磁性Fe3O4@PPy复合纳米材料的制备方法与流程
本发明涉及聚合物材料领域,具体地指一种磁性fe3o4@ppy复合纳米材料的制备方法。
背景技术:
磁性复合材料由于与一般材料而言有着特殊的性能而受到研究者的热捧,其在化学和物理等方面表现出的特殊性能而广受关注。由于磁性的存在,纳米磁性复合材料主要表现在超顺磁性,磁化率和高矫顽力方面。而fe3o4磁性微纳米材料容易产生聚沉现象,因为其相互之间存在磁性吸引以及范德华力的作用,再加上四氧化三铁胶体溶液的抗氧化性差,很容易被空气氧化生成γ-fe2o3粒子,更加导致了fe3o4磁性微纳米材料的聚集和沉淀。
磁性复合材料的制备方法有许多,其主要的原理是导电聚合物将磁性颗粒包裹在其中最后形成核壳结构,采取主要方法有溶胶-凝胶法、电化学合成法、原位聚合法等。溶胶-凝胶法制备纳米磁性复合材料的优点是反应容易进行,并且有较低的合成温度,缺点是凝胶中难免会有大量孔隙存在,在干燥过程中会有许多有害气体及有机物逸出会对材料的致密性与稳定性产生不同程度的影响。电化学合成法的优点是具有高度的选择性,可制得许多普通方法不易合成的化合物,缺点是无法进行大规模批量生产且所得产品的包覆率很低,在耐酸性能上还需要提高;原位聚合法的操作简单,缺点是获得的产品质量不好控制,容易使纳米胶体形成聚合改变了纳米颗粒的粒径,造成溶液产生聚集,另外,需要耗费大量水,从而导致水资源的浪费和回收处理。因此对这种磁性复合材料的核-壳形磁性纳米粒子的磁场诱导自组装行为的研究就显得具有重要意义。
技术实现要素:
本发明的目的就是要提供一种磁性fe3o4@ppy复合纳米材料的制备方法,该制备方法通过电解质介导法在fe3o4核心纳米颗粒表面包覆了一层致密的ppy层使得纳米颗粒在水相体系中的抗聚集性和耐无机酸腐蚀性得到了极大的增强,而且表现出优秀的超顺磁性,具有环境友好、无污染、包覆效果好等优点。
为实现上述目的,本发明所提供的一种磁性fe3o4@ppy复合纳米材料的制备方法,包括如下步骤:将无机盐电解质、fe3o4纳米颗粒、吡咯分散于蒸馏水与醇类溶剂的共混液中,搅拌均匀得到反应液;再通过恒电流电解法对反应液进行处理,得到混合液;然后将所得混合液依次进行过滤、洗涤处理;最后将洗涤后的固体颗粒进行干燥处理,即可得到磁性fe3o4@ppy复合纳米材料。
进一步地,所述无机盐电解质、fe3o4纳米颗粒、吡咯的摩尔比为0.01~0.1:1:0.1~1。
优选地,所述无机盐电解质、fe3o4纳米颗粒、吡咯的摩尔比为0.05~0.1:1:0.5~1。最佳地,所述无机盐电解质、fe3o4纳米颗粒、吡咯的摩尔比为0.07:1:0.5。
进一步地,所述无机盐电解质为nacl或者na2so4,所述fe3o4纳米颗粒的粒径为5~50nm。优选地,所述fe3o4纳米颗粒的粒径为25~35nm。
进一步地,所述蒸馏水与醇类溶剂的共混液中蒸馏水与醇类溶剂的摩尔比为1:0.01~1,所述无机盐电解质与醇类溶剂的摩尔比为1:0.1~1。
进一步地,所述醇类溶剂为乙醇或者异丙醇。
进一步地,所述搅拌为机械搅拌或者超声搅拌。
进一步地,所述机械搅拌的转速为50~5000rpm,搅拌的时间为5~150min。
进一步地,所述超声搅拌的转速为500~1000rpm,搅拌的时间为5~15min。
再进一步地,所述恒电流电解法处理中电流密度为0.1~3.0ma/cm2,反应温度为15~60℃,反应时间为10~300min。
更进一步地,所述干燥处理的温度为50~75℃,干燥的时间为8~36h。
与现有技术相比,本发明具有如下优点:
其一,本发明利用了一种新的电解质介导法在四氧化三铁纳米颗粒上包覆聚吡咯,在反应液中加入无机盐电解质,通过在fe3o4核心纳米颗粒表面包覆了一层致密的ppy层,使得纳米颗粒在水相体系中的抗聚集性和耐无机酸腐蚀性得到了极大的增强,而且ppy包覆层对核心fe3o4的磁性影响很小,fe3o4@ppy复合纳米材料的饱和磁化强度ms为4.35emu/g,表现出优秀的超顺磁性,赋予磁性微纳米材料优异的物理化学性质。
其二,本发明采用电解质介导法使磁性粒子包覆在导电聚合物中,极大的减弱了纳米粒子的团聚现象,可以避免fe3o4纳米胶体形成聚合,不会改变纳米颗粒的粒径,造成溶液产生聚集,使得每一个fe3o4纳米颗粒表面都包覆了一层致密的ppy层,有助于形成完整的核壳结构。
其三,本发明的电解质介导法操作简单,能够进行大规模批量生产,而且所得fe3o4@ppy复合纳米材料的包覆率很高,在耐酸性能上性能优越,本发明方法为新型复合纳米材料的开发和应用提供了新的制备方法和手段,同时为磁性复合纳米材料在医疗卫生和工业生产等领域的应用提供了重要的实验和理论依据。
附图说明
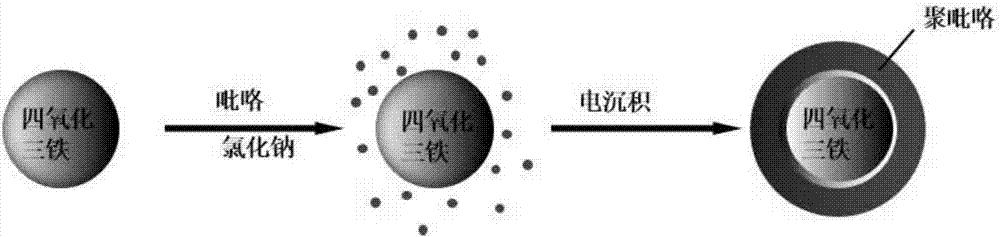
图1为实施例1制备fe3o4@ppy的复合纳米材料的反应流程图;
图2为实施例2所制备的fe3o4@ppy的复合纳米材料与吡咯的红外光谱对比图;
图3为实施例3所制备的fe3o4@ppy的复合纳米材料的透射电镜与扫描透射电镜-能量色散谱联用技术对纳米颗粒的表征示意图;其中,a是一般视野下投射电镜照片;b是能谱视野下电镜照片;c是能谱聚焦位点;d是碳元素分布图;e是氮元素分布图;f是铁元素分布图;g铁元素分布图;
图4为实施例3所制备的fe3o4@ppy的复合纳米材料与fe3o4纳米颗粒的粒径分布示意图;
图5为实施例3所制备的fe3o4@ppy的复合纳米材料的耐盐酸测试结果示意图;
图6为实施例3所制备的fe3o4@ppy的复合纳米材料在盐酸中的稳定性测试结果示意图;
图7为实施例3所制备的fe3o4@ppy的复合纳米材料与fe3o4纳米颗粒的磁化曲线对比示意图。
具体实施方式
下面结合附图和具体实施例对本发明作进一步的详细说明。
实施例1
本发明磁性fe3o4@ppy复合纳米材料的制备方法,包括如下步骤:
(1)将nacl(0.01mmol)、fe3o4纳米颗粒(1.0mmol)、吡咯(1.0mmol)分散于蒸馏水(0.01mmol)和乙醇(0.01mmol)的共混液中,搅拌均匀得到反应液,超声搅拌的转速为1000rpm,搅拌的时间为5min;
(2)将反应液通过恒电流电解法处理,以1.0macm-2的电流密度在50℃的反应条件下对混合物体系处理30min;
(3)然后将所得分散体系进行过滤,用蒸馏水洗涤滤出物多次;
(4)将滤出的固体颗粒置于烘箱中,在70℃条件下干燥24h,取出后即为fe3o4@ppy的复合纳米材料,具体过程见实施例图1,该方法在制备过程中无污染,环境友好。
实施例2
本发明磁性fe3o4@ppy复合纳米材料的制备方法,包括如下步骤:
(1)将na2so4(0.1mmol)、fe3o4纳米颗粒(1.0mmol)、吡咯(0.1mmol)分散于蒸馏水(0.1mmol)和乙醇(0.01mmol)中,搅拌均匀得到反应液,机械搅拌的转速为5000rpm,搅拌的时间为150min;
(2)将反应液通过恒电流电解法处理,以0.1macm-2的电流密度在60℃的反应条件下对混合物体系处理300min;
(3)然后将所得分散体系进行过滤,用蒸馏水洗涤滤出物多次;
(4)将滤出的固体颗粒置于烘箱中,在50℃条件下干燥36h,取出后即为fe3o4@ppy的复合纳米材料。将所得产物进行红外光谱测试,其结果见图2。从图2中可以看出,其fe3o4的特征峰和ppy的特征峰都非常明显,说明产物中的成份复合预期。
实施例3
本发明磁性fe3o4@ppy复合纳米材料的制备方法,包括如下步骤:
(1)将nacl(0.07mmol)、fe3o4纳米颗粒(1mmol)、吡咯(0.5mmol)分散于蒸馏水(0.07mmol)和异丙醇(0.07mmol)中,搅拌均匀得到反应液,超声搅拌的转速为500rpm,搅拌的时间为15min;
(2)将反应液通过恒电流电解法处理,以3.0macm-2的电流密度在50℃的反应条件下对混合物体系处理10min;
(3)然后将所得分散体系进行过滤,用蒸馏水洗涤滤出物多次;
(4)将滤出的固体颗粒置于烘箱中,在70℃条件下干燥24小时,取出后即为fe3o4@ppy的复合纳米材料。
对产物进行透射电镜与扫描透射电镜-能量色散谱联用技术对纳米颗粒的表征观察核元素分析,结果见图3。从图3中可以看出,颗粒呈现良好的分散性。对产物进行粒径分析,结果见图4。从图4中可以看出,包覆前和包覆后磁性铁颗粒的粒径发生了较大的变化,这是由于ppy包覆层增加了粒径导致的。对产物进行耐酸性测试,结果见图5,从结果中可以看出,经过包覆的磁性颗粒在酸性溶液中能很好的保持稳定性,而没有包覆的磁性颗粒则溶解于酸性溶液,耐酸腐蚀性和化学稳定性方面具有非常大的提升。针对不同的ph值,包覆样品fe3o4@ppy和fe3o4在盐酸中的稳定性结果见图6,可以看出包覆样品具有很好的稳定性。对产物进行顺磁性测试,结果见图7,从图7中颗粒看出,磁性颗粒在包覆前后保持了良好的磁性。
实施例4
本发明磁性fe3o4@ppy复合纳米材料的制备方法,包括如下步骤:
(1)将na2so4(0.01mmol)、fe3o4纳米颗粒(1.0mmol)、吡咯(1.0mmol)分散于蒸馏水(0.1mmol)和乙醇(0.01mmol)中,搅拌均匀得到反应液,机械搅拌的转速为5000rpm,搅拌的时间为150min;
(2)将反应液通过恒电流电解法处理,以0.5macm-2的电流密度在50℃的反应条件下对混合物体系处理60min;
(3)然后将所得分散体系进行过滤,用蒸馏水洗涤滤出物多次;
(4)将滤出的固体颗粒置于烘箱中,在70℃条件下干燥24h,取出后即为fe3o4@ppy的复合纳米材料。
实施例5
本发明磁性fe3o4@ppy复合纳米材料的制备方法,包括如下步骤:
(1)将na2so4(0.05mmol)、fe3o4纳米颗粒(1.0mmol)、吡咯(0.5mmol)分散于蒸馏水(0.1mmol)和乙醇(0.01mmol)中,搅拌均匀得到反应液,机械搅拌的转速为50rpm,搅拌的时间为5min;
(2)将反应液通过恒电流电解法,以3.0macm-2的电流密度在45℃的反应条件下对混合物体系处理200min;
(3)然后将所得分散体系进行过滤,用蒸馏水洗涤滤出物多次;
(4)将滤出的固体颗粒置于烘箱中,在70℃条件下干燥22h,取出后即为fe3o4@ppy的复合纳米材料。
上述实施案例只为说明本发明的技术方案及特点,其目的在于更好的让熟悉该技术的人士予以实施,并不能以此限制本发明的保护范围,凡根据本发明精神实质所作的等效变化或修饰,均在本发明保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种高结晶度植物纤维制备微纳纤丝的预处理方法与制造工艺
- 氮掺杂碳化细菌纤维素/石墨烯/铂复合纳米材料及其制备方法与制造工艺
- 一种高稳定性锂离子电池电极用硅/氮化碳/碳复合纳米材料的制造方法与工艺
- 二维片状MoS2@石墨烯复合纳米材料及其制备方法与制造工艺
- 一种球形等级结构二氧化锡/氧化铜复合纳米材料的制备方法与制造工艺
- 一种基于罗丹明衍生物的重金属离子多重检测传感器制备方法与制造工艺
- 一种石墨相碳化氮纳米片/四氧化三钴纳米片鱼鳞状结构复合纳米材料及制备方法和应用与制造工艺
- 一种硫化钼‑四氧化三铁复合纳米抑菌材料的制备与应用的制造方法与工艺
- 铂‑磺化石墨烯复合纳米材料模拟过氧化物酶的制造方法与工艺
- 一种用于空气净化的复合纳米材料及其制备方法与制造工艺