;电喷雾电压:2000-5000v;鞘气压力:10-50arb;辅助气压力:10-30arb。此质谱条件下,可以在10min内完成一次检测,检测灵敏度高,线性关系较好,方法回收率高,稳定性良好,抗干扰能力强,最低检出限达到10ppm以下,适用于各类表面活性剂的检测。具体地,所述液相色谱仪为超高效液相色谱仪。超高效液相色谱仪借助于hplc的理论及原理,增加了分析的通量、灵敏度及色谱峰容量,有利于提高本方法的检测速率和检测灵敏度。具体地,所述色谱条件为,流动相a:0.1%甲酸水溶液;流动相b:0.1%甲酸乙腈溶液;流速:0.1-0.5ml/min;柱温:30℃;进样量:10μl;色谱柱:acquitycshtmc18,1.7μm,2.1x100mm。此色谱条件下,可以在10min内完成一次检测,检测灵敏度高,线性关系较好,方法回收率高,稳定性良好,抗干扰能力强,最低检出限达到10ppm以下,适用于各类表面活性剂的检测。优选地,所述液相洗脱梯度为,时间/min05.00-5.505.51-6.50流动相a(%)50-95550-95流动相b(%)5-5080-955-50。进一步地,所述表面活性剂为硫酸类阴离子型表面活性剂、季胺类阳离子型表面活性剂、甜菜碱型两性离子型表面活性剂、多元醇类非离子型表面活性剂、以及聚氧乙烯型非离子型表面活性剂。优选地,所述表面活性剂为十二烷基硫酸钠、苯扎溴铵、3-磺丙基十四烷基二甲甜菜碱、吐温20、吐温80、曲拉通x-100、以及曲拉通x-114。综上所述,本发明的有益技术效果为:通过进行液相色谱串联质谱检测并建立线性关系,将液相色谱的分离能力和质谱的高选择性、高灵敏度结合起来,在最初的线性回归并确定线性关系、检出限和定量限后,只需对原料药样品进行一次液相色谱串联质谱检测,即可较快地计算出表面活性剂的含量,达到了能较快地对多种表面活性剂进行定量分析的目的。附图说明图1是本发明提供的检测方法的流程图。图2是本发明的实施例1的十二烷基硫酸钠的mrm色谱图。图3是本发明的实施例1的十二烷基硫酸钠的标准曲线图。图4是本发明的实施例2的苯扎溴铵的mrm色谱图。图5是本发明的实施例2的苯扎溴铵的标准曲线图。图6是本发明的实施例3的3-磺丙基十四烷基二甲甜菜碱的mrm色谱图。图7是本发明的实施例3的3-磺丙基十四烷基二甲甜菜碱的标准曲线图。图8是本发明的实施例4的曲拉通x-100的mrm色谱图。图9是本发明的实施例4的曲拉通x-100的标准曲线图。图10是本发明的实施例5的曲拉通x-114的mrm色谱图。图11是本发明的实施例5的曲拉通x-114的标准曲线图。具体实施方式为了使本发明实现的技术手段、创作特征、达成目的与作用更加清楚及易于了解,下面结合附图和具体实施方式对本发明作进一步阐述:制备例储备液的配制:使用电子分析天平精密称取10.0±0.1mg的十二烷基硫酸钠/苯扎溴铵/3-磺丙基十四烷基二甲甜菜碱/曲拉通x-100/曲拉通x-114标准品于10ml容量瓶中,分别用超纯水定容至刻度线,配制成四种1μg/ml的储备液,于4℃保存。标准溶液的配置:取十二烷基硫酸钠/苯扎溴铵/曲拉通x-100/曲拉通x-114的储备液,分别用超纯水稀释成50、100、200、400、600、800、1000ng/ml,作为标准溶液,于4℃保存。取3-磺丙基十四烷基二甲甜菜碱的储备液,用超纯水稀释成25、50、100、200、500、1000ng/ml,作为标准溶液,于4℃保存。原料药溶液的配制:使用电子分析天平精密称取100.0±0.1mg的利拉鲁肽于10ml容量瓶中,用水定容至刻度线,配制成10mg/ml的原料药溶液,于4℃保存。实验溶液的配置:取十二烷基硫酸钠/苯扎溴铵/曲拉通x-100/曲拉通x-114的储备液,用原料药溶液稀释成200、400、1000ng/ml,作为实验溶液,于4℃保存。取3-磺丙基十四烷基二甲甜菜碱的储备液,用原料药溶液稀释成100、200、1000ng/ml,作为实验溶液,于4℃保存。其中,所使用的仪器和原料的型号及生产厂家参照表1。表1原料/仪器成分/型号生产厂家乙腈色谱纯默克甲酸色谱纯ace利拉鲁肽cas:204656-20-2派金十二烷基硫酸钠sds;cas:151-21-3生工苯扎溴铵cas:7281-04-1生工3-磺丙基十四烷基二甲甜菜碱cas:14933-09-6生工曲拉通x-100cas:9002-93-1生工曲拉通x-114cas:9036-19-5生工三重四极杆液质联用仪thermotsqquantumultra赛默飞世尔科技超高效液相色谱仪watersacquitytmultraperformancelc美国沃特世公司电子分析天平mettlertoledome-204e梅特勒实施例实施例1:参照图1,为本发明公开的一种表面活性剂的微量快速检测方法,包括以下步骤,s1标准溶液前处理,选择制备例所得的十二烷基硫酸钠的标准溶液,准备将这些标准溶液分别按照优化后的色谱条件和质谱条件进行检测;s2质谱检测,根据十二烷基硫酸钠的相对分子质量,建立并优化质谱条件,然后使用三重四极杆液质联用仪依据优化后的质谱条件进行检测,找到响应值最大的一级离子、二级离子和对应的碰撞能量,同时保存源区参数;s3液相色谱检测,根据十二烷基硫酸钠在色谱柱上的保留时间,建立并优化色谱条件和液相洗脱梯度,然后使用超高效液相色谱仪依据优化后的色谱条件进行检测,计算色谱峰面积;s4标准曲线建立,根据检测结果,以十二烷基硫酸钠的特征离子质量对色谱峰面积的比值为纵坐标(y)、标准溶液浓度为横坐标(x)进行线性回归,得到十二烷基硫酸钠标准曲线,同时得到线性方程、r2、线性范围、定量限和检出限等参数;s5方法学验证,对原料药进行三水平添加回收率实验及精密度实验,验证s2-s3的方法准确度和重现性;s6若s5测得的结果符合条件,则可使用s1-s3的方法对药品中十二烷基硫酸钠进行检测,然后使用s4的标准曲线对药品中十二烷基硫酸钠进行定性定量分析。其中,所使用的仪器和原料的型号及生产厂家参照表1,s2-s5的具体实施方式如下。1.质谱条件离子源:电喷雾正离子扫描,esi+;监测反应:多反应监测扫描mrm;离子源温度:275℃;电喷雾电压:2000v;鞘气压力:30arb;辅助气压力:15arb;碰撞气:氩气。其中,十二烷基硫酸钠的一级离子、二级离子、碰撞能量、保留时间参照表2和图2。2.色谱条件流动相a:0.1%甲酸水溶液;流动相b:0.1%甲酸乙腈溶液;流速:0.1ml/min;柱温:30℃;进样量:10μl;色谱柱:acquitycshtmc18,1.7μm,2.1x100mm。其中,液相洗脱梯度参照表3。3.标准曲线建立参照图3,以十二烷基硫酸钠的特征离子质量对色谱峰面积的比值为纵坐标(y)、标准溶液浓度为横坐标(x)进行线性回归,绘制得十二烷基硫酸钠标准曲线图,同时得到线性方程、r2、线性范围、定量限等参数。然后按照定量限的浓度,取对应浓度的十二烷基硫酸钠的标准溶液,用超纯水多次稀释,并分别按照本实施例的方法进行测定,测得检出限。其中,十二烷基硫酸钠标准曲线的相关数据和检测限参照表4。4.方法学验证4.1回收率实验采用制备例的十二烷基硫酸钠的标准溶液和实验溶液,按照本实施例的方法进行测定,平行测定6次并计算平均值,同时计算回收率,计算结果参照表5。4.2精密度实验取25ng/ml的十二烷基硫酸钠的标准溶液,按照本实施例的方法进行测定,平行测定6次并计算保留时间,同时计算峰面积的rsd值,计算结果参照表6。实施例2:参照图1,为本发明公开的一种表面活性剂的微量快速检测方法,与实施例1的不同之处在于,选择制备例所得的苯扎溴铵的相关溶液进行检测,检测条件如下。1.质谱条件离子源:电喷雾正离子扫描,esi+;监测反应:多反应监测扫描mrm;离子源温度:320℃;电喷雾电压:4500v;鞘气压力:50arb;辅助气压力:20arb;碰撞气:氩气。其中,十二烷基硫酸钠的一级离子、二级离子、碰撞能量、保留时间参照表2和图4。2.色谱条件流动相a:0.1%甲酸水溶液;流动相b:0.1%甲酸乙腈溶液;流速:0.3ml/min;柱温:30℃;进样量:10μl;色谱柱:acquitycshtmc18,1.7μm,2.1x100mm。其中,液相洗脱梯度参照表3。参照图5,另外,标准曲线和方法学验证的相关数据参照表4-6。实施例3:参照图1,为本发明公开的一种表面活性剂的微量快速检测方法,与实施例1的不同之处在于,选择制备例所得的3-磺丙基十四烷基二甲甜菜碱的相关溶液进行检测,检测条件如下。1.质谱条件离子源:电喷雾正离子扫描,esi+;监测反应:多反应监测扫描mrm;离子源温度:300℃;电喷雾电压:3800v;鞘气压力:10arb;辅助气压力:20arb;碰撞气:氩气。其中,3-磺丙基十四烷基二甲甜菜碱的一级离子、二级离子、碰撞能量、保留时间参照表2和图6。2.色谱条件流动相a:0.1%甲酸水溶液;流动相b:0.1%甲酸乙腈溶液;流速:0.5ml/min;柱温:30℃;进样量:10μl;色谱柱:acquitycshtmc18,1.7μm,2.1x100mm。其中,液相洗脱梯度参照表3。参照图7,另外,标准曲线和方法学验证的相关数据参照表4-6。实施例4:参照图1,为本发明公开的一种表面活性剂的微量快速检测方法,与实施例1的不同之处在于,选择制备例所得的曲拉通x-100的相关溶液进行检测,检测条件如下。1.质谱条件离子源:电喷雾正离子扫描,esi+;监测反应:多反应监测扫描mrm;离子源温度:200℃;电喷雾电压:4200v;鞘气压力:40arb;辅助气压力:30arb;碰撞气:氩气。其中,3-磺丙基十四烷基二甲甜菜碱的一级离子、二级离子、碰撞能量、保留时间参照表2和图8。2.色谱条件流动相a:0.1%甲酸水溶液;流动相b:0.1%甲酸乙腈溶液;流速:0.3ml/min;柱温:30℃;进样量:10μl;色谱柱:acquitycshtmc18,1.7μm,2.1x100mm。其中,液相洗脱梯度参照表3。参照图9,另外,标准曲线和方法学验证的相关数据参照表4-6。实施例5:参照图1,为本发明公开的一种表面活性剂的微量快速检测方法,与实施例4的不同之处在于,选择制备例所得的曲拉通x-114的相关溶液进行检测。1.质谱条件离子源:电喷雾正离子扫描,esi+;监测反应:多反应监测扫描mrm;离子源温度:400℃;电喷雾电压:5000v;鞘气压力:40arb;辅助气压力:20arb;碰撞气:氩气。其中,3-磺丙基十四烷基二甲甜菜碱的一级离子、二级离子、碰撞能量、保留时间参照表2和图10。2.色谱条件流动相a:0.1%甲酸水溶液;流动相b:0.1%甲酸乙腈溶液;流速:0.3ml/min;柱温:30℃;进样量:10μl;色谱柱:acquitycshtmc18,1.7μm,2.1x100mm。其中,液相洗脱梯度参照表3。参照图11,另外,标准曲线和方法学验证的相关数据参照表4-6。表2一级离子(m/z)二级离子(m/z)碰撞能量(v)保留时间(min)实施例1266.0236.082.29实施例2304.190.9322.33实施例3364.2168.1254.08实施例4647.8133.0212.25实施例5427.5315.3112.40表3表4线性方程r2线性范围(ng/ml)定量限(ng/ml)检出限(ng/ml)实施例1y=82.78x–376.710.99100-100010050实施例2y=102.4x–783.10.9950-10005025实施例3y=3823.7x–1487740.9925-500256.25实施例4y=41.577x–20.7210.99100-100010050实施例5y=501.3x–125270.99100-100010050从表4可知,十二烷基硫酸钠的检出限为50ng/ml,在100-1000ng/ml范围内有线性关系;苯扎溴铵检出限为25ng/ml,在50-1000ng/ml范围内有线性关系;3-磺丙基十四烷基二甲甜菜碱检出限为6.25ng/ml,在25-500ng/ml范围内有线性关系;曲拉通x-100检出限为50ng/ml,在100-1000ng/ml范围内有线性关系;曲拉通x-114检出限为50ng/ml,在100-1000ng/ml范围内有线性关系。由此可知,采用本发明的检测方法检测各类表面活性剂的标准溶液,能获得较好的线性关系。表5表6从表5和表6可知,1.在高、中、低浓度下,十二烷基硫酸钠的回收率在93.20%-100.23%内,平均回收率96.78%,方法准确度高;同时重复分析6次,峰面积和保留时间相对标准偏差分别为0.28%和2.76%,小于3.0%,方法重现性好;2.在高、中、低浓度下,苯扎溴铵的回收率在96.41%-98.02%内,平均回收率97.35%,方法准确度高;同时重复分析6次,峰面积和保留时间相对标准偏差分别为0.27%和1.77%,小于2.0%,方法重现性好;3.在高、中、低浓度下,3-磺丙基十四烷基二甲甜菜碱的回收率在100.21%-115.50%内,平均回收率105.74%,方法准确度高;同时重复分析6次,峰面积和保留时间相对标准偏差分别为0.19%和4.51%,小于5.0%,方法重现性好;4.在高、中、低浓度下,曲拉通x-100的回收率在93.56%-97.18%内,平均回收率95.78%,方法准确度高;同时重复分析6次,峰面积和保留时间相对标准偏差分别为0.73%和2.56%,小于3.0%,方法重现性好;5.在高、中、低浓度下,曲拉通x-114的回收率在95.00%-98.19%内,平均回收率96.31%,方法准确度高;同时重复分析6次,峰面积和保留时间相对标准偏差分别为0.93%和0.35%,小于1.0%,方法重现性好。另外,各类表面活性剂进行一次质谱检测和液相色谱检测的时间均低于10min。综上所述,采用本发明的检测方法检测各类表面活性剂的标准溶液,检出限均达到10ppm以下,在检测时间短的同时,能获得较好的线性关系,且方法准确度和重现性较佳。这是因为优化后的质谱条件、色谱条件和液相洗脱梯度均有利于达成本检测方法快速微量精准检测的目的,同时利用三重四极杆液质联用仪和超高效液相色谱仪组成液相色谱-三重四级杆质谱联用系统,将液相色谱的分离能力和质谱的高选择性、高灵敏度结合起来,具有提供相对分子质量和结构信息,同时,能够快速微量精准检测的优点,为表面活性剂的残留检测提供了一种高效准确的分析方法。在此条件下,在最初的线性回归并确定线性关系、检出限和定量限后,只需对原料药样品进行一次液相色谱串联质谱检测,即可较快地计算出表面活性剂的含量,解决现有技术检测适用的表面活性剂的种类单一的问题。最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1