本发明涉及一种碳氮材料的制备方法,具体涉及一种氮掺杂胶囊结构碳材料的制备方法,属于材料制备技术领域。
背景技术:
超级电容器的储能途径具有两个方面,即双电层储能和赝电容储能,它具有其他储能器件无法比拟的优点,如较高的功率密度和能量密度,快速充放电,循环寿命长,可操作温度范围宽等。目前,对于超级电容器性能因素的探索,人们主要集中在电极材料方面。包括碳材料(多孔活性炭、碳纤维、碳纳米管、石墨烯和碳碳复合材料),过渡金属氧化物及氢氧化物和导电聚合物。其中碳材料作为超级电容器最早应用的材料,具有许多电化学优势。碳材料具有的高比表面积在超级电容器电极材料性能方面起了重要的作用,通过在电极和电解液界面之间的电子转移储存双电层能量,具有较高的功率密度,但其能量密度还有待提高。通过一些途径提高碳材料的比表面积,增加及优化孔结构,进行化学修饰得到含有羧基等基团的碳材料或制备含碳的复合材料是提高电化学性能的途径之一。
在导电聚合物赝电容材料中,聚苯胺明显的氧化还原可逆反应、低廉的价格、高电容和合成简便等优点,使其在制备超级电容器电极材料方面成为热点。聚苯胺含有的碳氮元素可以提供较多的碳氮基团,可以通过对其结构的调控改变表面的孔结构,提高比表面积来进一步提高材料的电化学性能。
技术实现要素:
为了克服现有技术的不足,本发明的目的在于提供一种多孔、高比表面积的氮掺杂胶囊结构碳材料的制备方法。该制备方法先通过水热法制备出氢氧化钴,然后加入多巴胺通过搅拌制备氢氧化钴/多巴胺复合物,再通过高温煅烧得到掺杂碳氮的钴氧化物,最后加入酸进行回流刻蚀得到氮掺杂的胶囊结构碳材料
本发明的技术方案具体介绍如下。
一种氮掺杂胶囊结构碳材料的制备方法,具体步骤如下:
(1)氯化钴溶液与六亚甲基四胺溶液进行超声得到混合溶液;
(2)将步骤(1)得到的混合溶液移入到聚四氟乙烯不锈钢反应釜,进行水热反应,然后降至室温并离心分离和冻干,得到氢氧化钴粉末;
(3)将氢氧化钴粉末加入到水中进行搅拌,然后加入氨水室温搅拌;
(4)向步骤(3)中得到的溶液中加入多巴胺进行室温搅拌,然后进行过滤洗涤和冻干;
(5)在管式炉氮气氛围保护下进行煅烧得到氮掺杂的钴氧化物粉末;
(6)向步骤(5)得到的粉末中加入酸溶液,进行加热回流刻蚀;反应结束后,过滤、洗涤、干燥得到氮掺杂胶囊结构碳材料。
本发明中,步骤(1)中,所述氯化钴溶液中的氯化钴和与水的质量体积比为20:1~50:1mg/ml;六亚甲基四胺溶液中的六亚甲基四胺和水的30:1~60:1mg/ml。
本发明中,步骤(1)中,氯化钴与六亚甲基四胺的质量比为1:2~1:5。
本发明中,步骤(2)中,水热反应温度为120~180℃;水热反应时间为12~24h。
本发明中,步骤(3)中,氢氧化钴与水的质量体积比为2:1~5:1mg/ml;氢氧化钴粉末在水中的搅拌时间为15~30min;氢氧化钴和氨水的质量比为5:1~20:1;加入氨水后的搅拌时间为15~30min。
本发明中,氢氧化钴粉末与多巴胺的质量比为3:1~6:1;室温搅拌时间为24~48h;过滤洗涤溶剂为乙醇与去离子水的混合液,乙醇和去离子水的体积比为5:1~1:5。
本发明中,步骤(5)中,所述煅烧温度为600~1000℃;煅烧时间为2~4h。
本发明中,步骤(6)中,酸溶液为盐酸和硫酸的混合溶液,其中盐酸与硫酸摩尔比为3:1;加热回流时间为6~12h。
本发明中,氮掺杂的钴氧化物粉末与酸溶液的质量比为3:1~1:2。
和现有技术相比,本发明的有益效果在于:
本发明通过上述制备方法制备得到的氮掺杂胶囊结构碳材料,表面粗糙,具有较多孔和较大的比表面积(710~920m2g-1),呈现胶囊结构,结构尺寸可以通过对条件的控制而调整。
本发明通过上述制备方法制备得到的氮掺杂胶囊结构碳材料,进一步制备成超级电容器电极材料,表现优异的电化学特征,是理想的能源材料之一。
附图说明
图1:a是实施例1通过水热法制备得到的氢氧化钴纳米六边形的扫描电镜图;b是实施例1制备得到的氮掺杂胶囊结构碳材料的扫描电镜图。
图2:实施例2所制得的氮掺杂胶囊结构碳材料的扫描电镜图。
图3:实施例3所制得的氮掺杂胶囊结构碳材料的xrd衍射图。
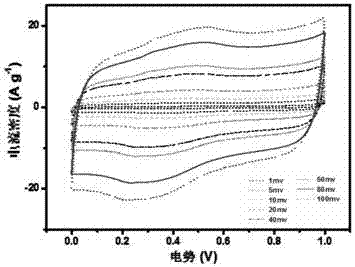
图4:实施例4所制得的氮掺杂胶囊结构碳材料在电化学测试中的循环伏安图。
具体实施方式
下面通过具体实施例并结合附图对本发明进一步阐述,但并不限制本发明。
实施例1
(1)称取400mg氯化钴加入到20ml去离子水中,超声分散15min,得到氯化钴溶液;
(2)称取800mg六亚甲基四胺加入到20ml去离子水中,进行超声分散15min,得到六亚甲基四胺溶液;
(3)将步骤(1)中分散得到的氯化钴溶液与步骤(2)中得到的六亚甲基四胺溶液进行超声15min,得到混合溶液;
(4)将步骤(4)得到的混合溶液全部移入到聚四氟乙烯不锈钢反应釜,在120℃下进行水热反应20h,然后降至室温并离心分离和冻干,得到氢氧化钴粉末,其扫描电镜图见图1a;
(5)200mg氢氧化钴粉末加入到100ml去离子水中进行搅拌15min,然后加入氨水20mg,室温搅拌30min;
(6)向步骤(5)中得到的溶液中加入50mg多巴胺进行室温搅拌24h,然后进行过滤洗涤和冻干;
(7)称取步骤(6)得到的冻干后的样品200mg,在600℃的管式炉,氮气氛围保护下进行高温煅烧3h,得到氮掺杂的钴氧化物粉末;
(8)向步骤(7)得到的粉末中加入100ml酸溶液(盐酸与硫酸摩尔比为3:1),进行加热回流刻蚀10h;
(9)去离子水过滤洗涤,放入烘箱干燥。
通过此实施例步骤(1)~(4)得到的氢氧化钴,其扫描电镜见图1a,呈现二维六边形片状结构;此实施例制备得到的氮掺杂胶囊结构碳材料的扫描电镜见图1b,通过上述酸刻蚀,氢氧化钴已被去除,材料呈现胶囊状结构,表面粗糙,空隙较多,比表面积大,为911.6m2g-1。
将该实施例制备得到的碳材料活性物质按照质量比为m(活性物质):m(ptfe)=9:1进行混合,加入1ml乙醇作为溶剂,磁力搅拌均匀,烘至呈黏糊状,取适量涂压在1cm*4cm泡沫镍上并真空干燥以备用。以1moll-1koh溶液作为电解液,hg电极作为参比电极,泡沫镍作为对照电极,样品材料作为工作电极,通过电化学工作站测定其电化学性能。结果表明:在电流密度为0.5ag-1时,其比电容为427.0fg-1;在10ag-1时,其比电容为318.7fg-1,电容保持率为74.6%,具有较好的倍率性能。
实施例2
(1)称取600mg氯化钴加入到20ml去离子水中,超声分散30min,得到氯化钴溶液;
(2)称取1800mg六亚甲基四胺加入到80ml去离子水中,进行超声分散30min,得到六亚甲基四胺溶液;
(3)将步骤(1)中分散得到的氯化钴溶液与步骤(2)中得到的六亚甲基四胺溶液进行超声15min,得到混合溶液;
(4)将步骤(4)得到的混合溶液全部移入到聚四氟乙烯不锈钢反应釜,在120℃下进行水热反应24h,然后降至室温并离心分离和冻干,得到氢氧化钴粉末;
(5)取200mg氢氧化钴粉末加入到100ml去离子水中进行搅拌15min,然后加入氨水20mg,室温搅拌30min;
(6)向步骤(5)中得到的溶液中加入60mg多巴胺进行室温搅拌48h,然后进行过滤洗涤和冻干;
(7)称取步骤(6)得到的冻干后的样品200mg,在800℃的管式炉,氮气氛围保护下进行高温煅烧4h,得到氮掺杂的钴氧化物粉末;
(8)向步骤(7)得到的粉末中加入100ml酸溶液(盐酸与硫酸摩尔比为3:1),进行加热回流刻蚀12h;
(9)去离子水过滤洗涤,放入烘箱干燥。
通过此实施例制备得到的氮掺杂胶囊结构碳材料的扫描电镜见图2,材料呈胶囊状结构,具有粗糙的表面结构,空隙较多。
将该实施例制备得到的碳材料活性物质按照质量比为m(活性物质):m(ptfe)=9:1进行混合,加入1ml乙醇作为溶剂,磁力搅拌均匀,烘至呈黏糊状,取适量涂压在1cm*4cm泡沫镍上并真空干燥以备用。以1moll-1koh溶液作为电解液,hg电极作为参比电极,泡沫镍作为对照电极,样品材料作为工作电极,通过电化学工作站测定其电化学性能。结果表明,在电流密度为0.5ag-1时,其比电容为400.8fg-1;在10ag-1时,其比电容为315.1fg-1,电容保持率为78.6%,倍率性能好。
实施例3
(1)称取400mg氯化钴加入到20ml去离子水中,超声分散30min,得到氯化钴溶液;
(2)称取900mg六亚甲基四胺加入到30ml去离子水中,进行超声分散30min,得到六亚甲基四胺溶液;
(3)将步骤(1)中分散得到的氯化钴溶液与步骤(2)中得到的六亚甲基四胺溶液进行超声30min,得到混合溶液;
(4)将步骤(4)得到的混合溶液全部移入到聚四氟乙烯不锈钢反应釜,在120℃下进行水热反应24h,然后降至室温并离心分离和冻干,得到氢氧化钴粉末;
(5)取200mg氢氧化钴粉末加入到100ml去离子水中进行搅拌30min,然后加入氨水40mg,室温搅拌30min;
(6)向步骤(5)中得到的溶液中加入40mg多巴胺进行室温搅拌48h,然后进行过滤洗涤和冻干;
(7)称取步骤(6)得到的冻干后的样品200mg,在700℃的管式炉,氮气氛围保护下进行高温煅烧3h,得到氮掺杂的钴氧化物粉末;
(8)向步骤(7)得到的粉末中加入100ml酸溶液(盐酸与硫酸摩尔比为3:1),进行加热回流刻蚀12h;
(9)去离子水过滤洗涤,放入烘箱干燥。
通过此实施例制备得到的氮掺杂胶囊结构碳材料的xrd衍射图见图3,在25.2°和42.3°处具有两个明显宽峰,为碳材料的特征峰;材料为胶囊状,表面较为粗糙,空隙多,比表面积为863.9m2g-1。
将该实施例制备得到的碳材料活性物质按照质量比为m(活性物质):m(ptfe)=9:1进行混合,加入1ml乙醇作为溶剂,磁力搅拌均匀,烘至呈黏糊状,取适量涂压在1cm*4cm泡沫镍上并真空干燥以备用。以1moll-1koh溶液作为电解液,hg电极作为参比电极,泡沫镍作为对照电极,样品材料作为工作电极,通过电化学工作站测定其电化学性能。结果表明:在电流密度为0.5ag-1时,其比电容为388.3fg-1;在10ag-1时,其比电容为306.5fg-1,电容保持率为78.9%。
实施例4
(1)称取600mg氯化钴加入到20ml去离子水中,超声分散30min,得到氯化钴溶液;
(2)称取1200mg六亚甲基四胺加入到60ml去离子水中,进行超声分散30min,得到六亚甲基四胺溶液;
(3)将步骤(1)中分散得到的氯化钴溶液与步骤(2)中得到的六亚甲基四胺溶液进行超声15min,得到混合溶液;
(4)将步骤(4)得到的混合溶液全部移入到聚四氟乙烯不锈钢反应釜,在140℃下进行水热反应24h,然后降至室温并离心分离和冻干,得到氢氧化钴粉末;
(5)取200mg氢氧化钴粉末加入到100ml去离子水中进行搅拌30min,然后加入氨水40mg,室温搅拌30min;
(6)向步骤(5)中得到的溶液中加入70mg多巴胺进行室温搅拌48h,然后进行过滤洗涤和冻干;
(7)称取步骤(6)得到的冻干后的样品200mg,在700℃的管式炉,氮气氛围保护下进行高温煅烧4h,得到氮掺杂的钴氧化物粉末;
(8)向步骤(7)得到的粉末中加入200ml酸溶液(盐酸与硫酸摩尔比为2:1),进行加热回流刻蚀12h;
(9)去离子水过滤洗涤,放入烘箱干燥。
通过此实施例制备得到的氮掺杂胶囊结构碳材料呈现胶囊状结构,表面粗糙,空隙较多,比表面积大。
将该实施例制备得到的碳材料活性物质按照质量比为m(活性物质):m(ptfe)=9:1进行混合,加入1ml乙醇作为溶剂,磁力搅拌均匀,烘至呈黏糊状,取适量涂压在1cm*4cm泡沫镍上并真空干燥以备用。以1moll-1koh溶液作为电解液,hg电极作为参比电极,泡沫镍作为对照电极,样品材料作为工作电极,通过电化学工作站测定其电化学性能。其循环伏安图见图4。在电流密度为0.5ag-1时,其比电容为412.4fg-1;在10ag-1时,其比电容为316.9fg-1,电容保持率为76.8%。
实施例5
(1)称取400mg氯化钴加入到20ml去离子水中,超声分散30min,得到氯化钴溶液;
(2)称取1200mg六亚甲基四胺加入到30ml去离子水中,进行超声分散30min,得到六亚甲基四胺溶液;
(3)将步骤(1)中分散得到的氯化钴溶液与步骤(2)中得到的六亚甲基四胺溶液进行超声30min,得到混合溶液;
(4)将步骤(4)得到的混合溶液全部移入到聚四氟乙烯不锈钢反应釜,在160℃下进行水热反应24h,然后降至室温并离心分离和冻干,得到氢氧化钴粉末;
(5)取400mg氢氧化钴粉末加入到100ml去离子水中进行搅拌30min,然后加入氨水80mg,室温搅拌30min;
(6)向步骤(5)中得到的溶液中加入120mg多巴胺进行室温搅拌48h,然后进行过滤洗涤和冻干;
(7)称取步骤(6)得到的冻干后的样品200mg,在800℃的管式炉,氮气氛围保护下进行高温煅烧3h,得到氮掺杂的钴氧化物粉末;
(8)向步骤(7)得到的粉末中加入200ml酸溶液(盐酸与硫酸摩尔比为2:1),进行加热回流刻蚀12h;
(9)去离子水过滤洗涤,放入烘箱干燥。
通过此实施例制备得到的氮掺杂胶囊结构碳材料呈现胶囊状结构,表面粗糙。
将该实施例制备得到的碳材料活性物质按照质量比为m(活性物质):m(ptfe)=9:1进行混合,加入1ml乙醇作为溶剂,磁力搅拌均匀,烘至呈黏糊状,取适量涂压在1cm*4cm泡沫镍上并真空干燥以备用。以1moll-1koh溶液作为电解液,hg电极作为参比电极,泡沫镍作为对照电极,样品材料作为工作电极,通过电化学工作站测定其电化学性能。结果表明:在电流密度为0.5ag-1时,其比电容为527.7fg-1;在10ag-1时,其比电容为427.5fg-1;循环2000圈后,其比电容为484.9fg-1,循环稳定性较好。
实施例6
(1)称取400mg氯化钴加入到20ml去离子水中,超声分散30min,得到氯化钴溶液;
(2)称取1600mg六亚甲基四胺加入到100ml去离子水中,进行超声分散30min,得到六亚甲基四胺溶液;
(3)将步骤(1)中分散得到的氯化钴溶液与步骤(2)中得到的六亚甲基四胺溶液进行超声30min,得到混合溶液;
(4)将步骤(4)得到的混合溶液全部移入到聚四氟乙烯不锈钢反应釜,在120℃下进行水热反应24h,然后降至室温并离心分离和冻干,得到氢氧化钴粉末;
(5)取400mg氢氧化钴粉末加入到100ml去离子水中进行搅拌30min,然后加入氨水60mg,室温搅拌30min;
(6)向步骤(5)中得到的溶液中加入150mg多巴胺进行室温搅拌48h,然后进行过滤洗涤和冻干;
(7)称取步骤(6)得到的冻干后的样品200mg,在800℃的管式炉,氮气氛围保护下进行高温煅烧4h,得到氮掺杂的钴氧化物粉末;
(8)向步骤(7)得到的粉末中加入150ml酸溶液(盐酸与硫酸摩尔比为1:1),进行加热回流刻蚀12h;
(9)去离子水过滤洗涤,放入烘箱干燥。
通过此实施例制备得到的氮掺杂胶囊结构碳材料具有胶囊状结构和粗糙的表面,空隙明显,比表面积为717.5m2g-1。
将制备得到的该实施例的碳材料活性物质按照质量比为m(活性物质):m(ptfe)=9:1进行混合,加入1ml乙醇作为溶剂,磁力搅拌均匀,烘至呈黏糊状,取适量涂压在1cm*4cm泡沫镍上并真空干燥以备用。以1moll-1koh溶液作为电解液,hg电极作为参比电极,泡沫镍作为对照电极,样品材料作为工作电极,通过电化学工作站测定其电化学性能。结果表明:在电流密度为0.5ag-1时,比电容达到455.6fg-1。
上述内容仅为本发明的实施方式的具体列举,而依据本发明的技术方案所作的任何等效变换,均应属于本发明的保护范围。