本发明属于分子生物学检测领域,具体涉及用于循环肿瘤DNA拷贝数变异检测的装置及方法。
背景技术:
早在1948年,Mandel和Metais就从正常人血液中检测到游离的DNA片段(cell-free,cfDNA)。1977年Leon等人发现肿瘤患者体内cfDNA含量明显高于健康个体,而晚期肿瘤患者含量更高。随着研究的不断深入,研究者发现在肿瘤患者的血浆和血清cfDNA中存在与肿瘤基因改变相同的DNA片段,命名为ctDNA(circulating tumor DNA)。ctDNA是肿瘤细胞向外周血液中释放的基因组DNA。由于外周血循环DNA半衰期短,因此循环肿瘤DNA能够真实反映患者病变组织基因突变的真实情况。有文献报道称,癌变人群血浆中游离100~400bp大小的DNA片段浓度明显高于正常人,可以作为筛查的标志物。循环肿瘤DNA在恶性肿瘤诊疗中的应用越来越受到关注和重视,作为研究的热点和突破口将有可能为临床肿瘤的早期诊断、预后判定及疗效监测等提供一系列方便、快捷、特异、无创的分子生物学检测手段。
基因的拷贝数变异(Copy Number Variation,CNV)是一类在临床上非常重要的结构变异,与多种肿瘤的预后,靶向药物的敏感性相关。可靠的CNV检测结果可以为临床用药以及病情评估等提供十分重要的依据。目前临床上所使用的CNV检测技术大多为基于PCR或免疫组化的实验手段(如FISH,IHC等)。此类方法通常基于肿瘤组织样本的基因组DNA设计优化,单次检测仅可覆盖一个基因,且检测结果灵敏度较低。肿瘤组织样本通常由手术或者穿刺获得,这种方法具有侵入性和一定风险,而且比较昂贵。对于肿瘤演化产生异质性和抗药性,以及转移期患者体内存在多个肿瘤病灶,单次原位活检存在很大局限。将传统实验手段用于ctDNA样本的CNV检测,检测性能将无法得到保障。
基于新一代测序(Next-Generation Sequencing,NGS)平台的CNV检测,可以在保证检测性能的前提下一次性给出多个基因的CNV检测结果。传统的NGS平台CNV检测技术大多基于全基因组测序技术平台完成研发,随着NGS技术的不断进步,基于目标区域捕获的高深度测序技术在临床检测的应用场景下逐渐表现出优势。
但是,由于全基因组测序数据与目标区域捕获测序数据存在根本差别,目前传统NGS平台的CNV检测方法对于目标区域捕获测序数据并不适用,在检测CNV的准确性上难以保证,且检测灵敏度有待提高。血浆中游离DNA含量极微,片段化严重,且循环肿瘤DNA仅占血浆游离DNA总量的0.02%~50%,加之ctDNA的释放量会受到患者病情,癌种,分期,用药情况等各类综合因素的影响,导致这一问题在肿瘤循环DNA样本中表现尤为明显。此外,由肿瘤细胞所释放的携带CNV突变的ctDNA所占比例也较低,这也进一步加大的检测的难度。因此,如何提高循环肿瘤DNA样本CNV检测体系的鲁棒性、灵敏度和准确性成为本领域亟待解决的技术问题。
技术实现要素:
鉴于上述现有技术中存在的问题,本发明的目的在于提供一种用于循环肿瘤DNA样本的CNV的检测灵敏度更高的检测装置及检测方法。
本发明的发明人为解决上述技术问题进行了深入研究,结果发现:在循环肿瘤DNA样本的CNV检测方法中,是否对数据进行合理的降噪处理,是否使用了合适的背景库,会直接影响到检测结果。通过更为合理全面的降噪处理,动态背景库的应用,能够提高循环肿瘤DNA样本CNV检测的灵敏度,从而完成了本发明。
即,本发明包括:
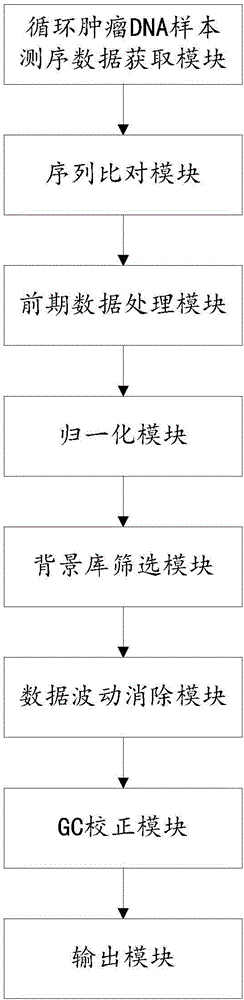
一种循环肿瘤DNA拷贝数变异(所述拷贝数变异可以发生在基因区域,也可以发生在非基因区域)检测装置,其包括:
测序数据获取模块,用于获取来自待检循环肿瘤DNA样本的捕获测序数据以及来自健康人群样本的测序数据,所述健康人群样本为多个健康人(健康正常人)样本;
序列比对模块,其与所述测序数据获取模块连接,用于将所述测序数据获取模块获得的测序数据与参考基因组序列进行比对,得到比对结果(包含例如,每条可以与参考基因组比对上的短序列所在的染色体,坐标,短序列与参考基因组的匹配情况等信息),根据该比对结果计算每一个位点(指基因组上的每个位点,但捕获测序中可能有一些位点的深度值为0)的深度值;
前期数据处理模块,其与所述序列比对模块连接,用于将目标区域(100k~100M,全基因组或者重点关注区域)划分为一定长度(50~1000bp)的有重叠(10~70%)的窗口,去掉窗口内位点的深度极值(极大值和极小值)并计算深度均值或中值,且计算该窗口内的参考基因组序列的GC含量;
归一化模块,其与所述前期数据处理模块连接,用于对所述前期数据处理模块所得到的每一个窗口内的深度均值或中值进行归一化,计算得到待检循环肿瘤DNA样本和健康人群样本每个窗口内的Z值;
背景库筛选模块,其与所述归一化模块连接,用于根据待检循环肿瘤DNA样本与健康人群样本的Z值,筛选出n个健康人样本(每个健康人样本对应一个健康人),得到n个健康人样本的背景库样本集,然后使用该n个健康人样本在m个窗口内的Z值构建m行n列的矩阵Xm×n;
数据波动消除模块,其与所述背景库筛选模块连接,用于消除捕获测序带来的固有数据波动;
GC校正模块,其与所述数据波动消除模块连接,用于根据各窗口内的GC含量进行GC矫正;
输出模块,其与所述GC校正模块连接,用于输出CNV检测结果(包括例如,用于展示CNV检测结果的图,CNV变异的阴性/阳性的判定结果等)。
本发明的循环肿瘤DNA拷贝数变异检测装置的测序数据获取模块采用二代测序方法对待检循环肿瘤DNA样本中的DNA进行测序而得到的测序数据。二代测序的主流平台一般均采用边合成边测序(Sequencing By Synthesis,SBS)技术进行核酸测序。测序前,需要对核酸(DNA或RNA)样本进行测序文库的构建,基本流程如下:首先将片段化后的DNA进行片段的末端修复,之后在修复后的片段3'端加“A”碱基,然后将上述DNA片段与含有测序引物结合位点的DNA接头(Adapter)连接,最后通过PCR进行扩增,完成测序文库构建。对于具体的二代测序方法没有特殊限制,可以采用任何本领域技术人员已知的二代测序方法。
优选地,所述测序数据是采用捕获测序方法获得的测序数据;
所述捕获测序的目标基因可以因不同的目标疾病而异。所述目标疾病可以是例如实体癌(例如胃癌、乳腺、结肠直肠癌、肺癌等)。
具体例如,在所述目标疾病是乳腺癌的情况下,所述目标基因可以是例如EGFR基因、ERBB2基因、FGFR1基因、KIT基因、PIK3CA基因或/和PTEN基因;在所述目标疾病是结肠直肠癌的情况下,所述目标基因可以是例如EGFR基因、ERBB2基因、FGFR2基因、KRAS基因、MET基因、PTEN基因;在所述目标疾病是胃癌的情况下,所述目标基因可以是例如EGFR基因、ERBB2基因、FGFR1基因、FGFR2基因、KRAS基因、MET基因、PIK3CA基因或/和PTEN基因;在所述目标疾病是肺癌的情况下,所述目标基因可以是例如ALK基因、BRAF基因、EGFR基因、ERBB2基因、FGFR1基因、KRAS基因、MET基因、PIK3CA或/和PTEN。
优选地,所述前期数据处理模块采用滑动窗口法划分所述窗口。
优选地,所述归一化模块依据下述公式(1)计算得到待检样本每个窗口内的Z值,公式(1)中Zi表示第i个窗口的Z值,
Zi=trimScale(Zi,Zi)……(1)。
优选地,定义公式(2):
定义
其中,chr表示染色体,St表示待检生物样本,SN表示健康人群样本;
所述背景库筛选模块根据待检循环肿瘤DNA样本与健康人群样本的Z值,筛选出使得所述d值最小的n个健康人样本,得到筛选后的背景库样本集S1,S2,S3,…,Sn(N和n均为自然数且n<N)。
优选地,所述数据波动消除模块对背景库矩阵Xm×n做奇异值分解,得到m行r列因子矩阵Um×r,r为因子个数,然后取贡献率最大的k个因子(即排名靠前的k个因子,k一般为4~10)进行LOESS回归,得到残差Zp。
优选地,所述GC校正模块根据各窗口内的GC含量,对Zp基于LOESS回归做GC矫正,得到残差Zpg。
优选地,所述循环肿瘤DNA样本拷贝数变异检测装置还包括:
数据质检模块,其与所述测序模块和所述序列比对模块连接,用于对所述测序模块获得的测序数据进行质检。质检包括但不限于例如去除低质量的短序列、去除N含量较高的短序列、去除与Adapter相关的短序列、并最终统计各项相关的质控指标。
此外,本发明还包括:
一种循环肿瘤DNA样本拷贝数变异(所述拷贝数变异可以发生在基因区域,也可以发生在非基因区域)检测方法,其包括:
测序数据获取步骤,获取来自待检循环肿瘤DNA样本的捕获测序数据以及来自健康人群样本的测序数据,所述健康人群样本为多个健康人样本;
序列比对步骤,将所述测序数据获取步骤获得的测序数据与参考基因组序列进行比对,得到比对结果(包含例如,每条可以与参考基因组比对上的短序列所在的染色体,坐标,短序列与参考基因组的匹配情况等信息),根据该比对结果计算每一个位点(指基因组上的每个位点,但捕获测序中可能有一些位点的深度值为0)的深度值;
前期数据处理步骤,将目标区域(100k~100M,全基因组或者重点关注区域)划分为一定长度(50~1000bp)的有重叠(10~70%)的窗口,去掉窗口内位点的深度极值(极大值和极小值)并计算深度均值或中值,且计算该窗口内的参考基因组序列的GC含量;
归一化步骤,对前期数据处理步骤所得到的每一个窗口内的深度均值或中值进行归一化,计算得到待检循环肿瘤DNA样本和健康人群样本每个窗口内的Z值;
背景库筛选步骤,根据待检循环肿瘤DNA样本与健康人群样本的Z值,筛选出n个健康人样本(健康人样本,每个背景库样本对应一个健康人),得到背景库样本集,然后使用该n个健康人样本在m个窗口内的Z值构建m行n列的矩阵Xm×n;
数据波动消除步骤,消除捕获测序带来的固有数据波动;
GC校正步骤,根据各窗口内的GC含量进行GC矫正;以及
输出步骤,输出CNV检测结果(包括例如,用于展示CNV检测结果的图,CNV变异的阴性/阳性的判定结果等)。
本发明的循环肿瘤DNA样本拷贝数变异检测方法的测序数据获取步骤获取采用二代测序方法对待检循环肿瘤DNA样本中的DNA进行测序而得到的测序数据。二代测序的主流平台一般均采用边合成边测序(Sequencing By Synthesis,SBS)技术进行核酸测序。测序前,需要对核酸(DNA或RNA)样本进行测序文库的构建,基本流程如下:首先将片段化后的DNA进行片段的末端修复,之后在修复后的片段3'端加“A”碱基,然后将上述DNA片段与含有测序引物结合位点的DNA接头(Adapter)连接,最后通过PCR进行扩增,完成测序文库构建。对于具体的二代测序方法没有特殊限制,可以采用任何本领域技术人员已知的二代测序方法。
优选地,所述测序数据是采用捕获测序方法获得的测序数据;
所述捕获测序的目标基因可以因不同的目标疾病而异。所述目标疾病可以是例如实体癌(例如胃癌、乳腺、结肠直肠癌、肺癌等)。
具体例如,在所述目标疾病是乳腺癌的情况下,所述目标基因可以是例如EGFR基因、ERBB2基因、FGFR1基因、KIT基因、PIK3CA基因或/和PTEN基因;在所述目标疾病是结肠直肠癌的情况下,所述目标基因可以是例如EGFR基因、ERBB2基因、FGFR2基因、KRAS基因、MET基因、PTEN基因;在所述目标疾病是胃癌的情况下,所述目标基因可以是例如EGFR基因、ERBB2基因、FGFR1基因、FGFR2基因、KRAS基因、MET基因、PIK3CA基因或/和PTEN基因;在所述目标疾病是肺癌的情况下,所述目标基因可以是例如ALK基因、BRAF基因、EGFR基因、ERBB2基因、FGFR1基因、KRAS基因、MET基因、PIK3CA或/和PTEN。
优选地,所述前期数据处理步骤采用滑动窗口法划分所述窗口。
优选地,所述归一化步骤依据下述公式(1)计算得到待检样本每个窗口内的Z值,公式(1)中Zi表示第i个窗口的Z值,
Zi=trimScale(Zi,Zi)……(1)。
优选地,定义公式(2):
定义
其中,chr表示染色体,ST表示待检循环肿瘤DNA样本,SN表示健康人群样本;
所述背景库筛选步骤根据待检循环肿瘤DNA样本与健康人群样本的Z值,筛选出使得所述d值最小的n个健康人样本,得到筛选后的背景库样本集S1,S2,S3,…,Sn(N,n均为自然数且n<N)。
优选地,所述数据波动消除步骤对背景库矩阵Xm×n做奇异值分解,得到m行r列因子矩阵Um×r,r为因子个数,然后取贡献率最大的k个因子(即排名靠前的k个因子,k一般为4~10)进行LOESS回归,得到残差Zp。
优选地,所述GC校正步骤根据各窗口内的GC含量,对Zp基于LOESS回归做GC矫正,得到残差Zpg。
优选地,所述拷贝数变异检测方法还包括:
数据质检步骤,对所述测序步骤获得的测序数据进行质检。质检包括但不限于例如去除低质量的短序列、去除N含量较高的短序列、去除与Adapter相关的短序列、并最终统计各项相关的质控指标。
其中,上述各步骤的优选实施方式可参照前述。
根据本发明,提供一种对循环肿瘤DNA样本CNV的检测灵敏度更高的检测装置及检测方法。
附图说明
图1为本发明的循环肿瘤DNA样本拷贝数变异检测装置的示意图。
图2为实施例1对乳腺癌多个基因的CNV检测结果图。
发明的具体实施方式
本说明书中提及的科技术语具有与本领域技术人员通常理解的含义相同的含义,如有冲突以本说明书中的定义为准。
定义
参考基因组:一个细胞或者生物体所携带的一套完整的单倍体序列,包括全套基因和间隔序列。
比对:一般指序列比对,指为确定两个或多个序列之间的相似性以至于同源性,而将它们按照一定的规律排列的过程。
深度值:对于基因组上的某个位点,根据比对结果,覆盖到该位点的短序列数量即为该位点的深度值。
窗口(滑动窗口):一般指基因组上的一段固定长度的区域。
背景库:由多例(一般认为≥20例)健康人样本所组成的样本库。
捕获测序:通过预先设计好的探针,对基因组上的特定区域(感兴趣的区域)进行DNA片段抓取,并最终对抓取到的DNA片段进行NGS测序的过程。
NGS(高通量测序):高通量测序技术(High-throughput sequencing)又称“下一代”测序技术("Next-generation"sequencing technology),以能一次并行对几十万到几百万条DNA分子进行序列测定和一般读长较短等为标志。
归一化(Z值):
trimScale(w,v):定义w为某个需要进行归一化的值,v为某个数据集
a.去掉v上下一定百分比的数据得到
b.计算的均值μ和标准差σ
c.计算得到作为最终结果
SVD(奇异值分解):SVD是线性代数中一种重要的矩阵分解,是矩阵分析中正规矩阵酉对角化的推广。在信号处理、统计学等领域有重要应用。其作用是把数据集映射到低维空间中去。数据集的特征值(在SVD中用奇异值表征)按照重要性排列,降维的过程就是舍弃不重要的特征向量的过程,而剩下的特征向量组成的空间即为降维后的空间。
实施例
以下通过实施例对本发明进行更具体的说明。应当理解,此处所描述的实施例是用于解释本发明,而非用于限定本发明。
实施例1:
采用本发明的循环肿瘤DNA样本拷贝数变异检测装置对一例女性乳腺癌患者的外周血样本的CNV情况进行检测。
1.1提取外周血样本的cfDNA
采用MagMAX Cell-Free DNA Isolation Kit试剂盒(Life公司)提取血液cfDNA,得到提取的cfDNA,提取方法参照使用手册。
1.2末端修复(End Repair)
(1)预先从-20℃保存的试剂盒中取出所需试剂,单个样本配制量参见表1。
表1
(2)末端修复反应:加入DNA样本后将1.5mL离心管置于Thermomixer中20℃温浴30分钟。反应结束后使用1.8×核酸纯化磁珠回收纯化反应体系中的DNA,溶于32μLEB。
1.3末端加“A”(A-Tailing)
(1)预先从-20℃保存的试剂盒中取出所需试剂,单个样本配制量参见表2:
表2
(2)末端加“A”反应:加入32μL上一步纯化回收的DNA后将1.5mL离心管置于Thermomixer中37℃温浴30分钟。使用1.8×核酸纯化磁珠回收纯化反应体系中的DNA,溶于18μL EB中。
1.4接头的连接(Adapter Ligation)
(1)预先从-20℃保存的试剂盒中取出所需试剂,单个样本配制量参见表3:
表3
(2)接头的连接反应:加入18μL上一步纯化回收的DNA后将样本管置于Thermomixer中20℃温浴15分钟。使用1.8×核酸纯化磁珠回收纯化反应体系中的DNA,溶于30μL的EB中。
1.5PCR反应
(1)从-20℃保存的试剂盒中取出所需试剂,2mL的PCR管中配制PCR反应体系:
表4
(2)设定PCR程序,PCR反应的程序设定如下:
反应结束及时将样品取出放入4℃冰箱保存并按要求退出或关闭仪器。
(3)用0.9×核酸纯化磁珠回收纯化反应体系中的DNA,纯化后的文库溶于20μL的ddH2O中。对文库进行Qubit检测,将文库送检安捷伦2100。
1.6乳腺癌目标区域捕获芯片文库杂交
(1)本实验中,用于提供杂交捕获反应的离子环境的缓冲液、以及用于洗脱物理吸附或非特异性杂交的清洗液、漂洗液均可从商业途径获得。
(2)准备杂交文库:将待杂交的DNA文库在冰上融化,取总质量1μg(在后续操作步骤中将此DNA文库称为样本文库)。
(3)制备Ann引物Pool:将样本文库Index对应的标签引物In1(100μM)及公共引物(1000μM)各取1000pmol混合,(在后续操作步骤中将此混合物称为Ann引物pool)。
(4)杂交样本的制备:向1.5mL EP管中加入5μL COT DNA(Human Cot-1DNA,Life technologies,1mg/mL)、1μg样本文库、Ann引物pool。用封口膜密封制备好的杂交样本EP管,将盛有样本文库pool/COT DNA/Ann引物pool的EP管置于真空装置中直到完全干燥。
(5)杂交样本的溶液:向样本文库pool/COT DNA/Ann引物pool的干粉中加入:
7.5μL 2×杂交缓冲液
3μL杂交组分A
(6)充分混匀后将上述混合物置于预先准备好的95℃加热模块上变性10分钟。
(7)将上述混合物转移至含有4.5μL捕获芯片的0.2mL平盖PCR管中。充分涡旋震荡3秒,将杂交样品混合物置于47℃加热模块上16小时。加热模块的热盖温度需设定为57℃,杂交后产物需进行后续洗脱回收操作。
(8)将10×清洗液(Ⅰ,Ⅱ与Ⅲ)、10×漂洗液和2.5×磁珠清洗液配置成1×工作液。
表5
(9)将下列试剂在47℃加热模块中预热:
400μL 1×漂洗液
100μL 1×清洗液I
1.7制备亲和吸附磁珠
(1)将链霉亲和素磁珠(Dynabeads M-280Streptavidin,以下简称磁珠)在室温下平衡30分钟后,将磁珠充分涡旋混匀15秒。
(2)向1.5mL离心管中分装100μL磁珠,将盛有100μL磁珠的离心管置于磁力架上,约5分钟后小心吸弃上清,加两倍于磁珠初始体积的1×磁珠清洗液,涡旋混匀10秒。将盛有磁珠的离心管放回磁力架,吸附磁珠。待溶液澄清,吸弃上清。重复次步骤,共洗涤两次。
(3)洗涤完毕后吸弃磁珠清洗液,用磁珠初始体积的1×磁珠清洗液涡旋重悬磁珠转入0.2mL的PCR管中。将PCR管置于磁力架上吸附磁珠澄清后吸弃上清。
1.8DNA与亲和吸附磁珠的结合及漂洗
(1)将杂交的样本文库转入盛有亲和吸附磁珠的0.2mL PCR管中,涡旋振荡混匀。
(2)将0.2mL PCR管置于47℃加热模块45分钟,每隔15分钟涡旋混匀一次,使DNA与磁珠结合。
(3)45分钟孵育后,向15μL捕获的DNA样本中加入47℃预热的1×清洗液I 100μL。涡旋混匀10秒。将0.2mL PCR管中的全部组分转入1.5mL离心管中。将1.5mL离心管置于磁力架上吸附磁珠,弃上清。
(4)将1.5mL离心管从磁力架上取下,加入200μL预热47℃的1×漂洗液。吸打混匀10次(需迅速操作,防止试剂、样品温度低于47℃)。混匀后样本置于47℃加热模块上5分钟。重复此步骤,用47℃的1×漂洗液共洗涤两次。将1.5mL的离心管置于磁力架上,吸附磁珠,弃上清。
(5)向上述1.5mL离心管中加入200μL室温的1×清洗液I,涡旋混匀2分钟。将离心管置于磁力架上,吸附磁珠,弃上清。向上述1.5mL离心管中加入200μL室温的1×清洗液Ⅱ,涡旋混匀1分钟。将离心管置于磁力架上,吸附磁珠,弃上清。向上述1.5mL离心管中加入200μL室温的1×清洗液Ⅲ,涡旋混匀30秒。将离心管置于磁力架上,吸附磁珠,弃上清。
(6)1.5mL离心管从磁力架上取下,加入45μL PCR水,溶解洗脱磁珠捕获样本。
1.9捕获DNA的PCR扩增
(1)按下表制备捕获后PCR mix,制备好后涡旋震荡混匀。富集引物F和富集引物R均购自英潍捷基公司。
(2)磁珠吸附DNA PCR的扩增程序设定如下:
(3)杂交捕获DNA PCR产物的回收纯化:用核酸纯化磁珠回收纯化反应体系中的DNA,磁珠使用量为0.9×,纯化后的文库溶于30μL的ddH2O中。
1.10文库定量
对文库进行2100Bio Analyzer(Agilent)/LabChip GX(Caliper)及QPCR检测,记录文库浓度。
1.11文库上机测序
构建好的文库用NextSeq 550AR进行测序。
1.12数据处理及分析
采用本发明的FFPE样本拷贝数变异检测装置对1.11文库上机测序的结果进行处理分析。
实施例1的FFPE样本拷贝数变异检测装置包括下述模块。
测序数据获取模块:
获取了使用乳腺癌目标区域捕获芯片对待检测的基因组DNA进行捕获测序获得测序数据。
数据质检模块:
对测序数据进行数据质检,过滤掉平均质量值低的短序列,过滤掉N含量高的短序列,过滤掉与Adapter相关的短序列,得到过滤的测序数据C。
序列比对模块:
使用经过过滤的测序数据C,与人参考基因组HG19进行短序列比对,获得比对结果A。根据该比对结果A计算基因组上的每个位点的深度值,得到结果D。
前期数据处理模块:
将癌症目标区域划分为一定长度且有重叠的窗口,去掉窗口内的深度极值并计算深度中值,且计算该窗口内的参考基因组序列的GC含量,得到结果X。
归一化模块:
结合结果X与D,依据公式Zi=trimScale(Zi,Zi)计算得到待检测基因组DNA每个窗口内的Z值。
背景库筛选模块:
定义
chr是染色体的意思,St表示待检测样本,Sn表示背景库样本。
根据待检基因组DNA与背景库的Z值,筛选出使得d值最小的背景库样本,得到筛选后的背景库样本集S1,S2,S3,…,Sn。
使用这n个样本在m个窗口内的Z值构建m行n列矩阵Xm×n作为背景库待用。
数据波动消除模块:
对背景库矩阵Xm×n做奇异值分解,得到m行n列因子矩阵Um×n,n为因子个数。取贡献率最大的几个因子进行LOESS回归,得到残差Zp。
GC校正模块:
根据m个窗口内的GC含量,对Zp基于LOESS回归做GC矫正,得到残差Zpg。
输出模块:
输出模块用于展示CNV检测结果的图。
检测结果如图2所示,图中的每一个小圆点为一个窗口的Zpg值。其中,PIK3CA与ERBB2两个基因均检出拷贝数增加。
1.13结果验证
使用同一患者剩余的cfDNA样本,使用数字PCR方法验证ERBB2基因的表达量是否升高(数字PCR法测扩增,现在有文献摸索了HER2的扩增条件,认为ERBB2和内参基因的比值大于1.25/1.3的为阳性),检测出结果表明ERBB2基因与内参基因的比值为1.43,大于1.25的阈值,表明该患者发生ERBB2基因的扩增,验证结果与1.12检测结果一致。本发明的检测装置能够成功检出肿瘤循环DNA样本的拷贝数变异。
工业实用性
本发明的循环肿瘤DNA样本的CNV检测装置及检测方法能够显著地提高CNV的检测灵敏度。