钴基双金属硒化物/氮掺杂碳复合材料及其制备方法与流程
1.本发明涉及一种钴基双金属硒化物/氮掺杂碳复合材料及其制备方法,属于新材料技术领域。
背景技术:
2.近年来,锂离子电池作为电能和化学能的存储与转换装置越来越受到人们的关注。高性能电极的设计是实现高能量密度锂离子电池的关键,负极材料在很大程度上决定了电池的电化学性能。商用石墨负极由于容量有限(372mah g
‑1)不能满足锂离子电池的要求。因此,探索适用于锂离子电池的高性能负极材料势在必行。金属硒化物因其较高的理论比容量和合适的电位而得到了研究者们的青睐。其中,铁磁性金属(铁、锌、镍)硒化物在金属硒化合物家族中最能展现出成本竞争力,因为该类金属分布广泛,可直接从天然矿石中获取。然而,许多因素包括导电性差、体积膨胀和产物在电解液中溶解及反应等问题限制其在当前电极材料中的实际应用。而碳材料改性、构建双金属硒化物等方法是解决这些问题的有效途径。
3.碳材料改性不仅可有效地提高金属硒化物的导电性,还可以抑制其体积膨胀。同时,碳材料的本征属性可赋予电极材料附加的有益性质,比如高吸附性可以促进电解液吸附及抑制中间产物的穿梭。此外,由于双金属硒化物的协同作用,双金属硒化物比单金属硒化物具有更多的氧化还原位、更大的晶粒尺寸、更高的离子扩散动力学和更好的导电性。具体的,双金属化合物通常通过晶相界面的协同耦合及相应的原子排列和局部电子结构变化,来促进氧化还原过程中的界面电子转移反应,使金属硒化物暴露更多反应活性位点、促进电化学反应动力学并缓解体积膨胀问题,进而达到提升储锂性能的目的。但现实情况是,由于各组分的物理化学性质和晶格结构不同,设计和合成具有最佳协同作用的双金属硒化物具有很大的难度。针对这一问题,人们投入了大量的精力,利用外延生长、溶液化学过程和合成后修饰等方法开发双金属硒化物。然而,这些合成方法往往较为复杂和费时。高效制备具有超长的循环寿命、高的可逆容量和稳定循环的双金属硒化物和碳的复合材料是一个巨大的技术难题。
技术实现要素:
4.本发明的目的在于提供一种钴基双金属硒化物/氮掺杂碳复合材料及其制备方法,利用氢氧化钾辅助合成钴基双金属有机框架材料,解决现有技术合成步骤复杂、费时且产率低的问题。以双金属有机框架材料作为前驱体材料硒化后得到了钴基双金属硒化物/氮掺杂碳复合材料,以其作为锂离子电池负极材料,提高导电性,抑制体积膨胀,提升储锂性能。
5.本发明的目的通过以下技术方案予以实现:
6.一种钴基双金属硒化物/氮掺杂碳复合材料,为钴基双金属硒化物和氮掺杂碳均匀复合的材料,钴基双金属硒化物颗粒均匀分散在多孔多面体氮掺杂碳基底上,钴基双金
属硒化物/氮掺杂碳多面体的大小为600~1000nm,钴基双金属硒化物颗粒的大小为50~200nm;钴基双金属硒化物占复合材料质量的68.5~72.6wt%,氮掺杂碳占复合材料质量的27.4~31.5wt%。
7.前述钴基双金属硒化物/氮掺杂碳复合材料,钴基双金属硒化物为铁钴双金属硒化物或锌钴双金属硒化物或镍钴双金属硒化物。
8.一种钴基双金属硒化物/氮掺杂碳复合材料的制备方法,包括以下步骤:
9.步骤一、钴基双金属有机框架的制备:
10.首先,分别配制氢氧化钾和2
‑
甲基咪唑的混合溶液a,无水三氯化铁或硝酸锌或硝酸镍的溶液b,硝酸钴溶液c,依次将溶液b、c加入到溶液a中,溶液a、溶液b、溶液c混合后的溶液中2
‑
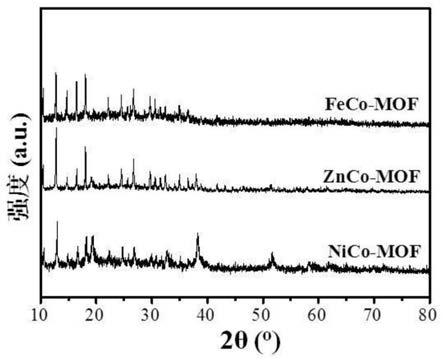
甲基咪唑、无水三氯化铁或硝酸锌或硝酸镍、硝酸钴、氢氧化钾的物质的量浓度比为1:0.014:0.0084:(0.027~1.642);然后,搅拌4~6h,干燥后获得钴基双金属有机框架材料feco
‑
mof或znco
‑
mof或nico
‑
mof;
11.步骤二、钴基双金属/氮掺杂碳复合材料的制备:
12.将钴基双金属有机框架材料置于管式炉中,400~600℃保温0.5~2h,制得钴基双金属/氮掺杂碳复合材料feco/nc或znco/nc或nico/nc;
13.步骤三、钴基双金属硒化物/氮掺杂碳复合材料的制备:
14.将硒粉与钴基双金属/氮掺杂碳复合材料feco/nc或znco/nc或nico/nc分别置于瓷舟的上下游,硒粉与钴基双金属/氮掺杂碳复合材料的质量比为2:1,在350℃保温3h,400~600℃保温0.5~2h,制得钴基双金属硒化物/氮掺杂碳复合材料fe
‑
co
‑
se/nc或zn
‑
co
‑
se/nc或ni
‑
co
‑
se/nc。
15.优选地,步骤二中所述将管式炉在400~600℃保温0.5~2h的工艺方法为:在n2的氛围下,以1~5℃/min的速率从室温升至400~600℃保温0.5~2h,然后自然冷却。
16.优选地,步骤三中所述将管式炉在350℃保温3h,400~600℃保温0.5~2h的工艺方法为:在n2氛围下,以1~5℃/min的速率从室温升至350℃保温3h,再以1~5℃/min的速率升温至400~600℃并保温0.5~2h,然后自然冷却。
17.与现有技术相比,本发明的有益效果是:本发明在水系溶剂中,利用氢氧化钾辅助一步合成钴基双金属有机框架材料。此外,相比于外延生长和表面活性剂辅助策略的步骤复杂、费时且产率低,其反应时间需要24小时,产率也只有40%以下;而本发明方法更加简单、绿色高效和低成本,具体来说,6小时就能完成反应,其产率为65~75%,有利于规模化制备。进一步地,以双金属有机框架材料作为前驱体材料硒化后得到了钴基双金属硒化物/氮掺杂碳复合材料。钴基双金属硒化物/氮掺杂碳复合材料形貌均一且均匀分散,避免了双金属硒化物团聚的问题,这些双金属硒化物颗粒均匀分散在多孔多面体氮掺杂碳基底上,展现出优异的储锂性能,钴基双金属硒化物/氮掺杂碳复合材料展现出优异的电化学性能,具有优异比容量、循环稳定性和倍率性能。实验结果证明构建双金属硒化物和氮掺杂碳复合材料可以提供丰富的氧化还原位点,缓解体积膨胀并促进电化学反应动力学。
附图说明
18.图1为本发明实施例的feco
‑
mof、znco
‑
mof和nico
‑
mof的x射线衍射谱图;其中,实施例1的为feco
‑
mof,实施例2的为znco
‑
mof,实施例3的为nico
‑
mof;
19.图2为本发明实施例1的feco/nc、实施例2的znco/nc和实施例3的nico/nc的x射线衍射(xrd)谱图;
20.图3为本发明实施例1的fe
‑
co
‑
se/nc的xrd谱图;
21.图4为本发明实施例2的zn
‑
co
‑
se/nc的xrd谱图;
22.图5为本发明实施例3的ni
‑
co
‑
se/nc的xrd谱图;
23.图6(a)、(b)、(c)为本发明实施例1的feco
‑
mof的扫描电子显微图像(sem图);
24.图7(a)、(b)、(c)为本发明实施例1的feco/nc的sem图;
25.图8(a)、(b)、(c)为本发明实施例1的fe
‑
co
‑
se/nc的sem图;
26.图9为本发明实施例zn
‑
co
‑
se/nc的sem图;
27.图10为本发明实施例ni
‑
co
‑
se/nc的sem图;
28.图11(a)、(b)为本发明实施例1的fe
‑
co
‑
se/nc的高分辨透射电子显微图像(hr tem);
29.图12(a)为本发明实施例的fe
‑
co
‑
se/nc、zn
‑
co
‑
se/nc和ni
‑
co
‑
se/nc的n2吸附
‑
脱附等温线图,图12(b)为本发明实施例的fe
‑
co
‑
se/nc、zn
‑
co
‑
se/nc和ni
‑
co
‑
se/nc的孔径分布图;
30.图13为本发明实施例1的fe
‑
co
‑
se/nc的x射线光电子谱图(xps):其中(a)为全谱图;(b)为fe 2p高分辨率谱图;(c)为co 2p高分辨率谱图;(d)为se 2p高分辨率谱图;(e)为c1s高分辨率谱图,(f)为n1s高分辨率谱图;
31.图14为本发明实施例的fe
‑
co
‑
se/nc、zn
‑
co
‑
se/nc和ni
‑
co
‑
se/nc的拉曼光谱图;
32.图15为本发明实施例的fe
‑
co
‑
se/nc、zn
‑
co
‑
se/nc和ni
‑
co
‑
se/nc的电化学性能:(a)fe
‑
co
‑
se/nc的循环伏安曲线;(b)fe
‑
co
‑
se/nc在0.1ag
‑1下的充放电曲线;(c)fe
‑
co
‑
se/nc、zn
‑
co
‑
se/nc和ni
‑
co
‑
se/nc的倍率性能图;(d)fe
‑
co
‑
se/nc在不同电流密度下的充放电曲线;(e)0.1ag
‑1下fe
‑
co
‑
se/nc、zn
‑
co
‑
se/nc和ni
‑
co
‑
se/nc的循环性能图;(f)fe
‑
co
‑
se/nc在1ag
‑1下的充放电曲线;(g)1ag
‑1下fe
‑
co
‑
se/nc、zn
‑
co
‑
se/nc和ni
‑
co
‑
se/nc的循环性能图;
33.图16为本发明实施例fe
‑
co
‑
se/nc、zn
‑
co
‑
se/nc和ni
‑
co
‑
se/nc的循环前的电化学阻抗图;
34.图17为实施例4的sem图;
35.图18为实施例5的sem图;
36.图19为实施例6的sem图;
37.图20为实施例7的sem图。
具体实施方式
38.下面结合附图和具体实施例对本发明作进一步说明。
39.实施例1:fe
‑
co
‑
se/nc复合材料的制备
40.配制30ml 6mol l
‑1的氢氧化钾,然后加入1.2g 2
‑
甲基咪唑(c4h6n2),经过超声和搅拌后得到混合溶液a;取0.249g三氯化铁溶于15ml去离子水中得到溶液b,取0.269g六水合硝酸钴溶于15ml去离子水中形成溶液c;然后,依次将溶液b、c加入到溶液a中,搅拌4~6h,获的样品记为feco
‑
mof。将上述的产品feco
‑
mof置于管式炉中,在n2的氛围下以2℃min
‑1升温速率升至500℃保温1h,自然冷却后得碳化产物feco/nc。然后,将硒粉与feco/nc(质量比为2:1)分别置于瓷舟的上下游,在氮气的氛围下升温至350℃保温3h,500℃保温1h,自然冷却制得铁钴双金属多相硒化物和氮掺杂碳的复合材料,记为fe
‑
co
‑
se/nc。
41.图1给出了实施例1所制备的feco
‑
mof的x射线衍射谱图可见,feco
‑
mof的特征峰与已报道的水溶液中合成的该类mofs的特征峰相一致。
42.由图2中实施例1所制备的feco/nc的x射线衍射谱图可见,经过碳化过后,feco/nc复合材料在26.40
°
处有个碳的(002)晶面的特征峰,而在44.8
°
和65.3
°
处的特征峰分别对应cofe(jcpds no.44
‑
1433)的(110)和(200)晶面。
43.由图3中实施例1所制备的fe
‑
co
‑
se/nc的xrd谱图可见,fe
‑
co
‑
se/nc的所有特征峰与o
‑
fese2(orthorhombic,pnnm,jcpds no.21
‑
0432)、o
‑
cose2(orthorhombic,pnnm,jcpds no.53
‑
0449)和c
‑
cose2(cubic,pa
‑
3,jcpds no.09
‑
0234)一致,并且尖锐的衍射峰表明其表面具有较高的结晶度。其中位于31.1
°
、34.9
°
、48.2
°
、54.1
°
和64.1
°
处的特征峰分别对应于o
‑
fese2的(101)、(111)、(211)、(031)和(122)晶面。其余位于30.8
°
、34.5
°
、36.0
°
、47.7
°
、65.0
°
和63.3
°
处的特征峰则分别对应于o
‑
cose2的(101)、(111)、(120)、(211)、(311)和(122)晶面。位于34.2
°
、37.6
°
、51.8
°
、56.5
°
、58.8
°
和74.0
°
处的特征峰分别对应于c
‑
cose2的(210)、(211)、(311)、(230)、(321)和(421)晶面。证明fe
‑
co
‑
se/nc中的铁钴硒化物是一个正交相fese2(o
‑
fese2)、正交相cose2(o
‑
cose2)和立方相cose2(c
‑
cose2)并存的双金属硒化物。
44.图6为实施例1所制备的feco
‑
mof的sem图,结果表明,该双金属mof为多面体结构,直径约为1μm。图7为实施例1所制备的feco/nc的sem图,可以看出多面体的形貌得到保留,但略有收缩且表面粗糙的小颗粒出现,直径约为900nm。图8为实施例1所制备的fe
‑
co
‑
se/nc的sem图,结果表明其的形貌发生变化,多面体结构发生膨胀,从收缩状态变得饱满,并且表面出现向外突出的纳米结块,分布较为均匀。结构膨胀与多面体内部或表面在硒化反应过程中发生的相转变有关。而纳米结块的形成是由于一部分金属离子在硒化过程中向外逸出,与硒离子结合形成金属硒化物晶体团簇。
45.图11为实施例1所制备的fe
‑
co
‑
se/nc的tem图和hrtem图,结果表明,fe
‑
co
‑
se纳米颗粒均匀分布在碳基体上;高分辨电镜图像显示fe
‑
co
‑
se纳米颗粒的晶格条纹清晰,说明其结晶度好。
46.图12(a)给出了实施例1所制备的fe
‑
co
‑
se/nc的氮气吸附脱附曲线,结果表明,fe
‑
co
‑
se/nc的比表面积分别为7.6m2g
‑1,孔隙体积为0.038cm
3 g
‑1。图12(b)给出了fe
‑
co
‑
se/nc的孔径分布图,孔径集中在7
‑
100nm之间,具有由中孔(<50nm)和大孔(>50nm)组成的分级多孔结构,测试结果显示fe
‑
co
‑
se/nc的平均孔径分别为20.4nm。丰富的孔隙结构有利于电解质的扩散和离子/电子的输运,为体积膨胀提供缓冲空间。
47.图13为实施例1所制备的fe
‑
co
‑
se/nc的xps谱图。图13(a)的结果表明,在整个测量光谱中可以观察到c、n、o、se、fe和co元素的特征峰。图13(b)是xps高分辨fe 2p光谱,可以看出拟合显示了fe2p
1/2
和fe2p
3/2
轨道的特征峰,在725.2ev和712.6ev处的两个特征峰对应于fe
2+
,而位于717.4ev处的峰对应于fe
3+
。在高分辨co 2p光谱(图13(c))中,可以看出在fe原子诱导下,co 2p光谱的键能值较高。在778.7ev处的峰对应co0,位于782.2ev和797.5ev处两个特征峰对应co
2+
,而处于780.7ev和793.9ev处的两个峰则归属于co
3+
。se 3d
光谱(图13(d))可分解为位于54.7、55.6和59.2ev处的三个峰,分别对应于se 3d
5/2
(fe
‑
se和co
‑
se键)、se 3d
3/2
(se
‑
se键)和seo
x
。c 1s的高分辨光谱(图13(e))中出现三个拟合峰,分别位于284.8、285.5和286.5ev处,与c
‑
c、c=n键和c
‑
o键一一对应,其中c
‑
n键的存在证实了n掺杂。其余位于283.5ev和296.1ev处的两个峰分别归属于c
‑
co和c
‑
se健。图13(f)是高分辨n1s光谱,三个峰特征峰分别归因于氮掺杂碳材料中存在的吡啶型n(398.4ev)、吡咯型n(399.3ev)和石墨型n(400.9ev)。n掺杂改性不仅可以提高碳基体的导电性,还可以引入更多的缺陷,为li
+
的插入提供大量的活性中心,有利于储锂性能的提升。
48.图14给出实施例1所制备的fe
‑
co
‑
se/nc的拉曼光谱图,可以看出fe
‑
co
‑
se/nc在1357和1576cm
‑1附近的两个特征峰,与氮掺杂碳中的d带(非晶碳)和g带(石墨碳)相对应。fe
‑
co
‑
se/nc的i
d
/i
g
(强度比)值为0.93,表明fe
‑
co
‑
se/nc中存在着更多的碳缺陷,这会加快电子转移速率,并增加锂离子的储存位置,有利于储锂性能的提升。
49.图15(a)为实施案例1所制备的fe
‑
co
‑
se/nc复合材料作为锂离子电池负极材料在0.2mv s
‑1扫速下的循环伏安曲线,在首圈的阴极扫描中可以观察到三个还原峰。其中位于~1.3v处的峰对应活性物质中锂的插层反应和li
x
fese2/li
x
cose2的形成,以及电解质的分解和固体电解质界面膜的形成。而位于~0.95和0.46v处的两个还原峰则对应于转换反应,伴随着fese、cose和lise2,和随后fe、co和li2se的生成。在首圈阳极扫描中,可以观察到三个氧化峰分别位于1.41v、2.14v和2.32v处,这三个氧化峰分别对应着fese、cose、中间产物lixfese2/lixcose2和最终产物fese2/cose2的形成。在随后的循环中,循环伏安曲线保持稳定且重叠良好,说明fe
‑
co
‑
se/nc的电化学反应是可逆和稳定的。图15(b)为实施案例1所制备的fe
‑
co
‑
se/nc复合材料作为锂离子电池负极材料在0.1ag
‑1时的恒流充放电曲线,充放电平台和循环伏安曲线中氧化还原峰的位置保持一致。第一个循环过程中,放电电压平台约为1.35v和0.79v,充电电压平台约为1.38、2.01v和2.29v。在fe
‑
co
‑
se/nc的第一次放电和充电过程中,电极的充放电比容量分别达到838和1233mah g
‑1。第一次循环充放电比容量相差较大,初始库仑效率为68%,较低的初始库伦效率与电解质的分解及在电极/电解液界面处固体电解质界面膜的形成有关。图15(c)给出了不同电流密度下的倍率性能,可以看出fe
‑
co
‑
se/nc作为锂离子电池负极材料,在0.1、0.2、0.5、1、2和5ag
‑1下的平均容量为1066、1040、952、876、700和393mah g
‑1。当电流恢复至0.1a g
‑1时比电容为1071mah g
‑1,容量保持率接近100%,表明fe、co硒化物展现出最佳的协同效应,这归因于o
‑
fese2、o
‑
cose2和c
‑
cose2之间存在着丰富的相界面,大大地增加了电化学反应活性位点,减轻了体积膨胀并且形成了稳定的固体电解质界面膜。图15(d)是fe
‑
co
‑
se/nc在不同电流下的充放电图,可以看出充放电平台的电势差随着电流的增大而逐渐增加,即使在5ag
‑1的大电流密度下,仍有着平稳的充放平台,表明fe
‑
co
‑
se/nc具有良好的结构稳定性可以承受充放电过程中发生的体积膨胀。图15(e)显示了在fe
‑
co
‑
se/nc在0.1ag
‑1下的循环性能图,在100次循环过程中,电极材料不断活化,经过稳步地增长,其容量稳定在1326mah g
‑1,库仑效率接近100%。更重要的是,转化反应的动力学过程较慢,往往导致较低的库伦效率和循环稳定性,而fe
‑
co
‑
se/nc电极循环的库伦效率为99%左右,表明其具有增强的离子扩散行为。图15(f)是fe
‑
co
‑
se/nc在1a g
‑1下的充放电曲线,前三圈进行了小电流下的活化,可以看出第四到七圈的曲线重合度仍然很好,证明初始状态下具有良好的可逆性,经过550圈循环后,比容量明显变大,充放电平台变长且平稳,说明经过550次循环后,fe
‑
co
‑
se/nc的氧化还原反应更
加平稳持久,这归因于电极内部形成了稳定的固体电解质界面膜,并且有丰富的氧化还原位点。图15(g)是1a g
‑1的电流密度下的长循环图,可以看出fe
‑
co
‑
se/nc具有高的比电容和循环稳定性,在550次循环后,其比容量为1247mah g
‑1。此外可以发现在循环过程中出现了比电容先衰减后增加的趋势,初期的衰减与体积膨胀有关,而后续的回升和增长是由于在反复充放电过程中电极材料发生了纳米化,使得更多的活性位点得到暴露。此外,氮掺杂碳的存在也有效地增加了活性位点。总之,fe
‑
co
‑
se/nc优异的储锂性能归因于双金属硒化物和氮掺杂碳的协同效应提供了丰富的氧化还原位点,并抑制了体积膨胀。并且其独特的三维多孔结构为电子提供了有效的传输通道,缩短了锂离子的扩散路径,也同时加快了电极/电解质界面处的电荷转移过程。
50.图16给出了fe
‑
co
‑
se/nc在循环前的电化学阻抗图,通过阻抗分析进一步了解电荷转移动力学。可以看出阻抗曲线由一个高频区的半圆和一个低频的斜线组成,半圆和斜线分别表示电荷转移阻抗和扩散阻抗。结果表明,可以看出fe
‑
co
‑
se/nc展现出相对最小的半圆,表明电荷转移阻抗较小,它可以抑制过厚的sei层形成,有利于电子转移。
51.实施例2:zn
‑
co
‑
se/nc复合材料的制备
52.首先,配制30ml 6mol l
‑1的氢氧化钾,然后加入1.2g 2
‑
甲基咪唑,经过超声和搅拌后得到混合溶液a;取0.467g六水合硝酸锌溶于15ml去离子水中得到溶液b,取0.269g六水合硝酸钴溶于15ml去离子水中形成溶液c;先将溶液b加入到溶液a中,搅拌30分钟后,再加入溶液c,继续搅拌4.5小时后,经去离子水洗涤和干燥后得的样品标记为znco
‑
mof。将上述获得的znco
‑
mof置于管式炉中,在n2的氛围下以2℃min
‑1升温速率升至500℃保温1h小时,自然冷却后得碳化产物znco/nc。最后,将硒粉与znco/nc(质量比为2:1)分别置于瓷舟的上下游,在氮气的氛围下升温至350℃保温3小时,500℃保温1小时,自然冷却后,得到锌钴的双金属多相硒化物和氮掺杂碳的复合材料,记为zn
‑
co
‑
se/nc。
53.图1给出了实施例2所制备的znco
‑
mof的x射线衍射谱图,结果表明,znco
‑
mof的特征峰与已报道的该类mofs的特征峰相一致。
54.图2给出了实施例1所制备的znco/nc的xrd谱图可见,经过碳化过后,znco/nc复合材料在26.40
°
处有个碳的(002)晶面的特征峰,在41.9
°
和48.8
°
处出现的特征峰对应znco的(111)和(200)晶面(jcpds no.29
‑
0524)。
55.图4为实施例2所制备的zn
‑
co
‑
se/nc的xrd谱图。结果表明,位于30.8
°
、34.5
°
、36.0
°
、47.7
°
、65.0
°
和63.3
°
处的特征峰则分别对应于o
‑
cose2的(101)、(111)、(120)、(211)、(311)和(122)晶面,位于34.2
°
、37.6
°
、51.8
°
、56.5
°
、58.8
°
和74.0
°
处的特征峰分别对应于c
‑
cose2的(210)、(211)、(311)、(230)、(321)和(421)晶面;其余在27.2
°
、45.2
°
和53.6
°
处的特征峰分别对应于c
‑
znse(jcpds no.65
‑
9602)的(111)、(220)和(311)晶面。
56.图9为实施例2所制备的zn
‑
co
‑
se/nc的sem图,可以看出硒化后zn
‑
co
‑
se/nc显示出笼状结构,这有利于电化学过程中电子和离子的转移。笼状结构的形成是因为zn和co金属在煅烧过程中对配体的分解起到催化作用,促使较多的配体快速分解而形成较大的孔洞。
57.图12(a)给出了实施例2所制备的zn
‑
co
‑
se/nc的氮气吸附脱附曲线,结果表明,zn
‑
co
‑
se/nc的比表面积为10.3m2·
g
‑1,总孔隙体积为0.055cm3·
g
‑1。图12(b)给出了zn
‑
co
‑
se/nc的孔径分布图,可以看出zn
‑
co
‑
se/nc的孔径在7
‑
100nm之间,具有由中孔(<50nm)
和大孔(>50nm)组成的分级多孔结构,测试结果显示fe
‑
co
‑
se/nc的平均孔径为18.8nm。
58.图14给出实施例2所制备的zn
‑
co
‑
se/nc的拉曼光谱图,可以看出fe
‑
co
‑
se/nc在1357和1576cm
‑1附近的两个特征峰,与氮掺杂碳中的d带(非晶碳)和g带(石墨碳)相对应。zn
‑
co
‑
se/nc的i
d
/i
g
(强度比)值为0.86,表明zn
‑
co
‑
se/nc中存在着更多的碳缺陷,这会加快电子转移速率,并增加锂离子的储存位置,有利于储锂性能的提升。
59.图15(c)给出了是不同电流密度下的zn
‑
co
‑
se/nc倍率性能,可以看出zn
‑
co
‑
se/nc展示出良好的倍率性能,该电极在0.1、0.2、0.5、1、2和5a g
‑1下的平均容量为979、984、920、844、684和358mah g
‑1。当电流恢复至0.1a g
‑1时比电容为944mah g
‑1,容量保持率接近100%,表明zn、co硒化物展现出最佳的协同效应。图15(e)给出了zn
‑
co
‑
se/nc在0.1a g
‑1下的循环性能图,在100次循环过程中,电极材料不断活化,经过稳步地增长,其容量从787mah g
‑1增长到1131mah g
‑1,库仑效率在98%以上。图15(g)给出了zn
‑
co
‑
se/nc在1a g
‑1的电流密度下的长循环图,可以看出zn
‑
co
‑
se/nc具有优异的比容量和循环稳定性,在550次循环后,其比容量为947mah g
‑1,高于大多数已报道的金属硒化物的储锂性能。
60.图16给出了zn
‑
co
‑
se/nc在循环前的电化学阻抗图,通过阻抗分析进一步了解电荷转移动力学。可以看出阻抗曲线由一个高频区的半圆和一个低频的斜线组成,半圆和斜线分别表示电荷转移阻抗和扩散阻抗。结果表明,可以看出zn
‑
co
‑
se/nc展现出较小的半圆,表明电荷转移阻抗较小,有利于电子转移。
61.实施例3:ni
‑
co
‑
se/nc复合材料的制备
62.首先配制30ml 6mol l
‑1的氢氧化钾,然后加入1.2g 2
‑
甲基咪唑(c4h6n2),经过超声和搅拌后得到混合溶液a;取0.446g六水合硝酸镍溶于15ml去离子水中得到溶液b,取0.269g六水合硝酸钴溶于15ml去离子水中形成溶液c;先将溶液b加入到溶液a中,搅拌30分钟后,然后将溶液c加入其中,继续搅拌4.5小时后,经去离子水洗涤和干燥后得的样品标记为nico
‑
mof。将上述的产品nico
‑
mof置于管式炉中,在n2的氛围下以2℃min
‑1升温速率升至500℃保温1小时,自然冷却后得碳化产物nico/nc。然后,将硒粉与nico/nc(质量比为2:1)分别置于瓷舟的上下游,在氮气氛围下升温至350℃保温3小时,500℃保温1小时,自然冷却后得镍钴的双金属多相硒化物和氮掺杂碳的复合材料,记为ni
‑
co
‑
se/nc。
63.图1给出了实施例3所制备的nico
‑
mof的xrd谱图,结果表明,nico
‑
mof的特征峰与已报道的水溶液中合成的该类mofs的特征峰相一致。
64.图2给出了实施例3所制备的nico/nc的xrd谱图可见,经过碳化过后,nico/nc复合材料在26.40
°
处有个碳的(002)晶面的特征峰,nico/nc的其它特征峰与co(jcpds no.15
‑
0806)和ni(jcpds no.04
‑
0850)的(111)和(200)晶面相对应。
65.图5为实施例3所制备的ni
‑
co
‑
se/nc复合材料的xrd谱图。结果表明,ni
‑
co
‑
se/nc的特征峰位于c
‑
cose2(jcpds no.09
‑
0234)和c
‑
nise2(jcpds no.65
‑
5015)之间,证实是钴镍双金属硒化物。ni
‑
co
‑
se/nc没有发生相转变可能是由于nise2和c
‑
cose2具有相似的晶体能带结构,阻碍了c
‑
cose2(100)发生相转变。
66.图10是为实施例3所制备的ni
‑
co
‑
se/nc复合材料的fesem图,可以看出ni
‑
co
‑
se/nc的形貌没有fe
‑
co
‑
se/nc和zn
‑
co
‑
se/nc的均匀,并且分散性稍差,同时存在着多面体结构和球状结构,但硒化后收缩形态得到保留,这归因于ni
‑
co
‑
se/nc在硒化反应过程中没有发生相转变。此外,其表面也出现了突出的颗粒状结构,是因为部分金属离子的逸出然后在
表面形成硒化物晶体团簇。
67.图12(a)给出了实施例3所制备的ni
‑
co
‑
se/nc的氮气吸附脱附曲线,结果表明,ni
‑
co
‑
se/nc的比表面积为7.5m2·
g
‑1,总孔隙体积为0.036cm3·
g
‑1。图12(b)给出了ni
‑
co
‑
se/nc的孔径分布图,可以看出ni
‑
co
‑
se/nc的孔径都在7
‑
100nm之间,都具有由中孔(<50nm)和大孔(>50nm)组成的分级多孔结构,测试结果显示ni
‑
co
‑
se/nc的平均孔径分别为21.5nm。
68.图14给出实施例3所制备的ni
‑
co
‑
se/nc的拉曼光谱图,可以看出ni
‑
co
‑
se/nc在1357和1576cm
‑1附近的两个特征峰,与氮掺杂碳中的d带(非晶碳)和g带(石墨碳)相对应。ni
‑
co
‑
se/nc的i
d
/i
g
(强度比)值为0.76,表明ni
‑
co
‑
se/nc中存在着一些的碳缺陷,这会加快电子转移速率,并增加锂离子的储存位置,有利于储锂性能的提升。
69.实施例4:
70.与实施例1不同之处在于,将获得的feco
‑
mof在n2的氛围下以2℃min
‑1升温速率升至400℃保温1h。图17为样品的sem图,结果表明,该样品的形貌和feco
‑
mof的形貌相似,说明该样品在400℃没有完全碳化。
71.实施例5:
72.与实施例1不同之处在于,将获得的feco
‑
mof在n2的氛围下以2℃min
‑1升温速率升至600℃保温1h。图18为样品的sem图,结果表明,该样品大部分颗粒保持多面体形貌,少部分多面体颗粒破碎,这主要是因为温度过高导致。
73.实施例6:
74.与实施例1不同之处在于,将硒粉与feco/nc(质量比为2:1)分别置于瓷舟的上下游,在氮气的氛围下升温至350℃保温3h,400℃保温1h,自然冷却制得样品。图19为样品的sem图,结果表明,该样品的多面体颗粒仍未密实的结构,说明没有硒化或没有硒化完全,这主要是因为硒化温度较低导致。
75.实施例7:
76.与实施例1不同之处在于,将硒粉与feco/nc(质量比为2:1)分别置于瓷舟的上下游,在氮气的氛围下升温至350℃保温3h,600℃保温1h,自然冷却制得样品。图20为样品的sem图,结果表明,该样品的颗粒仍保持多面体形貌,且由大量颗粒蓬松地组装而成。从这个结果可以看出,在高温下,产物中碳含量较少,且结构松散不牢固,会导致材料的导电性和结构稳定性差。
77.以上显示和描述了本发明的基本原理和主要特征以及本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1