用于预测个体的吸烟状况的系统和方法与流程
本申请根据35U.S.C.§119要求2013年12月16日提交的名称为“用于预测个体的吸烟状况的系统和方法(Systems and Methods for Predicting a Smoking Status of an Individual)”的美国临时专利申请61/916,443的优先权,所述美国临时专利申请全文并入本文中。
背景技术:
全基因组微阵列被用作测量全基因组表达水平和获得对各种病况的生物见解的实际手段。这种方法也被用来评定暴露于活性物质时的身体反应和预测所得表现型。可以检测到吸烟者的大气道细胞的转录组响应于香烟暴露所发生的分子改变,即使在没有明显的组织学异常时仍然可以检测到。这个观察结果表明,转录组数据也许可以用来评定生物系统在暴露于各种物质时的反应。
在很多的产品风险评定研究中,从所要的原发部位(如气道)获取样品是侵袭性的并且不方便。作为一个替代方案,外周采血是微创性的并且被广泛用于普通人群。因此,人们关注于寻找和建立可以在充当替代组织的外周血中可靠地使用的生物标志物。
探索分子生物标志物的前期尝试聚焦于鉴别案例与对照群体之间的差异表达的基因。最新的方法致力于越来越多地预测新案例,从而促使增强诊断、改良预后并且推进个体化用药。然而,对于临床应用来说稳固并且通用的计算方法的研发仍然具有挑战性。关于吸烟相关的疾病,已鉴别了外周血样品中的诊断标签。至少两个研究已经显示,差异表达基因可以区分患有早期非小细胞肺癌的受试者与对照受试者或患有非恶性肺病的受试者(Rotunno,M.,Hu,N.,Su,H.,Wang,C.,Goldstein,A.M.,Bergen,A.W.,Consonni,D.,Pesatori,A.C.,Bertazzi,P.A.,Wacholder,S.等人(2011).来自外周全血的用于I期肺腺癌的基因表达标签(A gene expression signature from peripheral whole blood for stage I lung adenocarcinoma).癌症预防研究(Cancer Prev Res)(Phila)4,1599-1608;Showe,M.K.,Vachani,A.,Kossenkov,A.V.,Yousef,M.,Nichols,C.,Nikonova,E.V.,Chang,C.,Kucharczuk,J.,Tran,B.,Wakeam,E.等人(2009).外周血单核细胞中的基因表达谱可以区分患有非小细胞肺癌的患者与患有非恶性肺病的患者(Gene expression profiles in peripheral blood mononuclear cells can distinguish patients with non-small cell lung cancer from patients with nonmalignant lung disease).癌症研究(Cancer Res)69,9202-9210)。
技术实现要素:
提供用于鉴别基于血液的稳固的基因标签的计算系统和方法,所述基因标签可以用来预测个体的吸烟者状况。本文所述的基因标签能够区分目前在吸烟的受试者与从不吸烟或已戒烟的受试者,从而能够准确地预测个体的吸烟者状况。
在某些方面,本公开内容的系统和方法提供用于评定从受试者获得的样品的计算机化方法。计算机化方法包括通过接收电路接收与样品相关的数据集,所述数据集包括LRRN3、CDKN1C、PALLD、SASH1、RGL1以及TNFRSF17的定量表达数据。处理器基于接收到的数据集产生评分,所述评分指示所预测的受试者的吸烟状况。所预测的吸烟状况可以将受试者归类为当前吸烟者或非当前吸烟者。
在某些具体实施中,数据集进一步包括IGJ、RRM2、SERPING1、FUCA1以及ID3的定量表达数据。在某些具体实施中,评分是向所述数据集应用的分类方案的结果,其中所述分类方案是基于数据集中的定量表达数据确定的。
在某些具体实施中,所述方法进一步包括计算LRRN3、CDKN1C、PALLD、SASH1、RGL1以及TNFRSF17中的每一个的倍数变化值,并且确定每个倍数变化值满足至少一个准则。所述准则可以要求对于至少两个独立群体数据集来说,每个各自计算的倍数变化值超过预定阈值。
在某些方面,本公开内容的系统和方法提供用于评定从受试者获得的样品的计算机化方法。设备包括用于检测测试样品中基因标签中的基因的表达水平的装置,所述基因标签包括LRRN3、CDKN1C、PALLD、SASH1、RGL1以及TNFRSF17。所述设备还包括用于使表达水平与吸烟者状况的分类相关联的装置,和用于输出吸烟者状况的分类结果作为受试者的吸烟者状况的预测的装置。
在某些方面,本公开内容的系统和方法提供一种用于预测个体的吸烟者状况的试剂盒。所述试剂盒包括:一组检测测试样品中基因标签中的基因的表达水平的试剂,所述基因标签包括LRRN3、CDKN1C、PALLD、SASH1、RGL1以及TNFRSF17;和使用所述试剂盒预测个体的吸烟者状况的说明书。
在某些方面,本公开内容的系统和方法提供一种用于评定吸烟产品的替代物对个体的影响的试剂盒。所述试剂盒包括:一组检测测试样品中基因标签中的基因的表达水平的试剂,所述基因标签包括LRRN3、CDKN1C、PALLD、SASH1、RGL1以及TNFRSF17;和使用所述试剂盒评定所述替代物对个体的影响的说明书。吸烟产品的替代物可以是加热式烟草产品(heated tobacco product,HTP),并且替代物对个体的影响可以是将个体归类为非吸烟者。
在某些方面,本公开内容的系统和方法提供一种用于评定从受试者获得的样品的方法。所述方法包括通过接收电路接收与样品相关的数据集。数据集包括选自由以下组成的群组的至少五个标志物的定量表达数据:LRRN3、CDKN1C、PALLD、SASH1、RGL1、TNFRSF17、IGJ、RRM2、SERPING1、FUCA1以及ID3。所述方法进一步包括通过处理器,基于接收到的数据集产生评分,所述评分指示所预测的受试者的吸烟状况。所预测的受试者的吸烟状况可以将受试者归类为当前吸烟者或非当前吸烟者。
评分可以是向数据集应用的分类方案的结果,其中所述分类方案是基于数据集中的定量表达数据确定的。所述方法可以进一步包括计算LRRN3、CDKN1C、PALLD、SASH1、RGL1以及TNFRSF17中的每一个的倍数变化值,并且确定每个倍数变化值满足至少一个准则。所述准则可以要求对于至少两个独立群体数据集来说,每个各自计算的倍数变化值超过预定阈值。
在某些方面,本公开内容的系统和方法提供一种用于评定从受试者获得的样品的设备。所述设备包括用于检测测试样品中基因标签中的基因的表达水平的装置,所述基因标签包括至少五个选自由以下组成的群组的标志物:LRRN3、CDKN1C、PALLD、SASH1、RGL1、TNFRSF17、IGJ、RRM2、SERPING1、FUCA1以及ID3。所述设备进一步包括用于使表达水平与吸烟者状况的分类相关联的装置,和用于输出吸烟者状况的分类结果作为受试者的吸烟者状况的预测的装置。
在某些方面,本公开内容的系统和方法提供一种用于预测个体的吸烟者状况的试剂盒。所述试剂盒包括:一组检测测试样品中基因标签中的基因的表达水平的试剂,所述基因标签包括至少五个选自由以下组成的群组的标志物:LRRN3、CDKN1C、PALLD、SASH1、RGL1、TNFRSF17、IGJ、RRM2、SERPING1、FUCA1以及ID3;和使用所述试剂盒预测个体的吸烟者状况的说明书。
在某些方面,本公开内容的系统和方法提供一种用于评定吸烟产品的替代物对个体的影响的试剂盒。所述试剂盒包括:一组检测测试样品中基因标签中的基因的表达水平的试剂,所述基因标签包括至少五个选自由以下组成的群组的标志物:LRRN3、CDKN1C、PALLD、SASH1、RGL1、TNFRSF17、IGJ、RRM2、SERPING1、FUCA1以及ID3;和使用所述试剂盒评定替代物对个体的影响的说明书。吸烟产品的替代物可以是HTP,并且替代物对个体的影响可以是将个体归类为非吸烟者。
附图说明
在结合附图考虑以下详细描述之后,本公开内容的更多特征、其性质和各种优点将变得显而易见,在附图中同样的参考符号在所有附图中指代相同的部分,并且在附图中:
图1是用于鉴别一组基因并且基于这组基因获得分类模型的方法的流程图。
图2是用于评定从受试者获得的样品的方法的流程图。
图3是示例性计算设备的框图,所述计算设备可用来实现本文所述计算机化系统中的任一个中的任一个部件。
图4A、4B和4C是样品数据集中的差异表达基因的火山图。
图5A、5B、5C、5D、5E和5F是指示不同研究的分类方案的各种盒形图。
具体实施方式
本文描述了用于鉴别基于血液的稳固的基因标签的计算系统和方法,所述基因标签可以用来预测个体的吸烟者状况。具体来说,本文所述的基因标签能够区分目前在吸烟的受试者与从不吸烟或已戒烟的受试者。
如本文所用,“稳固的”基因标签是能跨越研究、实验室、样品来源以及其它人口因素维持强大性能的基因标签。重要的是,稳固标签应该是可检测的,即使是在一组包括大的个体差异的群体数据中仍然是可检测的。为了避免过于乐观地报告标签的性能,还应该恰当地验证在整个数据集中的稳固性。
本公开内容的一个目标是获得可以准确地预测个体的吸烟者状况的基因标签。为了评估基因标签的性能,本文在下表中示出了预测结果,下表在各行中展示了所预测的状况并且在各列中展示了真正的状况。下表1示出了展示预测结果的一种方式的实例。表格第一行指出了经过预测样品应该与当前吸烟者相关的真正的当前吸烟者和非当前吸烟者人群,并且表格第二行指出了经过预测样品应该与非当前吸烟者相关的真正的当前吸烟者和非当前吸烟者人群。
表1
完美预测器是将所有当前吸烟者准确地预测为当前吸烟者(真阳性将是100%并且假阴性将是0%),并且将所有非当前吸烟者准确地预测为非当前吸烟者(真阴性将是100%并且假阳性将是0%)。如本文所述,根据吸烟状况对个体进行分类(例如,当前吸烟者、非当前吸烟者、既往吸烟者、从不吸烟者等),但一般来说,本领域的普通技术人员将理解,本文所述的系统和方法适用于任何分类方案。
为了评估预测器的强度,可以使用基于预测结果表中的值的各种度量标准。在本文中,一种度量标准称为“灵敏度”,其为当前吸烟者组中被准确地归类为当前吸烟者的个体的比例。换句话说,灵敏度度量标准等于真阳性的数量除以真阳性和假阴性的总和或TP/(TP+FN)。灵敏度值1表示当前吸烟者的完美分类。在本文中,另一种度量标准称为“特异性”,其为非当前吸烟者组中被准确地归类为非当前吸烟者的个体的比例。换句话说,特异性度量标准等于真阴性的数量除以真阴性和假阳性的总和或TN/(TN+FP)。特异性值1表示非当前吸烟者的完美分类。为了被认为是一种强预测器,期望灵敏度值和特异性值高。虽然本文使用灵敏度和特异性度量标准来评估预测器的性能,但是一般来说,也可以在不脱离本公开内容的范围的情况下使用任何其它度量标准,如阳性测试的预测值(TP/(TP+FP))或阴性测试的预测值(TN/(TN+FN))。
本文所述的系统和方法通过以下步骤构造了一种预测模型:首先从不同的训练数据集中鉴别出所展现的表达水平的倍数变化高的基因。然后,用独立的数据集验证所鉴别的这组基因。验证后,通过评估吸烟者状态已知的受试者的血液转录组并且针对具有一种吸烟者状况的个体与具有另一种吸烟者状况的个体比较所鉴别的那组基因的表达水平来测试这组基因。所得的这组经过成功验证和测试的基因在本文中称为“基因标签”。
基因标签可以用来将个体准确地分到特定的所预测的吸烟者状况组中。此外,通过能够准确地预测个体的吸烟者状况,基因标签能够通过比较使用HTP的个体的结果与吸常规香烟的个体的结果,检测到各种HTP的使用。可以在需要关于吸烟行为的遵从性的情况下使用基因标签。在一个实例中,所预测的个体的吸烟者状况(如通过基因标签所确定)可以在HTP的临床试验中用于鉴别在个体转换到HTP之后,个体是否出现生物学变化或个体何时出现生物学变化。一般来说,基因标签可以用于监测吸烟、戒烟或转换到HTP的任何与健康相关的研究中。
在一个实例中,从若干公开可用的基因表达数据集获得剖析当前吸烟者和非吸烟者或既往吸烟者的血液样品的数据。基于高倍数变化基因从各个独立研究中预选择基因是有利的,因为这么做增强了标签在不同研究中的稳固性并且确保预测模型不会因为单一数据集而有偏差。用独立数据集进行验证,所述独立数据集源自旨在探索COPD的新颖生物标志物的临床研究。另外,根据另一个临床研究,对连续5天从常规香烟(其燃烧烟草)转换到HTP(其不燃烧烟草;本文中称为烟草加热系统(Tobacco Heating System,THS)2.1)的吸烟者的血液转录组加以评估并将其与继续吸常规香烟的吸烟者进行比较。本文所述的标签在对当前吸烟者和非当前吸烟者进行分类方面表现非常好,如使用独立数据集由所述标签的性能所展示。另外,可在血液转录组中检测到持续5天转换到THS 2.1的影响,因为转换到THS 2.1的受试者被归类为非当前吸烟者。这说明,本文中的基因标签以及系统和方法不仅可以用于确定吸烟者状况,而且可以用于评估吸烟的短期影响。
使用基于有限数量的基因的标签相对于使用整个转录组来说就降低成本和工作量而言是有利的,因为分析最终将基于定量逆转录酶-聚合酶链式反应(quantitative reverse transcriptase-polymerase chain reaction,qRT-PCR)测量。使用qRT-PCR,设备和运行成本方面(如试剂)的投资比使用微阵列有利。
在一个实例中,在第一步中,获得不同的训练数据集以鉴别基因标签。确切地说,本文使用两个训练数据集:BLD-SMK-01和QASMC。然而,一般来说,可以在不脱离本公开内容的范围的情况下使用训练数据集的任何数量的任何组合。
关于BLD-SMK-01,从存储库(美国马里兰州贝茨维尔20705的博仕生物技术公司(BioServe Biotechnologies Ltd,Beltsville,MD 20705USA))获得使用PAXgene血液DNA试剂盒(凯杰(Qiagen))收集的血液样品。在采样时,受试者的年龄在23岁与65岁之间。排除无疾病史的受试者和正在服用处方药的受试者。当前吸烟者已经持续至少3年每天吸至少10根香烟。既往吸烟者在采样之前已戒烟至少2年并且在戒烟之前已有至少3年每天吸至少10根香烟。当前吸烟者和非吸烟者通过年龄和性别匹配。从当前吸烟者获得总共31份血液样品,从从不吸烟者获得30份血液样品,并且从既往吸烟者获得30份血液样品。
还从安妮女王街医疗中心(Queen Ann Street Medical Center,QASMC)临床研究获得血液样品,所述临床研究在英国伦敦的心肺中心(The Heart and Lung Centre)根据优质临床规范(Good Clinical Practice,GCP)进行并且已在ClinicalTrials.gov上以标识符NCT01780298注册。QASMC研究旨在鉴别能够区分患有COPD的受试者(吸烟史≥10包年(pack year)且处于GOLD阶段1或2的当前吸烟者)与三组对照的相匹配的非吸烟受试者(从不吸烟者、既往吸烟者和当前吸烟者)的生物标志物或一组生物标志物。在四组中的每一组中,从六十名受试者获得样品(总共240名受试者)。包括年龄介于40岁与70岁之间的男性和女性受试者。所有受试者都通过种族、性别和年龄(相差5岁以内)与所述研究中所招募的COPD受试者相匹配。将血液样品传送到丹麦奥尔胡斯的AROS应用生物技术公司(AROS Applied Biotechnology AS(Aarhus,Denmark)),所述血液样品在此公司经过进一步处理并且然后与昂飞(Affymetrix)人类基因组U133 Plus2.0基因芯片杂交,如下所述。
根据制造商的说明书,使用PAXgene血液miRNA试剂盒(目录编号763134;凯杰)分离总RNA(包括微小RNA)。使用UV分光光度计(NanoDrop ND1000;美国马萨诸塞州沃尔瑟姆的赛默飞世尔科技(Thermo Fisher Scientific,Waltham,MA,USA)),通过在230、260和280nm下测量吸光度来确定RNA样品的浓度和纯度。使用安捷伦(Agilent)2100生物分析仪进一步检查RNA的完整性。仅对RNA完整指数(RNA integrity number,RIN)大于6的RNA进行处理以用于进一步分析。
RNA制备和昂飞杂交.使用NuGENTMOvationTM全血试剂和NuGENTMOvationTMRNA扩增系统V2,由50ng RNA制备靶向转录物的3'端的昂飞探针组。用Nanodrop 1000或8000分光光度计(赛默飞世尔科技)或SpectraMax 384Plus(Molecular Devices)测量cDNA的数量。通过使用安捷伦2100生物分析仪评定未片段化cDNA的大小来确定cDNA的质量。还使用电泳图监测最终的片段化并且生物素化的产物的大小分布。在标记cDNA之后,根据制造商的指南,使所述片段与GeneChip Human Genome U133Plus 2.0阵列杂交。将用于目标制备的样品完全随机化以用于昂飞基因表达微阵列。
Taqman qRT-PCR分析.根据制造商的说明书,使用iScriptTMcDNA合成试剂盒(目录编号170-8890;美国加利福尼亚州埃库莱斯的伯乐公司(Bio-Rad,Hercules CA,USA)),用500ng起始RNA进行逆转录反应。然后,将cDNA精确地稀释到10ng/μL。向样品中添加商用人类通用RNA(human universal RNA,UHR)参考(目录编号740000,美国加利福尼亚州圣克拉拉的安捷伦技术(Agilent Technologies,Santa Clara,CA,USA))作为校准剂,以可靠地比较多个实验和仪器的数据。Taqman分析中所用的探针跨越外显子,并且选择五个管家基因(B2M、GAPDH、FARP1、A4GALT、GINS2)用于数据归一化步骤。使用分析和快速高级主混合物(目录:444963)进行qPCR步骤。简单来说,稀释cDNA,以允许在384孔板中每孔涂覆1.25ng。并行地,为每个Taqman分析制备主混合物(含Taqman分析试剂和Taqman高级混合物)。最终反应体积是10μL。使用Viia7仪器(生命技术(Life Technologies))运行qPCR并且应用自动基线和默认Ct阈值设定以便分析结果。在添加通用人类参考(Universal Human Reference,UHR)样品时,使每个基因的Ct值相对于UHR Ct值归一化(通过减法),并且然后相对于GAPDH管家基因值归一化(产生所谓的ΔΔCt值)。
从美国加利福尼亚州的生命技术获得Taqman引物。下表2列举了用于执行qRT-PCR的引物序列。
表2
微阵列分析-数据质量检查和归一化.在调查芯片图像以检测关于芯片扫描的矫作物之后,经由标准质量控制管道处理数据。简单来说,使用affy封装的ReadAffy功能(Gautier,L.,Cope,L.,Bolstad,B.M.和Irizarry,R.A.(2004).affy---在探针水平上分析昂飞基因芯片数据(affy---analysis of Affymetrix GeneChip data at the probe level).生物信息学(Bioinformatics)20,307-315)[6]读取原始数据文件,所述affy封装来自微阵列分析工具的Bioconductor套组(Gentleman,R.C.,Carey,V.J.,Bates,D.M.,Bolstad,B.,Dettling,M.,Dudoit,S.,Ellis,B.,Gautier,L.,Ge,Y.,Gentry,J.等人(2004).Bioconductor:计算生物学和生物信息学开放式软件开发(Bioconductor:open software development for computational biology and bioinformatics).基因组生物学(Genome Biol)5,R80),所述微阵列分析工具可用于R统计环境(R研发核心团队(R Development Core Team)(2007).R:一种用于统计计算的语言和环境(R:A Language and Environment for Statistical Computing))。通过产生并且检查以下各者来控制质量:RNA降解曲线(affy封装的AffyRNAdeg功能)、[09:42:29]归一化的未缩放的标准误差曲线、相对对数表达曲线(affyPLM封装(Brettschneider,J.,Collins,F.和Bolstad,B.M.(2008).短寡核苷酸微阵列数据的质量评定(Quality Assessment for Short Oligonucleotide Microarray Data).技术计量学(Technometrics)50,241-264))以及相对对数表达值的平均值。另外,用眼检查伪图像(探针水平模型的残留)来确保不存在空间效应。在质量控制检查时排除低于一组阈值的阵列进行进一步分析。
关于群体水平分析(即,平均倍数变化研究),随后使用GC-稳固微阵列分析(GC-Robust Microarray Analysis,GC-RMA)使数据归一化。使用背景校正和分位数归一化,从通过质量控制检查的所有阵列产生微阵列表达值(Irizarry,R.A.,Hobbs,B.,Collin,F.,Beazer-Barclay,Y.D.,Antonellis,K.J.,Scherf,U.和Speed,T.P.(2003).高密度寡核苷酸阵列探针水平数据的探索、归一化和总结(Exploration,normalization,and summaries of high density oligonucleotide array probe level data).生物统计学(Biostatistics)4,249-264)。关于个体标签预测模型,用MAS5(Affymetrix,I.(2002).统计学算法说明文件(Statistical algorithms description document).技术论文(Technical paper))使数据归一化。
统计建模-群体水平分析.关于每一个比较,拟合总体线性模型,基于适中的t统计法产生每个探针组在表达阵列上的原始p值。使用本亚明-霍赫贝格错误发现率(Benjamini-Hochberg False Discovery Rate,FDR)方法校正因为评估大量基因而出现的多个测试影响。
统计建模-个体样品预测建模.为了在预测模型中实现稳固性,从美国国家生物技术信息中心基因表达大棚车(Gene Expression Omnibus,GEO)(http://www.ncbi.nlm.nih.gov/gds/?term=GEO)获得来自血液的独立基因表达数据集(GSE15289)和PBMC(GSE42057)并加以处理。来自NOWAC研究的数据集(GSE15289)(Dumeaux,V.,Olsen,K.S.,Nuel,G.,Paulssen,R.H.,A.-L.和Lund,E.(2010a).解密正常血液基因表达变异:NOWAC后基因组研究(Deciphering normal blood gene expression variation—The NOWAC postgenome study).公共科学图书馆·遗传学(PLoS genetics)6,e1000873)包括来自年龄介于48岁与63岁之间的285名绝经后女性的全血样品,包括211名从不吸烟者和74名当前吸烟者。Bahr等人的数据集(GSE42057)(Bahr,T.M.,Hughes,G.J.,Armstrong,M.,Reisdorph,R.,Coldren,C.D.,Edwards,M.G.,Schnell,C.,Kedl,R.,LaFlamme,D.J.和Reisdorph,N.(2013).慢性阻塞性肺病中的外周血单核细胞基因表达(Peripheral Blood Mononuclear Cell Gene Expression in Chronic Obstructive Pulmonary Disease).美国呼吸道细胞和分子生物学杂志(American journal of respiratory cell and molecular biology))源自从36名当前吸烟者(其中22人患有COPD并且14人是健康的)和100名既往吸烟者(其中72人患有COPD并且28人是健康的)收集的外周血单核细胞(peripheral blood mononucleated cell,PBMC)样品。所有受试者都是非西班牙裔白人。
使用从GSE15289和GSE42057数据集中抽取的受试者数据来鉴别每个数据集中在吸烟者样品与从不吸烟者(或既往吸烟者)样品之间展现出高的平均表达变化的基因。使L1和L2为M个(此处,M=1000,但一般来说,M可以是任何值)相对于两个独立数据集(GSE15289和GSE42057)来说最高倍数变化基因的集合。为了获得清单L1,根据吸烟者状况(当前吸烟者和从不吸烟者)对数据集GSE15289进行分类,并且获得每组的平均基因表达水平。当前吸烟者组与从不吸烟者组之间在平均基因表达水平方面的差异在本文中称为倍数变化,并且在集合L1中包括M个具有最高倍数变化的基因。以类似方式,但针对当前吸烟者和既往吸烟者获得清单L2。
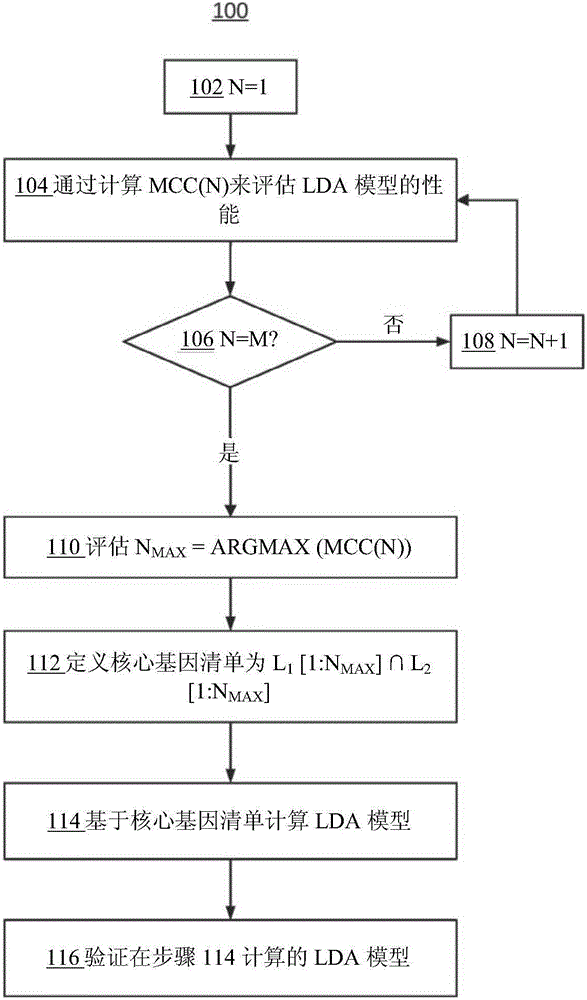
图1是用于鉴别一组基因并且基于这组基因获得分类模型的方法100的流程图。具体来说,方法100包括以下步骤:将计数器参数N初始化为1(步骤102),通过计算马修斯相关系数(Matthews Correlation Coefficient,MCC(N))评估线性判别分析(linear discriminant analysis,LDA)模型的性能(步骤104),并且判定计数器参数是否等于最大计数器值M(判定框106)。如果N小于M,那么方法100前进到步骤108以使N递增并且返回到步骤104以通过计算下一个系数MCC(N)来评估LDA模型的性能。当N达到M(判定框106)时,评估产生最大MCC值的N值(NMAX)(步骤110),并且将核心基因清单定义为两组基因L1[1:N]与L2[1:N]之间的交集(步骤112)。在鉴别了核心基因清单之后,基于核心基因清单计算LDA模型(步骤114)。
在步骤102,将计数器参数N初始化为1。计数器参数N从1变到最大值M并且在步骤108递增,直到在判定框106,N达到M为止。
在步骤104,通过计算系数MCC(N)评估LDA模型的性能。具体来说,可以对L1[1:N]∩L2[1:N]使用5折交叉验证(100次)来评估LDA模型的性能,交叉验证为集合L1中的最高倍数变化N和集合L2中的最高倍数变化N的交集。通过计算MCC(N)评估LDA模型。MCC度量法组合所有的真/假阳性和阴性率,并且因此提供单值公平度量值。MCC是可以用作复合材料性能评分的性能度量标准。MCC是介于-1与+1之间的值并且基本上是介于已知的二元分类与所预测的二元分类之间的相关系数。MCC可以使用以下方程式计算:
其中TP:真阳性;FP:假阳性;TN:真阴性;FN:假阴性。然而,一般来说,基于一组性能度量标准产生复合材料性能度量标准的任何合适的技术都可以用来评定LDA模型的性能。MCC值+1表示所述模型获得了完美的预测,MCC值0表示所述模型预测几乎就是随机的,并且MCC值-1表示所述模型预测完全不准确。MCC的优势在于当以仅类别预测可用的方式编码分类器函数时能够容易计算。相比之下,对于曲线下面积(area under the curve,AUC)计算,需要分类器函数提供数值评分。然而,一般来说,根据本公开内容,可以使用任何解释TP、FP、TN和FN的度量标准。
为了计算MCC,首先应该选择分类类别的集合。从从不吸烟者、既往吸烟者和当前吸烟者获得BLD-SMK-01数据集。图4A、4B和4C显示BLK-SMK-01样品中的差异表达基因的火山图。每个火山图显示所估算的log2(倍数变化)对比log10(经过调整的P值)。基于适中的t统计法计算P值并且通过本亚明霍赫贝格法加以调整。确切地说,图4A比较当前吸烟者与非吸烟者之间的基因表达谱,图4B比较当前吸烟者与既往吸烟者之间的基因表达谱,并且图4C比较既往吸烟者与从不吸烟者之间的基因表达谱。图4C中所示的火山图指出,在从不吸烟者与既往吸烟者之间无差异基因表达变化(即,在图4C中未观察到趋势),但图4A和4B指出,在当前吸烟者与从不吸烟者之间(图4A)并且在当前吸烟者与既往吸烟者之间(图4B)观察到了很多差异基因表达变化。
因此,BLD-SMK-01样品的群体水平转录组学分析表明,在从不吸烟者与既往吸烟者之间在全血中不存在差异基因表达变化,并且因此,基于血液转录组来区分既往吸烟者与从不吸烟者将是非常具有挑战性的。反之,在当前吸烟者与从不吸烟者之间和在当前吸烟者与既往吸烟者之间存在很多差异表达基因(图4A和4B)。因为在从不吸烟者群体与既往吸烟者群体之间未观察到差异,所以在步骤104仅使用两个类别来评估模型:当前吸烟者和非当前吸烟者。
确切地说,在步骤104,基因集合L1[1:N]∩L2[1:N]对应于两个独立数据集GSE15289和GSE42057的最高倍数变化N的交集。交叉验证基于L1[1:N]或L2[1:N]的每个预测模型,评定LDA模型的结果是否能推广到独立数据集。在一个实例中,为了对L1[1:N]基因集执行一例5折交叉验证,将L1[1:N]集合随机地分成五个子集:A、B、C、D和E。四个(A、B、C和D)子集被用来使用LDA技术训练分类器,并且第五个子集(E)被用来测试针对另外四个子集进行训练的分类器。训练和测试过程另外重复四次,其中其它子集(A、B、C和D)中的每一个都被用作测试子集来测试针对另外四个子集进行训练的分类器。
一般来说,LDA技术的准则是将描述n个特征的输入向量x归类为y类。所述分类是基于某个函数,所述函数是所观察到的特征的线性组合。基于数据的训练子集估算线性组合的系数。确切地说,为了使用LDA技术训练分类器,从四个训练子集中鉴别出数据中基因表达水平的线性组合。线性组合在本文中称为分类器并且在所预测的吸烟者状况与所预测的非吸烟者状况之间限定边界。分类器用于获得测试子集中的每个个体的预测状况。这个过程另外重复四次,使得五个子集各自作为测试子集处理一次。在五个子集各自都已作为测试子集一次之后,一例5折交叉验证完成,并且训练数据观察结果(具有L1[1:N]∩L2[1:N]集合中的特征)被分成五个新的子集A’、B’、C’、D’和E’,从而引发第二例5折交叉验证。
本文所述的实例是100例5折交叉验证的结果,但一般来说,本领域的普通技术人员将理解,可以在不脱离本公开内容的范围的情况下使用任何例数的k折交叉验证。此外,本文所述的实例是LDA技术的结果,LDA技术基于基因表达水平的线性组合形成分类器。然而,一般来说,本领域的普通技术人员将理解,可以使用基因表达水平的任何函数来形成分类器,如二次函数、多项式函数、指数函数或可以在R^N中形成一维流形来定义分类器的任何其它合适的函数。
在步骤110,在N达到最大数目M之后,考虑MCC的M值的集合,并且将对应于MCC的最大值的N值评为Nmax=argmaxN(MCC(N))。如图1中所示,在已计算出MCC的所有M值之后执行Nmax的评估步骤。然而,一般来说,本领域的普通技术人员将理解,可替代地,可以将在步骤104计算的值MCC(N)与一些预定阈值进行比较,然后评估下一个值MCC(N+1)。在这种情况下,当发现值MCC超过预定阈值时,方法100可以直接前进到步骤110,将Nmax的值赋给当前的N值,不考虑其余的N值=Nmax+1到M。
在步骤112,标签的核心基因清单由交集L1[1:Nmax]∩L2[1:Nmax],或处于L1[1:Nmax]和L2[1:Nmax]两者中的基因的集合定义。如此实例中所述,仅使用两个数据集L1和L2。然而,一般来说,本领域的普通技术人员将理解,可以使用任何数量的数据集来计算MCC值和鉴别用于定义基因标签的核心基因集。具体来说,可以使用m个数据集的交集或成对交集的并集。
在步骤114,使用在步骤112确定的核心基因清单计算LDA模型。具体来说,基于核心基因清单计算的LDA模型可以通过进行100次5折交叉验证或任何数量的n折交叉验证来计算。
在一个实例中,应用关于步骤102到114所述的统计建模方法,鉴别出核心基因标签包括以下六个基因:LRRN3、SASH1、PALLD、RGL1、TNFRSF17以及CDKN1C。当对从当前吸烟者和从不吸烟者获得的样品进行分类时,此模型的5折交叉验证(100次)MCC是0.77(其中灵敏度评分(sensitivity score,Se)是0.91并且特异性评分(specificity score,Sp)是0.85)。通过所述方法的设计,标签中的核心基因在NOWAC(GSE15289)和Bahr等人(GSE42057)研究中均处于高倍数变化基因中,并且所述预测基于这两个GSE研究之间的所有77个共同基因,改良了LDA模型的性能(Se=0.73,Sp=0.81)。尽管所有六个基因LRRN3、SASH1、PALLD、RGL1、TNFRSF17以及CDKN1C在本文中全都称为核心基因标签,但是本领域的普通技术人员将理解,可以使用六个基因的任意组合作为核心基因标签,如六个基因中的三个、四个或五个的任意组合。
在一些实施例中,扩充标签中的基因以包括扩展的基因集合,所述扩展的基因集合包括不在核心集合中但与高特异性评分和高灵敏度评分相关的其它基因。具体来说,当研究通过单独地利用高倍数变化基因的每个清单获得的预测模型时,重复地将IGJ、RRM2、ID3、SERPING1以及FUCA1鉴别为具有高特异性和灵敏度的标签中的潜在候选者。这五个基因也在NOWAC(当前吸烟者对比从不吸烟者)和Bahr等人(当前吸烟者对比既往吸烟者)研究的血液转录组中的高倍数变化基因中并且被用来将核心基因标签扩展为扩展标签。当对当前吸烟者和从不吸烟者进行分类时,所述模型基于扩展标签(LRRN3、SASH1、PALLD、RGL1、TNFRSF17、CDKN1C、IGJ、RRM2、ID3、SERPING1以及FUCA1)的交叉验证MCC是0.73(Se=0.88,Sp=0.84)。尽管所有十一个基因LRRN3、SASH1、PALLD、RGL1、TNFRSF17、CDKN1C、IGJ、RRM2、ID3、SERPING1以及FUCA1在本文中全都称为扩展基因标签,但是本领域的普通技术人员将理解,可以使用十一个基因的任意组合作为核心基因标签,如十一个基因中的五个、六个、七个、八个、九个或十个的任意组合。此外,组合可以包括核心基因标签中的六个基因中的三个、四个或五个的组合和扩展基因标签中额外基因中的五个基因中的两个、三个或四个。
将在步骤114计算的LDA模型的结果与在单独从BLD-SMK-01(也就是说不使用两个公共数据集GSE15289和GSE42057)学习稀疏标签时获得的模型的预测交叉验证结果进行对比。在预测吸烟者对比非吸烟者时,此模型的5折交叉验证性能产生Sp=0.96和Se=0.93,所述性能略高于基于核心标签和扩展标签的模型性能。尽管用本文所述的方法衍生的预测模型的交叉验证特异性和灵敏度(Sp=0.88,Se=0.84)导致性能略低于不使用独立数据集获得的模型的性能(Sp=0.96,Se=0.93),但是本文中所衍生的预测模型是有利的,因为所述模型与更宽范围的应用相关。具体来说,根据本公开内容的方法衍生的预测模型是稳固的,如当验证所述模型时所展现,如关于步骤116详细地描述。
在步骤116,验证在步骤114计算的LDA模型。通过使用来自BLD-SMK-01研究的既往吸烟者组和来自QASMC研究的血液数据集执行LDA模型的验证。在对QASMC转录组学样品进行质量检查之后,52份COPD、58份当前吸烟者、58份既往吸烟者以及59份从不吸烟者CEL文件可供预测。为了评估核心标签和扩展标签的预测性能,将QASMC样品分为两组:当前吸烟者(COPD和健康的)和非当前吸烟者(包括既往吸烟者和从不吸烟者)。这两组允许评估所述标签相对于COPD状况的稳固性。使用针对核心基因标签或扩展标签构建的模型预测每个居中数据集。
表3显示对各种标签的独立数据集使用LDA模型所得的预测结果。表3的格式遵循表1的格式,其中在不同行中显示预测分类并且在不同列中显示实际分类。具体来说,表3中所示的预测结果包括以下各者的预测结果:核心基因标签(前三行)、扩展基因标签(中间三行)、仅源自BLD-SMK-01样品的标签(倒数第二行)以及基于Beineke等人(Beineke,P.,Fitch,K.,Tao,H.,Elashoff,M.R.,Rosenberg,S.,Kraus,W.E.和Wingrove,J.A.(2012).用于吸烟状态的基于全血基因表达的特征(A whole blood gene expression-based signature for smoking status).BMC药物基因组学(BMC medical genomics)5,58.)中所述的基因集的标签(最后一行)。如表3中所示,核心标签和扩展标签均使得灵敏度和特异性评分高于源自单独的BLD-SMK-01样品的标签和由Beineke鉴别的标签。
表3
所述标签针对QASMC研究的分类性能证实,无论COPD状况如何,所述模型都是稳固的(针对核心标签,Se=0.9,Sp=0.9;并且针对扩展标签,Se=0.91,Sp=0.90)。
此外,图5A、5B、5D和5E显示各个盒形图,所述盒形图指示不同研究的分类方案。具体来说,图5A和5B分别绘制了根据BLD-SMK-01研究和QASMC研究的LDA模型,某个样品被归类为当前吸烟者的后验概率的盒形图。图5D和5E分别绘制了关于BLD-SMK-01研究和QASMC研究,线性判别函数的预测评分的盒形图。具体来说,将具有负分的样品归类为当前吸烟者,并且将具有正分的样品归类为非当前吸烟者。
还检查了如性别和年龄等其它共变量的影响。BLD-SMK-01和QASMC研究相对于性别和年龄取平衡。在年龄或性别与吸烟状况之间不存在统计学关联,如通过统计学卡方检验(关于BLD-SMK-01,χ2(性别,吸烟状况)P值=1;并且关于QASMC,χ2(性别,吸烟状况)P值=0.9)和统计学t检验(关于BLD-SMK-01,t检验(年龄对比吸烟状况)P值=0.8;并且关于QASMC,t检验(年龄对比吸烟状况)P值=0.46)所指出。
另外,在BLD-SMK-01中,测试标签中的每个基因与性别和年龄的关联,并且没有一个基因的ANOVA P值低于0.05,但PALLD基因显示微弱的性别影响。先前鉴别的基因标签发现了性别和/或年龄的影响并且确定了必需针对这类因素加以调整。Beineke等人2012。具体来说,年龄是两个公共数据集(GSE15289和GSE42057)的重要共变量,因为吸烟者平均年龄大于从不吸烟者或既往吸烟者,所以此共变量不包括在预测器中,因为它在BLD-SMK-01研究中与吸烟状况并没有统计学关联。然而,除了如由特异性和灵敏度评分定义的较佳性能之外,本文所述的基因标签通常与性别或年龄无关。这一点说明,本文所述的核心标签和扩展标签提供一种优于已知基因标签的优势,所述优势在于不必针对这些因素加以调整,从而简化了计算方法。
为了确定所发现的标签是否能够被转译到基于qRT-PCR的暴露生物标志物中,使二十个随机选择的样品(十名当前吸烟者和十名从不吸烟者)的子集经历qRT-PCR,测量扩展标签中的基因的表达水平。基于扩展标签中的基因,针对归一化的qRT-PCR数据训练LDA模型,并且通过10折交叉验证(1000次,选择10折是因为样品大小小)评定,得到特异性0.85和灵敏度0.96(表4)。当对核心标签应用相同操作时,获得特异性0.62和较低的灵敏度0.80(表4)。
表4
本公开内容的一个目标是应用核心基因标签和扩展基因标签来判定是否可以使用所述标签检测转换到加热式烟草产品(heated tobacco product,HTP)的影响。为了促进这个目标,从REX-EX-01研究获得数据。REX-EX-01研究是开放标签、随机、对照、双臂平行组研究,所述研究招募了42名年龄介于23岁与65岁之间的健康吸烟者,包括两个性别。进行所述研究,比较常规香烟的吸烟者与近期连续5天转换到HTP(本文中称为烟草加热系统2.1(THS 2.1))的吸烟者。根据优质临床规范(Good Clinical Practices,GCP)进行研究并且所述研究已在ClinicalTrials.gov上以标识符NCT01780714注册。将血液样品储存在PAXgene管中并且传送到丹麦奥尔胡斯的AROS应用生物技术公司,所述血液样品在此公司进行进一步处理并且然后与昂飞人类基因组U133Plus 2.0基因芯片杂交。
为了测试本文所鉴别的基因标签是否提供了用于评定临床试验中的暴露反应的灵敏并且非侵袭性工具,将所述标签应用于THS 2.1数据,判定是否可以在五天后在全血转录组中检测到转换到HTP。本研究的假设是转换到THS 2.1的吸烟者的全血转录组与既往吸烟者的全血转录组的类似性多过与当前吸烟者的全血转录组的类似性。代替表征对转换五天具有特异性的HTP用户的基因表达谱(例如,通过从REX-EX-01研究数据中提取标签),期望鉴别出基于转录组的暴露反应标签,所述标签还可以充当更长期的转换模式的指示器。这一点通过确立核心基因标签和扩展基因标签实现,核心基因标签和扩展基因标签均能够区分当前吸烟者样品与非当前吸烟者样品。
在对REX-EX-01研究的CEL文件执行质量检查之后,常规香烟吸烟者和THS 2.1用户在第5天分别剩余16和18个文件。下表5显示REX-EX-01样品针对核心基因标签(前三行)和扩展基因标签(后三行)的预测结果。关于扩展基因标签,仍然吸常规香烟的个体(当前吸烟者)主要被归类为当前吸烟者(69%),而转换到THS 2.1的受试者主要被归类为非当前吸烟者(89%)。关于核心标签,当前吸烟者的正确率相同(69%),并且78%转换到THS 2.1的受试者被归类为非当前吸烟者。因此,核心基因标签和扩展基因标签均可预测从HTP用户获得的样品为非当前吸烟者的样品。
表5
表5中所示的结果与转换到THS的受试者的血液转录组开始类似于既往吸烟者而不是当前吸烟者的血液转录组这一初始假设一致,但仍存在以下事实:在THS 2.1与常规香烟之间在烟碱和可替宁(cotinine)暴露方面无显著差异(数据未示出)。
此外,图5C绘制了从LDA模型,基于REX-EX-01数据,某个样品被归类为当前吸烟者的后验概率的盒形图,并且图5F绘制了基于REX-EX-01数据,来自线性判别函数的预测评分的盒形图。具有负预测评分的样品被归类为当前吸烟者,而正预测评分指示非当前吸烟者状况。
与依赖于单一基因的测量的基因标签相比,基因表达谱分析提供了在正常情况和病理情况下的生物过程的全面并且更完整的视图。当将多个基因的表达趋势综合在一起时,还有可能从对于疾病状态的暴露反应推导出用于指定生理状态的标签或分类器。虽然受到主要影响的组织可提供更准确地代表正常状态、暴露的状态或病理状态的样品,但是使用组织活检对受试者进行分类常常是不实际的。因为使用微创技术采血方便,所以基于血液的标签在生物标志物探索方面有巨大的前景。在这一研究中,已鉴别出两组基于全血的生物标志物,其中任一者均可以充当身体对吸烟的反应的标签并且因此可以用作个体吸烟状况的强预测器。
在这一研究中强烈突出的基因是LRRN3。在当前吸烟者中,LRRN3的表达相比于在非当前吸烟者中有所增加。在REX-EX-01研究中,在转换到HTP的受试者的血液中,所述表达在0天与5天之间显著降低,并且在仍然吸常规香烟的受试者的血液中保持恒定。因此,LRRN3似乎是核心标签和扩展标签中用于测量从常规香烟转换到HTP的影响到重要基因。在一个实例中,如所述的基因标签仅包括LRRN3并且无其它基因,或包括LRRN3以及任何其它基因。具体来说,包括LRRN3的基因标签能够通过证明在转换之后在0天与5天之间,LRRN3表达降低,而检测到从吸常规香烟到使用HTP的转换。
本文所述的系统药理学方法允许构造一个或多个可以区分当前吸烟者与非当前吸烟者的基于全血的稳固吸烟者基因标签。本文所述的核心基因标签是基于六个基因,并且扩展基因标签是基于核心基因标签加另外五个基因。两种基因标签在预测个体的吸烟者状况方面均具有显著的准确性,如通过灵敏度和特异性评分所评定。当向来自REX-EX-01研究的样品应用时,所述标签基于全血转录组数据将使用THS 2.1五天后的受试者鉴别为非当前吸烟者。因此,本文所述的标签提供了一种使用微创采样评定暴露反应的灵敏并且特定的工具。
图2是根据本公开内容的示意性实施例,用于评定从受试者获得的样品的方法200的流程图。方法200包括以下步骤:接收与样品相关的数据集,所述数据集包括LRRN3、CDKN1C、PALLD、SASH1、RGL1以及TNFRSF17的定量表达数据(步骤202);并且基于接收到的数据集产生评分,其中所述评分指示受试者的预测吸烟状况(步骤204)。在一些实施例中,在步骤202接收到的数据集进一步包括IGJ、RRM2、SERPING1、FUCA1以及ID3的定量表达数据。在一些实施例中,在步骤202接收到的数据集进一步包括CLDND1、MUC1、GOPC以及LEF1中的一个或多个的定量表达数据。
在步骤204产生的评分是向数据集应用的分类方案的结果,其中所述分类方案是基于数据集中的定量表达数据确定的。具体来说,在本文所述的实例中,可以向在202接收到的数据集应用针对LDA模型加以训练的分类器,确定个体的预测分类法。
本文所述的基因标签可以在由计算机实施的方法中使用,用于评定从受试者获得的样品。具体来说,可以获得与样品相关的数据集,并且所述数据集可以包括LRRN3、CDKN1C、PALLD、SASH1、RGL1以及TNFRSF17的定量表达数据用于核心基因标签。可以基于接收到的数据集产生评分,其中所述评分指示所预测的受试者的吸烟状况。具体来说,所述评分可以基于使用本文所述的LDA模型方法构建的分类器。数据集可以进一步包括扩展基因标签中所包括的其它标志物IGJ、RRM2、SERPING1、FUCA1以及ID3的定量表达数据。数据集可以进一步包括CLDND1、MUC1、GOPC以及LEF1中的一个或多个的定量表达数据。
在一些实施例中,数据集包括标志物集合LRRN3、CDKN1C、PALLD、SASH1、RGL1、TNFRSF17、IGJ、RRM2、SERPING1、FUCA1、ID3、CLDND1、MUC1、GOPC以及LEF1的任何数量的任何子集。可以向标签中所包括的标志物应用一个或多个准则,所述标志物如包括LRRN3、CDKN1C、PALLD、SASH1、RGL1以及TNFRSF17中的至少三个(或任何其它合适的数量),IGJ、RRM2、SERPING1、FUCA1以及ID3中的至少两个(或任何其它合适的数量),以及CLDND1、MUC1、GOPC以及LEF1中的至少一个(或任何其它合适的数量)。一般来说,可以在不脱离本公开内容的范围的情况下使用任何使用这些标志物的组合的标签。
在一些实施例中,本文所述的标签中的基因用于组装用来预测个体的吸烟者状况的试剂盒。具体来说,所述试剂盒包括一组检测测试样品中基因标签中的基因的表达水平的试剂,和使用所述试剂盒预测个体的吸烟者状况的说明书。所述试剂盒可以用于评定戒烟或吸烟产品的替代物(如HTP)对个体的影响。
图3是用于执行本文所述方法(如关于图1和2所述的方法)中的任一种或用于存储本文所述的核心基因标签、扩展基因标签或任何其它基因标签的计算设备的框图。具体来说,存储在计算机可读介质上的基因标签包括LRRN3、CDKN1C、PALLD、SASH1、RGL1以及TNFRSF17的表达数据。在另一个实例中,计算机可读介质所包括的基因标签包括至少五个标志物的表达数据,所述至少五个标志物选自由以下组成的群组:LRRN3、CDKN1C、PALLD、SASH1、RGL1、TNFRSF17、IGJ、RRM2、SERPING1、FUCA1以及ID3。
在某些具体实施中,可以在若干计算设备300中实施部件和数据库。计算设备300包括至少一个通信接口单元、输入/输出控制器310、系统存储器、以及一个或多个数据存储设备。系统存储器包括至少一个随机存取存储器(RAM 302)和至少一个只读存储器(ROM 304)。这些元件全部与中央处理单元(CPU 306)连通,以有利于计算设备300的运作。计算设备300可以以许多不同的方式配置。例如,计算设备300可以是常规的独立式计算机,或可选地,计算设备300的功能可以被分布在多个计算机系统和架构中。计算设备300可被配置成执行建模、评分和聚合操作中的一部分或全部。在图3中,计算设备300经由网络或局部网络连接到其它服务器或系统。
计算设备300可以被配置成分布式架构,其中,数据库和处理器被容纳在单独的单元或位置中。一些这样的单元执行主要的处理功能,并且至少包含通用控制器或处理器和系统存储器。在这样的方面,这些单元中的每一个经由通信接口单元308附接到通信集线器或端口(未示出),所述集线器或端口用作与其它服务器、客户端或用户计算机和其它相关设备的主要通信链路。通信集线器或端口自身可具有最低的处理能力,主要用作通信路由器。各种通信协议可以是系统的一部分,包括但不限于:Ethernet、SAP、SASTM、ATP、BLUETOOTHTM、GSM和TCP/IP。
CPU 306包括处理器,例如,一个或多个常规的微处理器和用于从CPU 306卸载工作量的诸如数学协处理器的一个或多个辅助的协处理器。CPU 306与通信接口单308和输入/输出控制器310通信,CPU 306通过通信接口单元308和输入/输出控制器310与诸如其它服务器、用户终端或设备的其它设备通信。通信接口单元308和输入/输出控制器310可包括多个通信信道,以用于与例如其它处理器、服务器或客户终端同时通信。彼此通信的设备不需要连续地发送到彼此。相反,这样的设备仅需要在必要时发送到彼此,实际上可以在大部分时间抑制交换数据,并且可能需要执行若干步骤以在设备之间建立通信链路。
CPU 306也与数据存储设备通信。数据存储设备可包括磁性、光学或半导体存储器的适当组合,并且可包括例如RAM 302、ROM 304、闪存驱动器、诸如压缩盘的光盘或硬盘或硬盘或驱动器。CPU 306和数据存储设备均可以例如完全位于单个计算机或其它计算设备内;或由通信介质连接到彼此,通信介质为例如USB端口、串行端口电缆、同轴电缆、以太网式电缆、电话线、射频收发器或其它类似的无线或有线介质、或上述的组合。例如,CPU 306可以经由通信接口单元308连接到数据存储设备。CPU 306可被配置成执行一个或多个特定的处理功能。
数据存储设备可以存储例如:(i)用于计算设备300的操作系统312;(ii)一个或多个应用程序314(例如,计算机程序代码或计算机程序产品),其适于根据本文所述系统和方法并且特别地根据关于CPU 306详细描述的过程来指导CPU 306;或者(iii)适于存储信息的数据库316,其可以用来存储程序所需的信息。在一些方面,数据库包括存储实验数据和公布的文献模型的数据库。
操作系统312和应用程序314可以例如存储成压缩、未编译和加密的格式,并且可包括计算机程序代码。程序的指令可以从计算机可读介质而不是数据存储设备(例如,从ROM 304或从RAM 302)读入处理器的主存储器中。虽然在程序中的指令的序列的执行造成CPU 306执行本文所述过程步骤,但硬连线电路可以用来代替软件指令或与软件指令结合使用,以实现本公开内容的过程。因此,所描述的系统和方法不限于硬件和软件的任何具体组合。
可以提供合适的计算机程序代码来执行本文所述的一个或多个功能。程序也可包括程序元素,例如,操作系统312、数据库管理系统和“设备驱动程序”,这些程序元素允许处理器经由输入/输出控制器310与计算机外围设备(例如,视频显示器、键盘、计算机鼠标等)进行交互。
如本文所用,术语“计算机可读介质”是指任何非暂时性介质,其提供或参与提供指令给计算设备300的处理器(或本文所述设备的任何其它处理器)以执行。这样的介质可以采取许多形式,包括但不限于非易失性介质和易失性介质。非易失性介质包括例如光学、磁性、或光磁性盘、或诸如闪存存储器的集成电路存储器。易失性介质包括动态随机存取存储器(dynamic random access memory,DRAM),其通常构成主存储器。常见形式的计算机可读介质包括例如软盘、软磁盘、硬盘、磁带、任何其它磁性介质、CD-ROM、DVD、任何其它光学介质、穿孔卡、纸带、带有孔图案的任何其它物理介质、RAM、PROM、EPROM或EEPROM(电可擦除可编程只读存储器)、FLASH-EEPROM、任何其它存储芯片或盒、或计算机可从其读取的任何其它非暂时性介质。
各种形式的计算机可读介质可以参与将一个或多个指令的一个或多个序列传输到CPU 306(或本文所述设备的任何其它处理器)以用于执行。例如,指令可以初始地承载在远程计算机(未示出)的磁盘上。远程计算机可以将指令加载到其动态存储器中,并且通过以太网连接、电缆线路或甚至使用调制解调器的电话线发送指令。计算设备300(例如,服务器)本地的通信设备可以在相应的通信线路上接收数据,并且将数据置于用于处理器的系统总线上。系统总线将数据传输到主存储器,处理器从主存储器获取并执行指令。由主存储器接收的指令可以可选地在由处理器执行之前或之后存储在存储器中。此外,指令可以经由通信端口被接收为电信号、电磁信号或光信号,这些信号是载送各种类型的信息的无线通信或数据流的示例性形式。
本文引用的每篇参考文献均以引用方式全文并入本文中。
虽然已结合具体实例特别地示出和描述了本公开内容的具体实施,但本领域的技术人员应当理解,在不脱离由所附权利要求限定的本公开内容的范围的情况下,可以对这些具体实施做出形式和细节上的各种更改。因此,本公开内容的范围由所附权利要求表示,并且落入权利要求书的等同物的涵义和范围内的所有更改因此都旨在被涵盖。
- 还没有人留言评论。精彩留言会获得点赞!