有机电致发光器件的制作方法与工艺
本发明涉及新型的有机电子材料制备的有机电致非掺杂蓝光发光器件,属于有机电致发光器件显示技术领域。
背景技术:
有机电致发光器件作为一种新型的显示技术,具有自发光、宽视角、低能耗、效率高、薄、色彩丰富、响应速度快、适用温度范围广、低驱动电压、可制作柔性可弯曲与透明的显示面板以及环境友好等独特优点,因此,有机电致发光器件技术可以应用在平板显示器和新一代照明上,也可以作为LCD的背光源。有机电子发光器件为在两个金属电极之间通过旋涂或者沉积一层有机材料而制备的器件,一个经典的三层有机电致发光器件包含空穴传输层,发光层和电子传输层。由阳极产生的空穴经空穴传输层跟由阴极产生的电子经电子传输层结合在发光层形成激子,而后发光。有机电致发光器件可以通过改变发光层的材料来发射红光,绿光和蓝光。因而,稳定的,高效的和色彩纯的有机电致发光材料对有机电致发光器件的应用和推广具有重要作用,同时也是OLEDs大面积面板显示的应用推广的迫切需求。在三原色(红,蓝,绿)当中,红光和绿光材料最近已经取得了很大的发展,也符合面板的市场需求。对于蓝光材料,也有一系列的商品化的材料,其中早期用得比较多的为出光兴产(IdemitsuKosanCo.,Ltd)的二苯乙烯基联苯(DPVBi)类化合物,以这类化合物制备的器件具有较高的效率,但是往往这些材料的稳定性比较差,更有甚地,这类化合物的发光颜色属于天蓝光,往往CIE值中的y>0.15。所以由于其不好的温度性和不纯的颜色很大程度地限制了这类化合物在全彩显示器件中的应用。另外一类蓝光材料为柯达公司的ADN和四叔丁基苝,但是这些化合物的发光效率比较差,而且稳定性也不好,从而无法大量使用。
技术实现要素:
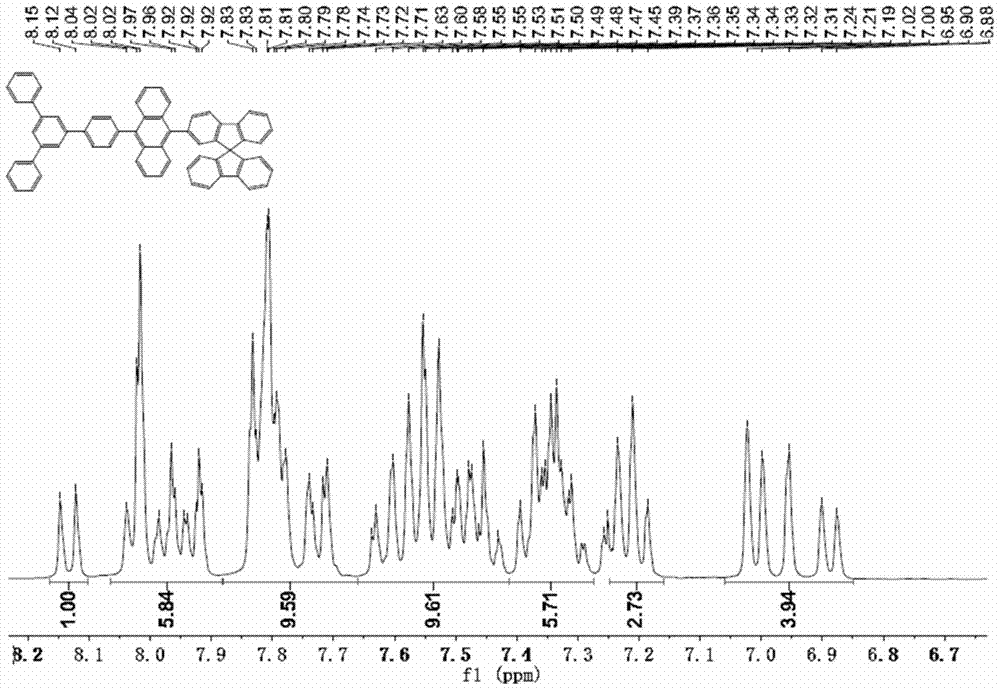
针对上述器件的缺陷,本发明提供一种电致发光效率良好和色纯度优异以及寿命长的有机电致非掺杂蓝光发光器件。一种有机电致发光器件,包含阳极,阴极,和有机层,所述有机层为空穴注入层、空穴传输层、电子注入层、电子传输层、发光层中至少包括发光层在内的一层或多层;所述发光层由具有式(I)所述结构的单一化合物组成,其中,R1-R17独立地表示为氢,氘原子,卤素,氰基,硝基,C1-C8烷基、C1-C8烷氧基,C6-C30的取代或者未取代的芳基,C3-C30的取代或者未取代的含有一个或者多个的杂原子芳基,C2-C8取代或者未取代的烯烷基,C2-C8取代或者未取代的炔烷基,其中,Ar1-Ar3独立地表示C6-C60取代或者未取代的芳基,C3-C60的取代或者未取代的带有一个或者多个杂原子的杂芳基,三芳香(C6-C30)胺基。优选:其中,R1-R17独立地表示为氢,卤素,氰基,硝基,C1-C8烷基、C1-C8烷氧基,C2-C8取代或者未取代的烯烷基,C2-C8取代或者未取代的炔烷基,C1-C4烷基取代或未取代的苯基,C1-C4烷基取代或未取代的萘基,或结合成C1-C4烷基取代或未取代的芴基;Ar1-Ar3独立地表示C1-C4烷基或者C6-C30芳基取代的苯基,C1-C4烷基或者C6-C30芳基取代的萘基,苯基,萘基,吡啶基,N-C6-C30的芳基或者C1-C4的烷基取代的咔唑基,二苯并噻吩基,二苯并呋喃基,蒽基,菲基,芘基,苝基,荧蒽基,(9,9-二烷基)芴基,(9,9-二烷基取代或未取代芳基)芴基,9,9-螺芴基。优选:其中,R1-R2可以独立地优选表示为氢,卤素,C1-C4的烷基,C1-C4烷基取代或未取代的苯基,C1-C4烷基取代或未取代的萘基,或结合成C1-C4烷基取代或未取代的芴基;其中,R3-R17可以独立地优选表示为氢,卤素,C1-C4的烷基,C1-C4烷基取代或未取代的苯基,C1-C4烷基取代或未取代的萘基,优选Ar1-Ar3为独立地表示苯基,甲苯基,二甲苯基,叔丁基苯基,萘基,吡啶基,甲基萘,联苯基,二苯基苯基,萘基苯基,二苯基联苯基,二芳香胺基苯基,N-苯基咔唑基,(9,9-二烷基)芴基,(9,9-二烷基取代或未取代苯基)芴基,9,9-螺芴基。优选:其中,R3-R17优选为氢,R1,R2可以独立优选表示为氢,甲基,乙基,丙基,异丙基,叔丁基,苯基,联苯基,萘基,或结合成芴基;Ar1-Ar3为独立地表示苯基,吡啶基,甲苯基,二甲苯基,萘基,甲基萘,联苯基,二苯基苯基,萘基苯基,二苯基联苯基,(9,9-二烷基)芴基,(9,9-二甲基取代或未取代苯基)芴基,9,9-螺芴基。优选:R3-R17优选为氢;R1,R2为独立地表示氢,甲基,或结合成芴基;Ar1,Ar2,Ar3为独立地表示苯基,萘基。优选:式(I)所的化合物为下列结构化合物所述有机层为空穴注入层,空穴传输层,发光层,电子注入层、电子传输层中的一层或多层。需要特别指出,上述有机层可以根据需要,这些有机层不必每层都存在,但至少有发光层。所述空穴传输层,电子传输层和/或发光层中含有式(I)所述的化合物。所述式(I)所述的化合物位于发光层。本发明的有机电致发光器件包含有一层发光层,该发光层的发光区域在440-490nm。所述发光层为无掺杂体系。本发明的电子器件有机层的总厚度为1-1000nm,优选1-500nm,更优选50-300nm。所述有机层可以通过蒸渡或旋涂形成薄膜。如上面提到的,本发明的式(I)所述的化合物如下,但不限于所列举的结构:本发明中的空穴传输层和空穴注入层,所需材料具有很好的空穴传输性能,能够有效地把空穴从阳极传输到有机发光层上。除本发明的化合物外,还可以包括小分子和高分子有机材料,可以包含如下,但是不限于这些,三芳香胺化合物,联苯二胺化合物,噻唑化合物,恶唑化合物,咪唑类化合物,芴类化合物,酞菁类化合物,六氰基六杂三苯(hexanitrilehexaazatriphenylene),2,3,5,6-四氟-7,7',8,8'-四氰二甲基对苯醌(F4-TCNQ),聚乙烯基咔唑,聚噻吩,聚乙烯,聚苯磺酸。本发明的有机电致发光层,采用本发明的结构式(I)化合物,不掺与其它材料。本发明的有机电子器件使用的有机电子传输材料要求具有很好的电子传输性能,能够有效地把电子从阴极传输到发光层中,除本发明的化合物外,还可以选择如下化合物,但是不限于此,氧杂恶唑,噻唑类化合物,三氮唑类化合物,三氮嗪类化合物,三氮杂苯类化合物,喔啉类化合物,二氮蒽类化合物,含硅杂环类化合物,喹啉类化合物,菲啰啉类化合物,金属螯合物,氟取代苯类化合物。本发明的有机电子器件根据需要,可以加入一层电子注入层,该电子注入层可以有效的把电子从阴极注入到有机层中,除本发明的化合物外,主要选自碱金属或者碱金属的化合物,或选自碱土金属或者碱土金属的化合物,可以选择如下化合物,但是不限于此,锂,氟化锂,氧化锂,氮化锂,8-羟基喹啉锂,铯,碳酸铯,8-羟基喹啉铯,钙,氟化钙,氧化钙,镁,氟化镁,碳酸镁,氧化镁。器件实验表明,本发明的有机电致发光器件具有电致发光效率良好和色纯度优异以及寿命长的优点。附图说明图1为本发明的器件结构图,其中10代表为玻璃基板,20代表为阳极,30代表为空穴注入层,40代表为空穴传输层,50代表为发光层,60代表为电子传输层,70代表为电子注入层,80代表为阴极。图2为化合物89的1HNMR图。图3为化合物89的13CNMR图。图4为化合物89的HPLC图。图5为化合物89的TGA图。图6为实施例4,实施例5及比较例1的电压–电流密度曲线图图7为实施例4,实施例5及比较例1的亮度–CIEy坐标图图8为实施例4,实施例5及比较例1的电致发光光谱图9为实施例4,实施例5及比较例1的电流密度–电流效率曲线图具体实施方式为了更详细叙述本发明,特举以下例子,但是不限于此。(其中下面的化合物1a、1b、1e、1h,3a、89a为市售的常用材料)实施例1中间体1c的合成向反应烧瓶中加入1a(240.00g,0.88mol),1b(496.32g,1.76mol),Pd(PPh3)4(20.35g,17.60mmol),碳酸钾(302.52g,2.20mol),甲苯(2400mL),纯水(1200mL)。抽放氮气三次后开启加热,待反应液温度达到95-105℃,保持此温度反应8-12h,取样TLC及HPLC,原料反应完全。停止加热,降温至20-30℃,抽滤,滤液分出有机层,水层再用乙酸乙酯萃取,合并有机层,再用水洗,无水硫酸镁干燥,抽滤,滤液浓缩得到暗黄色固体粗产品。石油醚重结晶,得到灰白色固体产品,收率90%,纯度95%。中间体1d的合成向反应烧瓶中加入相应比例的1c(302g,0.78mol),B(OEt)3(142g,0.97mol),n-BuLi/THF(1.6M,600mL),无水THF(3000mL),抽放氮气三次后冷却降温至反应液温度至-75~-65℃,缓慢滴加n-BuLi/THF溶液,控制反应液温度在-75~-65℃,滴加完毕后,继续保持此温度反应0.5-1h。后将一定量的B(OEt)3滴加进去,控制反应液温度在-75~-65℃,滴加完毕后,继续保持此温度反应0.5-1h,后反应液移至室温自然升温反应4-6h,然后加入2M稀盐酸,调节PH值至2-3,搅拌约1h,停止反应。加入乙酸乙酯萃取,水层再用EA萃取,合并有机层,无水硫酸镁干燥,抽滤,滤液浓缩得到灰白色固体产品,纯度95%,收率62.5%。中间体1f的合成向反应烧瓶中加入1d(150g,0.43mol),1e(500g,0.86mol),Pd(PPh3)4(5.0g,0.44mmol),碳酸钾(130g,0.92mol),甲苯(1000mL),纯水(500mL),抽放氮气三次开启加热,待反应液温度达到95-105℃,保持此温度反应8-12h,取样TLC及HPLC,原料反应完全。停止加热,降温至20-30℃,抽滤,滤液分出有机层,水层再用乙酸乙酯萃取,合并有机层,无水硫酸镁干燥,抽滤,滤液浓缩得到暗黄色固体粗产品,纯度80%,收率78.1%。中间体1g的合成向反应烧瓶中加入1f(210g,0.42mol),NBS(135g,0.71mol),DMF(5L)。抽放氮气三次开启加热,待反应液温度达到60-65℃,保持此温度反应6-8h,取样TLC及HPLC,原料反应完全。停止加热,降温至20-30℃,反应液倒入冰水中,析出暗黄色固体,抽滤得到黄色固体,烘干得到1g粗产品。粗产品加入DCM/MeOH至溶液稍微变混浊,继续搅拌约30min,析出大量固体,抽滤,得到浅黄色固体产品,收率约54.05%,纯度98.5%1HNMR(300MHz,CDCl3)δ8.64(d,J=8.8Hz,2H),7.99–7.90(m,4H),7.87(t,J=1.6Hz,1H),7.78(dd,J=9.3,2.3Hz,6H),7.61(ddd,J=8.8,6.5,1.1Hz,2H),7.56–7.48(m,6H),7.46–7.38(m,4H).13CNMR(76MHz,CDCl3)δ142.67(s),142.03(s),141.26(s),140.69(s),137.83(s),137.52(s),131.87(s),131.24(s),130.44(s),129.09(s),128.80(s),128.38–127.40(m),127.18(s),126.05–125.21(m),123.08(s),77.74(s),77.31(s),76.89(s),30.10(s).化合物1的合成向500ml三口烧瓶中依次加入1g(9.5g,16.92mmol),1h(6.41g,30.51mmol),Pd(PPh3)4(1.5g,1.3mmol),碳酸钾(5.84g,42.3mmol),甲苯(150mL),纯水(75mL)。抽放氮气三次后105℃下反应。由液相检测停反应时间,约12h左右。反应开始时反应液为催化剂的土黄色,之后慢慢变成黄色溶液,停反应后上层为清亮浅黄色,下层为水。停止反应后,过滤,用乙酸乙酯洗滤渣直至滤渣中无产物,收集滤液,旋干,大量的灰白色固体析出,收集滤渣干燥,得到目标产物,纯度98%。真空升华得到纯度为99.5%灰白色固体粉末。1H-NMR(300MHz,CDCl3)δ8.10–8.21(d,2H),7.96–7.98(dd,3H),7.87–7.89(m,2H),7.81–7.86(m,4H),7.78–7.81(d,4H),7.62–7.65(m,2H),7.59(s,1H),7.51–7.57(m,5H),7.45–7.48(m,2H),7.36–7.43(m,7H),3.88(s,2H).实施例2化合物3的合成向500ml三口烧瓶中依次加入1g(9.5g,16.92mmol),3a(7.25g,30.46mmol),Pd(PPh3)4(1.5g,1.3mmol),碳酸钾(5.84g,42.3mmol),甲苯(150mL),纯水(75mL)。抽放氮气三次后105℃下反应。由液相检测停反应时间,约12h左右。反应开始时反应液为催化剂的土黄色,之后慢慢变成黄色溶液,停反应后上层为清亮浅黄色,下层为水。停止反应后,过滤,用乙酸乙酯洗滤渣直至滤渣中无产物,收集滤液,旋干,大量的灰白色固体析出,收集滤渣干燥,得到目标产物,纯度98%。真空升华得到纯度为99.7%灰白色固体粉末。1H-NMR(300MHz,CDCl3)δ8.1–8.2(d,2H),7.96–7.99(dd,3H),7.88–7.89(m,2H),7.81–7.86(m,4H),7.78–7.81(d,4H),7.61–7.65(m,2H),7.59(s,1H),7.51–7.56(m,5H),7.46–7.48(m,2H),7.35–7.43(m,7H),1.61(s,6H).实施例3化合物89的合成向反应容器中依次加入1g(10.0g,17.8mmol),89a(7.1g,19.6mmol),Pd(PPh3)4(432.2mg,0.35mmol),K2CO3(6.14g,44.5mmol),甲苯(300mL)和水(150mL),对装置进行除氧、通入氮气保护,然后加热到100℃反应过夜。用DCM:PE=1:5的比例点板,产物点在365nm波长的紫外灯下发强烈的蓝光,Rf值在0.2左右。将反应液用硅胶抽滤,然后将滤饼用乙酸乙酯(100mL)洗涤两次,分液,用乙酸乙酯(100mL)萃取水层一次,合并有机层,再用水(200mL)洗涤一次有机相。旋干除去溶剂。粗品用120mlDCM/MeOH重结晶,抽滤得到黄色固体粉末13.1g,纯度为98.7%,收率92.2%。真空升华得到纯度为99.7%浅黄色固体粉末。m/z=797.从图2和图3可见化合物89的氢谱,碳谱与结构完全一致。从图4化合物89的高效液相色谱图可见根据本发明的合成方法制备的产物具有高纯度。从图5化合物89的热重分析图可见这类型化合物的分解温度高于摄氏400度,表明其非常高热稳定性。实施例4有机电致发光器件1的制备使用本发明的有机电子材料制备OLED首先,将透明导电ITO玻璃基板10(上面带有阳极20)依次经:洗涤剂溶液和去离子水,乙醇,丙酮,去离子水洗净,再用氧等离子处理30秒。然后,在ITO上蒸渡10nm厚的HAT-CN6作为空穴注入层30。然后,蒸渡NPB,形成30nm厚的空穴传输层40。然后,在空穴传输层上蒸渡30nm厚的化合物3作为发光层50。然后,在发光层上蒸渡15nm厚的TPBi作为电子传输层60。最后,蒸渡15nmBPhen:Li为电子注入层70和150nmAl作为器件阴极80。所制备的器件在20mA/cm2的工作电流密度下的电压为3.58V,电流效率达到3.21cd/A,在1000cd/m2亮度下CIEy坐标为0.0853,发射蓝光。器件中所述结构式实施例5有机电致发光器件2的制备方法同实施例4,将化合物3,换成化合物89,制作有机电致发光器件。所制备的器件在20mA/cm2的工作电流密度下的电压为3.84V,电流效率达到2.83cd/A,在1000cd/m2亮度下CIEy坐标为0.0888,发射蓝光。比较例1方法同实施例4,将化合物3替换成下列化合物TAT,制作对比用有机电致发光器件。TAT结构式所制备的器件在20mA/cm2的工作电流密度下的电压为4.00V,电流效率达到2.46cd/A,在1000cd/m2亮度下CIEy坐标为0.0952,发射蓝光。实施例4和5是本发明材料的具体应用,所制备的器件,本发明发射蓝光,效率和亮度都高于对比例。如上所述,本发明的材料具有高的稳定性,本发明制备的有机电致发光器件具有高的效率和光纯度。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1