本实用新型涉及纺织面料技术领域,尤其涉及一种光致变色布。
背景技术:
随着社会的发展,人们生活水平的不断提高,人们对于服装的功能性,舒适性和美感等提出了更为苛刻的要求。而目前,多数的面料缝制成的服装缺少动感,颜色单一,显得过于单调和呆板,无法给人们尤其是喜欢追求时尚元素的年轻人带来更多的美感与乐趣,单一颜色的面料满足不了市场的需求,也束缚了服装设计师对个性服装面料的选择。近来,在衣服基本功能不变的基础上,人们越来越多的注重衣物附加功效,不断的研发衣服的新材料,利用适当的工艺将不同材料的各种功效添加到衣料中,增加了衣服的功能性。
光致变色织物是指在太阳光或紫外光等光源的照射下颜色会发生变化的织物,当光线消失后又可逆的变回原来颜色。但是现有技术中制备的光致变色布的感光变色效果差,持久性不强,且透气性能、穿着舒适性不佳。
CN102477644A公开了一种光感变色面料的加工工艺,该加工工艺包括如下步骤:a)选材备料,b)制备功能纱线条,c)染经丝并还原清洗,d)染纬丝并还原清洗,e)浆纱,f)织布,g)退浆水洗,h)烧毛,i)烘培,j)成品定型。使制得的面料触感柔和、附有弹性,且面料能根据光线的强弱变化自动变换表面颜色。该实用新型在功能纱线条的制备中,经丝采用纳米竹碳纤维和天然棉纤维通过并条、粗纱、细纱、络筒工艺后加工成低捻复合纱线条,纬丝采用氨纶纤维和可变色感光纤维通过熔体纺丝法制成氨纶变色纤维,然后再通过清棉、梳棉工序制成条。其中的可变色感光纤维为光敏变色纤维材料,光敏变色纤维材料是通过涤纶纤维在具有光敏变色性化合物的溶液中采用浸渍工艺制得的,光敏变色性化合物材料很容易直接暴露在外界环境中,导致变色性能失效,且经过单纯的浸渍工艺,面料经多次清洗处理后会使光敏变色性化合物材料流失,使面料的变色寿命变短。
技术实现要素:
针对现有技术的不足,本实用新型的目的在于提供一种光致变色布,在日光、紫外光照射下感光变色效果好,持久性强。
为达此实用新型目的,本实用新型采用以下技术方案:
一种光致变色布,包括基布层、光致变色层、纳米二氧化硅层,所述光致变色层设置于所述基布层与所述纳米二氧化硅层之间,所述光致变色层由光致变色纤维层与光致变色微胶囊层交替层叠组成。光致变色微胶囊层使光致变色微胶囊囊内的光致变色材料与外界环境相隔离,避免了外界环境对光致变色材料的影响,光致变色微胶囊变色、褪色速度快,可逆变色次数高于上千次;光致变色纤维层手感好,穿着舒适,耐水洗,也使光致变色布具有持久的变色性能;光致变色纤维层与光致变色微胶囊层协同作用,共同使光致变色布在日光、紫外光的的照射下感光变色效果好,并且持久性强;纳米二氧化硅层中的纳米二氧化硅与光致变色纤维层和光致变色微胶囊层中的光敏变色物结合,使光致变色布更容易吸收日光、紫外光中的紫外线而更快、更容易显现出变色功能。
优选地,所述光致变色微胶囊层的厚度为所述光致变色纤维层的厚度的三分之一至二分之一。
优选地,所述光致变色纤维层与所述光致变色微胶囊层的层数相同,均为1~5层。所述光致变色纤维层是由光致变色纤维制备而成,所述光致变色纤维是将光敏变色性化合物与聚酯经熔融纺丝制得的。所述的光敏变色性化合物选自偶氮类化合物、螺噁嗪类化合物、螺吡喃类化合物中的一种或至少两种的混合物。所述的混合物典型但非限制性的实例为偶氮类化合物和螺噁嗪类化合物的混合物,偶氮类化合物和螺吡喃类化合物的混合物,螺噁嗪类化合物和螺吡喃类化合物的混合物。所述混合物也可以选择为偶氮类化合物、螺噁嗪类化合物和螺吡喃类化合物三种光敏变色性化合物的混合物。所述光致变色微胶囊层是将光致变色微胶囊固着于胶黏剂上制备得到的,可以为将光致变色微胶囊通过喷涂的方式固着于胶黏剂,然后将带有光致变色微胶囊的胶黏剂涂覆于光致变色纤维层,依次使得光致变色纤维层、光致变色微胶囊层交替层叠设置。所述光致变色微胶囊是以密胺树脂为壁材、螺噁嗪类化合物的四氯乙烯溶液为芯材,经原位聚合法制备得到的。
优选地,所述基布层与所述光致变色层之间还设置有透气层,透气层的设置使布具有良好的透气性能,透气层可以为聚丙烯、聚乙烯、聚酰胺、聚氨酯等材料经熔融纺丝制备而成,透气层上设置有若干孔洞,以增加透气层的透气性能,孔洞的形状可以为圆形、方形,也可以为其他形状。另外,为了增强光致变色布的透气性能,透气层上还可以设置有凸起,凸起使透气层与基布层与光致变色层的层与层之间形成小的空隙,增强透气性能。
优选地,所述基布层的厚度为0.5~5mm,例如为0.5mm、1mm、1.5mm、2mm、2.5mm、3mm、3.5mm、4mm、4.5mm、5mm。
优选地,所述光致变色层的厚度为0.5~2mm,例如0.5mm、0.6mm、0.7mm、0.8mm、0.9mm、1mm、1.1mm、1.2mm、1.5mm、1.6mm、1.8mm、2mm。
优选地,所述纳米二氧化硅层的厚度为0.1~0.5mm,例如0.1mm、0.2mm、0.3mm、0.4mm、0.5mm。
优选地,所述透气层的厚度为0.1~1mm,例如0.1mm、0.2mm、0.3mm、0.4mm、0.5mm、0.6mm、0.7mm、0.8mm、0.9mm、1mm。
其中,光致变色布的制备方法,制备方法简单,制备得到的光致变色布感光变色效果好,持久性强,包括如下步骤:
1)以氨纶纤维为原料制备基布层;
2)将光敏变色性化合物与聚酯共混后熔融纺丝制备光致变色纤维,以光致变色纤维为原料制备光致变色纤维层;
3)以密胺树脂为壁材、螺噁嗪类化合物的四氯乙烯溶液为芯材,经原位聚合法制备得到光致变色微胶囊,将光致变色微胶囊固着于胶黏剂上制备得到光致变色微胶囊层;
4)将步骤2)制备得到的所述光致变色纤维层与步骤3)制备得到的所述光致变色微胶囊层交替层叠设置,制备得到光致变色层;
5)在所述光致变色层的表面涂覆纳米二氧化硅层制备得到复合层;
6)将步骤5)制备的复合层置于步骤1)制备的所述基布层上,经热压粘合形成具有从上至下依次为纳米二氧化硅层、光致变色层、基布层的光致变色布。
步骤2)中,所述光敏变色性化合物与所述聚酯的质量比为(1~5):(30~50),例如光敏变色性化合物与聚酯的质量比为1:6、1:8、1:15、1:20、1:30、1:40、1:50、3:40、3:50、4:35、4:45。
步骤3)中,所述光致变色微胶囊的制备过程为,将5~10ml质量浓度为0.5~1%的螺噁嗪类化合物的四氯乙烯溶液加入到30~100ml质量浓度为0.1%的十二烷基硫酸钠的水溶液中,置于乳化机中10000~29000rpm转速下乳化20min,得到乳液;向制备得到的乳液中加入25~50ml质量浓度为20~50%的蜜胺树脂预聚物,并用盐酸调节pH值为4~5,再加入30~150ml质量浓度为0.05%~5%的聚乙二醇,升温至50~70℃,保温1~4h,停止反应,得到光致变色微胶囊;用去离子水清洗、干燥得到光致变色微胶囊。
光致变色材料很容易直接暴露在外界环境中,导致变色性能失效,变色寿命很短。随着微胶囊技术的快速发展,将光致变色材料制成微胶囊,使其与酸、碱、空气及杂质等化学环境隔开,大大提高了光致变色材料的耐疲劳性,延长了使用寿命。以四氯乙烯为溶剂将螺噁嗪类化合物溶解后作为芯材,与变色材料相容性较好的蜜胺树脂为壁材,利用原位聚合技术,将光致变色材料包覆在囊壁透明的微胶囊内部,制备在紫外和日光下具有光致变色特性的光致变色微胶囊。
当作为壁材的密胺树脂与作为芯材的螺噁嗪类化合物的四氯乙烯溶液用量比太低时,制备的微胶囊密封性较差,强度较低,在搅拌过程中容易破裂。当壁材与芯材用量比太高时,微胶囊的平均粒径增大,分布变宽,微胶囊的强度和密封性增加,但微胶囊表面粘附的粒子增多,导致微胶囊的透明度降低,难以显示微胶囊内部的颜色变化。因此,选择合适的壁材与芯材用量比,才能获得既透明又具有良好密封性的变色微胶囊。优选地,本发明的密胺树脂的质量与螺噁嗪类化合物的四氯乙烯溶液的质量配比为(3~5):1,可以为3:1、3.5:1、4:1、4.5:1、5:1。
十二烷基硫酸钠作为乳化剂,其质量分数不仅影响微胶囊的粒径及分布,也影响微胶囊的表面形貌。随着乳化剂质量分数的加大,分散相的界面张力逐渐变小,其在界面定向吸附形成的界面膜的强度增大,对分散液滴保护作用增强,降低了乳液液滴间的凝聚能力,从而使乳化液的稳定性增加,微胶囊平均粒径逐渐变小。但当乳化剂质量分数继续增加时,界面张力不再明显降低,但分散过程中形成大量的气泡,导致分散体系中有效分散相体积减少,因此乳化后的油相液滴粒径增大,导致最终制备的微胶囊粒径较大。
乳化速度太慢,光致变色微胶囊的粒径太大,乳化速度太快,又使形成的光致变色微胶囊的颗粒发生团聚、粘连。优选地,乳化搅拌速度为10000~29000rpm,例如10000rpm、15000rpm、18000rpm、20000rpm、25000rpm、29000rpm。
乳化时间太短,使得制备的光致变色微胶囊的粒径较大,囊壁厚度不均匀,随着乳化时间的延长,光致变色微胶囊的粒径趋于均匀,但是乳化时间太长,又会导致油滴之间聚集,使光致变色微胶囊的粒径太大。优选地,控制乳化时间为20min较合适。
分散剂聚乙二醇的加入影响了光致变色微胶囊的表面形貌及粒径分布。聚乙二醇的加入一定程度上抑制了蜜胺树脂的交联速度,使光致变色微胶囊的分布更均匀,形成的囊壁更透明。当聚乙二醇的用量过低时,反应体系分散不均匀,导致光致变色微胶囊结块、粘连,平均粒径过大;当聚乙二醇的用量过高时,导致光致变色微胶囊分散不均匀,产生团聚而产生大量的絮状物,使光致变色微胶囊的包覆率降低。优选地,聚乙二醇的质量浓度为0.05%~5%,例如0.05%、1%、1.5%、2%、2.5%、3%、3.5%、4%、4.5%、5%。
与现有技术相比,本实用新型的有益效果为:
本实用新型的光致变色布,光致变色层由光致变色纤维层与光致变色微胶囊层交替层叠组成;光致变色纤维层手感好,穿着舒适,耐水洗,也使光致变色布具有持久的变色性能;光致变色微胶囊层使光致变色微胶囊囊内的光致变色材料与外界环境相隔离,避免了外界环境对光致变色材料的影响,光致变色微胶囊变色、褪色速度快,可逆变色次数高于上千次;光致变色纤维层与光致变色微胶囊层协同作用,共同使光致变色布在日光、紫外光的的照射下感光变色效果好,并且持久性强;纳米二氧化硅层中的纳米二氧化硅与光致变色纤维层和光致变色微胶囊层中的光敏变色物结合,使光致变色布更容易吸收日光、紫外光中的紫外线而更快、更容易显现出变色功能,可保证数千次以上的光致变色可逆循环次数。
附图说明
图1为本实用新型的光致变色布的结构示意图;
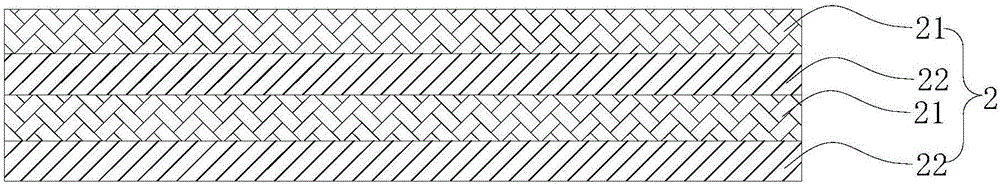
图2为图1的光致变色布中光致变色层的结构示意图;
图3为本实用新型的光致变色布的一个优选例的结构示意图。
附图标记如下:
1-基布层;2-光致变色层;21-光致变色纤维层;22-光致变色微胶囊层;3-纳米二氧化硅层;4-透气层。
具体实施方式
下面结合附图1、2、3,并通过具体实施方式来进一步说明本实用新型的技术方案。
如图1、2所示,本实用新型的一种光致变色布,包括基布层1、光致变色层2、纳米二氧化硅层3,光致变色层2设置于基布层1与纳米二氧化硅层3之间,光致变色层2由光致变色纤维层21与光致变色微胶囊层22交替层叠组成。光致变色纤维层21与光致变色微胶囊层22协同作用,共同使光致变色布在日光、紫外光的的照射下感光变色效果好,并且持久性强;纳米二氧化硅层3中的纳米二氧化硅与光致变色纤维层21和光致变色微胶囊层22中的光敏变色物结合,使光致变色布更容易吸收日光、紫外光中的紫外线而更快、更容易显现出变色功能;光致变色纤维层耐水洗,也使光致变色布具有持久的变色性能。
优选地,光致变色微胶囊层22的厚度为光致变色纤维层21的厚度的三分之一至二分之一,通过合理调节光致变色微胶囊层的厚度与光致变色纤维层的厚度,使得光致变色布既有较好的感光变色效果,还具有良好的手感和柔软度。进一步地,光致变色纤维层21与光致变色微胶囊层22的层数相同,均为1~5层,例如均为1层、2层、3层、4层、5层。
进一步地,光致变色纤维层21是由光致变色纤维制备而成,光致变色纤维是将光敏变色性化合物与聚酯经熔融纺丝制得的。
作为优选方案,光致变色微胶囊层22是将光致变色微胶囊固着于胶黏剂上制备得到的。光致变色微胶囊是以密胺树脂为壁材、螺噁嗪类化合物的四氯乙烯溶液为芯材,经原位聚合法制备得到的。
作为本实用新型的优选实施例,基布层1与光致变色层2之间还设置有透气层4,如图3所示,使制备的光致变色布具有良好的感光变色效果的同时,具有良好的透气性能,穿着舒适。更优选地,基布层1的厚度为0.5~5mm。光致变色层2的厚度为0.5~2mm。纳米二氧化硅层3的厚度为0.1~0.5mm。透气层4的厚度为0.1~1mm。
本实用新型的光致变色布的制备方法,包括如下步骤:
1)以氨纶纤维为原料制备基布层1;
2)将光敏变色性化合物与聚酯共混后熔融纺丝制备光致变色纤维,以光致变色纤维为原料制备光致变色纤维层21;
3)以密胺树脂为壁材、螺噁嗪类化合物的四氯乙烯溶液为芯材,经原位聚合法制备得到光致变色微胶囊,将光致变色微胶囊固着于胶黏剂上制备得到光致变色微胶囊层22;
4)将步骤2)制备得到的所述光致变色纤维层21与步骤3)制备得到的所述光致变色微胶囊层22交替层叠设置,制备得到光致变色层2;
5)在所述光致变色层2的表面涂覆纳米二氧化硅层3制备得到复合层;
6)将步骤5)制备的复合层置于步骤1)制备的所述基布层1上,经热压粘合形成具有从上至下依次设置的纳米二氧化硅层3、光致变色层2、基布层1的光致变色布。
按上述步骤制备不同的光致变色布作为实施例,每个实施例中不同之处在于步骤2)中光致变色纤维层21光致变色纤维的制备过程不同,以及步骤3)中光致变色微胶囊层22光致变色微胶囊的制备过程不同,其中具体过程如下:
实施例1
步骤2)中,光致变色纤维的制备原料中光敏变色性化合物与聚酯的质量比为1:30。
步骤3)中,光致变色微胶囊的制备过程为,将6ml质量浓度为0.5%的螺噁嗪类化合物的四氯乙烯溶液加入到50ml质量浓度为0.1%的十二烷基硫酸钠的水溶液中,置于乳化机中15000rpm转速下乳化20min,得到乳液;向制备得到的乳液中加入30ml质量浓度为35%的蜜胺树脂预聚物,并用盐酸调节pH值为4,再加入50ml质量浓度为4.5%的聚乙二醇,升温至60℃,保温4h,停止反应,得到光致变色微胶囊;用去离子水清洗、干燥得到光致变色微胶囊。
实施例2
步骤2)中,光致变色纤维的制备原料中光敏变色性化合物与聚酯的质量比为3:40。
步骤3)中,所述光致变色微胶囊的制备过程为,将10ml质量浓度为0.8%的螺噁嗪类化合物的四氯乙烯溶液加入到70ml质量浓度为0.1%的十二烷基硫酸钠的水溶液中,置于乳化机中20000rpm转速下乳化20min,得到乳液;向制备得到的乳液中加入50ml质量浓度为50%的蜜胺树脂预聚物,并用盐酸调节pH值为5,再加入100ml质量浓度为2%的聚乙二醇,升温至70℃,保温3h,停止反应,得到光致变色微胶囊;用去离子水清洗、干燥得到光致变色微胶囊。
实施例3
步骤2)中,光致变色纤维的制备原料中光敏变色性化合物与聚酯的质量比为5:42。
步骤3)中,光致变色微胶囊的制备过程为,将7ml质量浓度为0.6%的螺噁嗪类化合物的四氯乙烯溶液加入到80ml质量浓度为0.1%的十二烷基硫酸钠的水溶液中,置于乳化机中12000rpm转速下乳化20min,得到乳液;向制备得到的乳液中加入25ml质量浓度为40%的蜜胺树脂预聚物,并用盐酸调节pH值为4,再加入150ml质量浓度为0.5%的聚乙二醇,升温至65℃,保温4h,停止反应,得到光致变色微胶囊;用去离子水清洗、干燥得到光致变色微胶囊。
实施例4
步骤2)中,光致变色纤维的制备原料中光敏变色性化合物与聚酯的质量比为3:25。
步骤3)中,光致变色微胶囊的制备过程为,将10ml质量浓度为0.5%的螺噁嗪类化合物的四氯乙烯溶液加入到40ml质量浓度为0.1%的十二烷基硫酸钠的水溶液中,置于乳化机中25000rpm转速下乳化20min,得到乳液;向制备得到的乳液中加入35ml质量浓度为30%的蜜胺树脂预聚物,并用盐酸调节pH值为5,再加入70ml质量浓度为3%的聚乙二醇,升温至70℃,保温2h,停止反应,得到光致变色微胶囊;用去离子水清洗、干燥得到光致变色微胶囊。
将上述实施例1~4制备的光致变色纤维和光致变色微胶囊按照本实用新型的光致变色布的制备方法制备光致变色布。本实用新型制备得到的光致变色布在日光、紫外光的照射下感光变色效果好,可保证数千次以上的光致变色可逆循环次数,持久性强。
申请人声明,本实用新型通过上述实施例来说明本实用新型的详细方法,但本实用新型并不局限于上述详细方法,即不意味着本实用新型必须依赖上述详细方法才能实施。所属技术领域的技术人员应该明了,对本实用新型的任何改进,对本实用新型产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本实用新型的保护范围和公开范围之内。