2,6-二氯苯甲腈的制备方法与流程
2,6
‑
二氯苯甲腈的制备方法
技术领域
1.本发明整体涉及的技术领域是制备可用于杀虫和除草组合物中的化学物质的方法。更具体地讲,本发明涉及用于制备卤代苯甲腈的方法。
背景技术:
2.虽然已知许多化学物质是用于除草和杀虫应用的非常有效的原材料,但是许多化学物质需要对特殊设备和/或昂贵的原材料进行大量投资来生产它们。由于这可增加这些产品的商业化的显著成本,因此一直在寻找以更有效和高效的方式生成此类产品的方式。
3.2,6
‑
二氯苯甲腈,也称为二氯苄腈,是农业中广泛使用的除草剂。
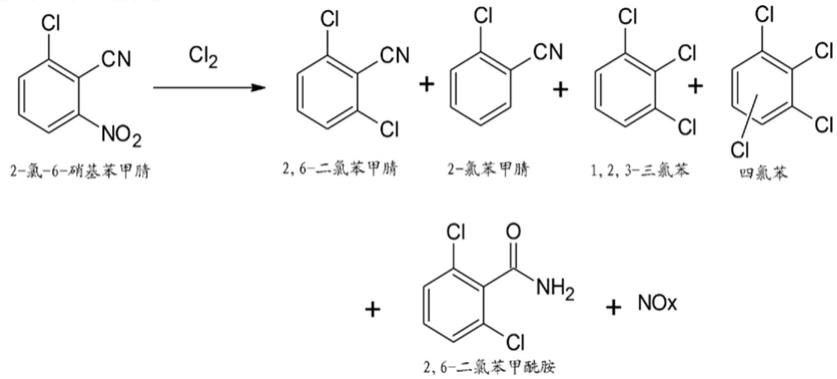
4.已知多种方法用于通过氨氧化反应合成2,6
‑
二氯苯甲腈。已知的用于生产此类材料的工业方法涉及诸如2,6
‑
二氯甲苯的气相催化氨氧化处理,需要对设备进行大量投资,并且昂贵的原材料2,6
‑
二氯甲苯的来源有限。
5.us4225534a公开了通过在非质子溶剂中与氯化锂或氯化锂和无水氯化铝的混合物或与氯化锂铝反应来制备2
‑
氯苯甲腈衍生物,使得其在工业上是不利的。
6.因此,本发明的目的是提供克服上述缺点的改进方法。
7.因此,本发明涉及利用特定反应参数改进方法,并且目的是提供期望产物的高选择性、收率和纯度。
技术实现要素:
:
8.在一个方面,本发明提供了一种用于生产卤代苯甲腈的简单且工业上可行的方法。
9.在另一方面,本发明提供了2,6
‑
二氯苯甲腈,其包括用氯气处理2
‑
氯
‑6‑
硝基苯甲腈。
10.在另一方面,本发明提供了在不存在溶剂的情况下使用氯对2
‑
氯
‑6‑
硝基苯甲腈的选择性脱硝基氯化合成。
11.在另一方面,本发明方法提供以下物质的受控形成:副产物,诸如no
x
、2
‑
氯苯甲腈、1,2,3
‑
三氯苯甲腈、四氯苯和2,6
‑
二氯苯甲酰胺,从而导致高收率和高纯度的期望产物2,6
‑
二氯苯甲腈。
12.在一个方面,本发明的方法以至少80%的高收率和至少99%的纯度提供2,6
‑
二氯苯甲腈。
13.这些以及附加实施方案将通过以下描述变得显而易见。
具体实施方式
14.本文所示的细节仅为示例,而且仅出于说明性地讨论本发明的各种实施方案的目的,并且提出这些细节是为了提供被认为是对本发明的原理和概念方面的最有用和最容易理解的描述。就这一点而言,没有试图比对本发明的基本理解所必需而更详细地示出本发
明的细节,该描述使本领域技术人员显而易见如何在实践中体现本发明的若干种形式。
15.现在将参考更详细的实施方案对本发明进行描述。然而,本发明可以以不同的形式体现,并且不应理解为限于本文所述的实施方案。相反,提供这些实施方案,使得本公开内容将是彻底和完整的,并且将本发明的范围充分传达给本领域的技术人员。
16.除非另有定义,否则本文所用的所有技术和科学术语具有与本发明所属领域的普通技术人员通常所理解相同的含义。在本文的本发明的描述中使用的术语仅用于描述特定实施方案,而无意于限制本发明。如在本发明的描述和权利要求书中所使用的,单数形式的“一个”、“一种”和“该(所述)”也意图包括复数形式,除非上下文另外明确指出。本文提及的所有出版物、专利申请、专利和其他参考文献均全文明确地以引用方式并入。
17.除非另外指出,否则在本说明书和权利要求书中使用的表示成分的量、反应条件等的所有数字均应理解为在所有情况下均由术语“约”修饰。因此,除非相反地指出,否则在以下说明书和权利要求书中提出的数值参数是近似值,其可以根据本发明试图获得的期望特性而变化。至少,并非试图将等同原则的应用限制于权利要求书的范围,应根据有效数位的数量和普通的舍入方法来解释每个数值参数。
18.除非另有定义,否则“卤代苯甲腈”意指带有一个或多个卤素部分(诸如氟、氯、溴或碘原子)的苯甲腈。具体示例为2,6
‑
二氯苯甲腈。
19.尽管阐述本发明的广泛范围的数值范围和参数是近似值,但是在具体示例中阐述的数值被尽可能精确地报道。然而,任何数值都固有地包含某些误差,这些误差必定是由其相应的测试测量中存在的标准偏差引起的。本说明书通篇给出的每个数值范围将包括落入这种较宽数值范围内的每个较窄数值范围,好像此类较窄数值范围均在本文中明确写出。
20.本发明的附加优点将在下面的描述部分地阐述,并且部分地将从描述中显而易见,或者可通过本发明的实践来了解。应当理解,前述一般描述和以下详细描述两者仅为示例性的和说明性的,并且不受权利要求书保护的本发明的限制。
21.本发明提供了用于制备卤代苯甲腈的方法。
22.因此,本发明提供了用于制备2,6
‑
二氯苯甲腈的有效且高效的方法。
23.本发明的方法是经济可行的,其非常适于大规模工业生产。
24.在一个实施方案中,用于制备2,6
‑
二氯苯甲腈的方法包括用氯气对2
‑
氯
‑6‑
硝基苯甲腈进行脱硝基氯化。
25.本发明的方法由以下反应方案表示。
[0026][0027]
方案1
[0028]
在一个实施方案中,用于制备2,6
‑
二氯苯甲腈的方法包括在不具有溶剂的情况下用氯气对2
‑
氯
‑6‑
硝基苯甲腈进行选择性脱硝基氯化。
[0029]
在一个实施方案中,2
‑
氯
‑6‑
硝基苯甲腈与氯气的比率在1:1至1:5的范围内。
[0030]
在一个实施方案中,2
‑
氯
‑6‑
硝基苯甲腈与氯气的比率在1:1或1:2或1:3或1:5的
范围内。
[0031]
在一个实施方案中,所述方法在约100℃至200℃的温度下进行。
[0032]
在一个实施方案中,所述方法在约150℃至200℃的温度下进行。
[0033]
在一个实施方案中,所述方法在150℃或155℃或160℃或165℃或170℃或175℃或180℃或185℃或190℃或195℃下进行。
[0034]
在一个实施方案中,所述方法进行约5小时至15小时。
[0035]
在一个实施方案中,所述方法进行约8小时至10小时。
[0036]
在一个实施方案中,所述方法在185℃至195℃下进行约8小时至10小时。
[0037]
上述方法(其中发生脱硝基氯化)也形成no
x
,其用酸例如浓硫酸处理以形成亚硝基硫酸。该亚硝基硫酸可用于不同的目的,例如重氮化和本领域技术人员已知的其他目的。
[0038]
在一个实施方案中,该方法提供对以下物质的过度形成的增强的控制:副产物,例如,过度氯化副产物,诸如1,2,3
‑
三氯苯和四氯苯(tcb);还原性脱氯,诸如2
‑
氯苯甲腈;以及水解产物,诸如2,6
‑
二氯苯甲酰胺,如方案2所示。
[0039][0040]
方案2
[0041]
因此,根据本发明获得的2,6
‑
二氯苯甲腈基本上不含杂质。
[0042]
通常,当其他化合物以不超过0.1%的量存在时,2,6
‑
二氯苯甲腈基本上不含另一种化合物。
[0043]
在一个实施方案中,所述方法中形成的每种副产物或杂质被限制为约小于2%,优选地小于1%,更优选地小于0.1%。
[0044]
在另一个实施方案中,1,2,3
‑
三氯苯是主要杂质,并且本发明的方法有利地将该杂质的形成限制为至多2%,所述杂质的形成可通过纯化方法进一步限制为小于0.1%。
[0045]
在一个实施方案中,根据本发明,通过在特定工艺参数下使用氯气对2
‑
氯
‑6‑
硝基苯甲腈进行选择性脱硝基氯化,以至少80%高收率生产的2,6
‑
二氯苯甲腈具有至少99%的高纯度。
[0046]
在一个实施方案中,反应物本身充当反应的溶剂,并且反应在不具有溶剂的情况下进行。
[0047]
反应在大约大气压下发生,当然这也可包括略高于和略低于大气压的条件。
[0048]
在2
‑
氯
‑6‑
硝基苯甲腈的脱硝基氯化过程中,控制氯气的引入也允许no
x
(诸如例
如no和no2)连续地离开该体系。
[0049]2‑
氯
‑6‑
硝基苯甲腈通过2,3
‑
二氯硝基苯的常规氰化物交换制备,其本身是3,4
‑
二氯硝基苯工艺的副产物,在批量产生下具有一次性使用或替代使用的问题。因此,本文所述的方法代表该前一种废品的可行商业用途。
[0050]
因此,本发明提供了非常高的选择性和收率,从而显著降低了工业生产成本并满足了大规模工业生产的需要。
[0051]
本文所述的目的是以一定速率限制氯吹扫,以将2
‑
氯
‑6‑
硝基苯甲腈选择性地转化为2,6
‑
二氯苯甲腈,并且限制其他副反应发生。附加的选择是通过反应蒸馏来改善选择性,以在2,6
‑
二氯苯甲腈一形成就将其从反应相中分离出来。
[0052]
表1展示出受控氯添加速率和不受控氯添加速率的比较结果。
[0053]
表1
[0054]
转化率不受控,%受控,%2,6
‑
二氯苯甲腈8591
‑
96tcb(三氯苯)8
‑
100.8
‑
2.0mncb(一硝基氯苯)1
‑
2<0.1附加杂质3
‑
40.9
‑2[0055]
在使用例如常规的溶剂洗剂技术,在适当的后处理以除去反应后上述含量的所有上述反应杂质后,获得收率为至少80%并且纯度水平大于99%(按重量计)的白色结晶固体2,6
‑
二氯苯甲腈。通过这种途径,这使得产品在许多水平上具有吸引力。
[0056]
将反应期间释放的nox洗涤到浓硫酸中以得到固体亚硝基硫酸,其用作可销售的重氮化试剂。
[0057][0058]
方案3
[0059]
因此,这代表了用于制备2,6
‑
二氯苯甲腈的工业上有利且经济上有利的方法。
[0060]
在另一方面,本发明提供了用于合成2,6
‑
二氯苯甲腈的方法,该方法包括以下步骤:
[0061]
a)制备2
‑
氯
‑6‑
硝基苯甲腈
[0062]
b)任选地纯化2
‑
氯
‑6‑
硝基苯甲腈
[0063]
c)制备2,6
‑
二氯苯甲腈
[0064]
d)任选地纯化2,6
‑
二氯苯甲腈
[0065]
在另一方面,本发明提供了2
‑
氯
‑6‑
硝基苯甲腈的制备,其包括在非质子酰胺的存在下,使1,2
‑
二氯
‑3‑
硝基苯与金属氰化物和/或金属氯化物反应以获得2
‑
氯
‑6‑
硝基苯甲腈。
[0066]
在一个实施方案中,氰化反应通过使1,2
‑
二氯
‑3‑
硝基苯与氰化钠和氰化铜或者氰化钠和氯化铜或氰化钠、氯化铜和氰化铜的混合物反应来进行。
[0067]
在一个实施方案中,所述非质子酰胺选自n,n
‑
二甲基甲酰胺、二甲基亚砜、n,n
‑
二甲基乙酰胺、六甲基磷酰胺和n
‑
甲基吡咯烷酮等。
[0068]
在一个实施方案中,所述非质子酰胺为n,n
‑
二甲基甲酰胺。
[0069]
在一个实施方案中,用于制备2
‑
氯
‑6‑
硝基苯甲腈的方法包括加热1,2
‑
二氯
‑3‑
硝基苯、金属氰化物和作为催化剂的非质子酰胺的混合物,以获得2
‑
氯
‑6‑
硝基苯甲腈。
[0070]
在另一个实施方案中,用于制备2
‑
氯
‑6‑
硝基苯甲腈的方法包括在100
‑
200℃的温度下加热1,2
‑
二氯
‑3‑
硝基苯、氰化钠和氰化铜以及二甲基甲酰胺的混合物,以获得2
‑
氯
‑6‑
硝基苯甲腈。
[0071]
在另一个实施方案中,用于制备2
‑
氯
‑6‑
硝基苯甲腈的方法包括在100
‑
200℃的温度下加热1,2
‑
二氯
‑3‑
硝基苯、氰化钠和氯化铜以及二甲基甲酰胺的混合物,以获得2
‑
氯
‑6‑
硝基苯甲腈。
[0072]
在另一个实施方案中,用于制备2
‑
氯
‑6‑
硝基苯甲腈的方法进行5
‑
10小时。
[0073]
在另一方面,本发明提供了2
‑
氯
‑6‑
硝基苯甲腈的制备,其包括在非质子酰胺的存在下,使1,2
‑
二氯
‑3‑
硝基苯与金属氰化物和/或金属氯化物反应以获得2
‑
氯
‑6‑
硝基苯甲腈。
[0074]
在一个实施方案中,用于制备2,6
‑
二氯苯甲腈的方法包括:
[0075]
a)在非质子酰胺的存在下,使1,2
‑
二氯
‑3‑
硝基苯与金属氰化物和/或金属氯化物或它们的混合物反应以获得2
‑
氯
‑6‑
硝基苯甲腈2
‑
氯
‑6‑
硝基苯甲腈,以及
[0076]
b)用氯气对2
‑
氯
‑6‑
硝基苯甲腈进行脱硝基氯化以获得2,6
‑
二氯苯甲腈。
[0077]
在另一个实施方案中,用于制备2,6
‑
二氯苯甲腈的方法包括:
[0078]
a)在非质子酰胺的存在下,使1,2
‑
二氯
‑3‑
硝基苯与金属氰化物和/或金属氯化物或它们的混合物反应以获得,以及
[0079]
b)在不具有溶剂的情况下用氯气对2
‑
氯
‑6‑
硝基苯甲腈进行脱硝基氯化以获得2,6
‑
二氯苯甲腈。
[0080]
工艺条件和参数如上所述。
[0081]
这些实施例仅是举例说明,并且不应理解为以任何方式限制本发明的范围和基础原理。除了本文所示和所述的那些之外,本发明的各种修改对本领域的技术人员而言将由以下实施例和前述说明而变得显而易见。此类修改也旨在落入权利要求书的范围内。
[0082]
实施例
[0083]
实施例1:
[0084]
该方法是以下实施例,其展示出温度对反应的影响。
[0085]
表2
[0086][0087]
在上述实施例中,以与释放的nox的摩尔当量从硫酸洗涤器中产生和分离固体形式的亚硝基硫酸。
[0088]
实施例2
[0089]
在具有顶置式搅拌器、tp、强力色谱柱且具有油浴中的浓h2so4洗涤器的1升4颈rbf中,装入2
‑
氯
‑6‑
硝基苯甲腈并升高温度。然后将氯气缓慢吹扫到烧瓶中以获得一致速率。
[0090]
分析所得反应物料的2,6
‑
cnbn转化率至≥99%;观察到2,6
‑
二氯苯甲腈为73
‑
80%;tcb为9.4
‑
10%。
[0091]
反应完成后,将反应物料冷却至100℃,并且用一氯苯稀释,通过用10%碳酸钠水溶液洗涤中和,并且用活性炭脱色。将该脱色的2,6
‑
二氯苯甲腈溶液在甲醇中回流并在10℃下结晶、过滤并在真空烘箱中干燥。
[0092]
表3
[0093][0094]
这些实施例展示出dcbn可通过利用受控的氯添加并且在不存在溶剂的情况下,在180
‑
190℃下进行cnbn的脱硝基氯化,以71
‑
76%的收率和>98%的纯度来制备。
[0095]
实施例3
‑6[0096]
将2
‑
氯
‑6‑
硝基苯甲腈(获自实施例7中所述的方法)装入配备有浓硫酸和苛性碱洗涤器、顶置式搅拌器和电加热油浴的rbf中,并且温度升至195℃。然后以受控的顺序以不同的速率吹扫氯气10
‑
16小时。分析所得反应物料的2,6
‑
cnbn转化率至≥99%,并且2,6
‑
二氯苯甲腈示出为92
‑
94%。在100℃下用一氯苯稀释反应物料;通过用10%碳酸钠水溶液洗涤中和;用活性炭脱色。
[0097]
将该脱色的2,6
‑
二氯苯甲腈溶液在甲醇中回流并在10℃下结晶、过滤并在真空烘箱中干燥。
[0098]
表4
[0099][0100]
该数据示出,如实施例7中所述制得的cnbn增加了产物含量和收率,因为它降低了
tcb形成的发生率,如上文实施例1和2所观察到的。
[0101]
实施例7
[0102]
用于制备2
‑
氯
‑6‑
硝基苯甲腈的方法
[0103][0104]
将2,3
‑
二氯硝基苯连同氰化钠和氰化铜或氰化钠或氯化铜一起加入配备有油浴、顶置式搅拌器、标称冷凝器、氮封和水洗涤器的1l rbf中。将反应物料缓慢加热至90℃,以获得熔融浆液物料,然后用搅拌器搅拌物料,并且持续缓慢加热。一旦反应物料在整个温度上升至反应转化温度(160℃)的过程中一次性或分批获得100℃,就加入n,n
‑
二甲基甲酰胺。在达到反应物料温度后,将其在160℃下保持5
‑
6小时,并且还加热至170℃另外5
‑
6小时,以获得至<5%的2,3
‑
二氯硝基苯转化率。将反应物料冷却至90℃;n,n
‑
二甲基甲酰胺通过真空蒸馏回收。在80℃用一氯苯稀释粗制的2
‑
氯
‑6‑
硝基苯甲腈;在该温度下,通过真空过滤除去无机盐。添加附加的热一氯苯以洗涤并从无机残余物中完全除去有机物料。包含有机滤液的产物在70℃下用5%氨水溶液洗涤1小时,分离层并用另外的10%hcl水溶液中和,并且在70℃下水洗
°
;在70℃下进行木炭处理
°
。使洗涤处理过的有机滤液经历一氯苯回收,并且将反应物料冷却至10℃,并且在真空过滤时过滤沉淀出的产物,并将其用冷溶剂洗涤。卸载湿饼并将其在80℃下干燥。
[0105]
表5:dmf催化剂比率对2,3
‑
dcnb向cnbn的转化率和杂质形成的影响
[0106][0107]
这些实施例展示出,将dmf载量从193g/mol的2,3
‑
dcnb降低至27
‑
28g/mol的2,3
‑
dcnb,显著改善了反应性能,即cnbn含量从41%提高至91
‑
96%。
[0108]
表6:使用氰化钠/氯化铜试剂进行cnbn转化
[0109][0110]
氰化铜是昂贵的,并且在大多数有机溶剂中的溶解性非常差,这使得其成为反应性的不良选择。通过用氯化铜替换它,该体系更易于以降低的成本进行商业制造。
[0111]
如本文所述,本领域中的这些问题和其他问题通过本文所述的本发明来解决。因此,本发明的范围应包括可落入权利要求书的范围内的所有修改形式和变型形式。通过考虑本文所公开的本发明的说明书和实践,本发明的其他实施方案对于本领域的技术人员将是显而易见的。旨在仅将说明书和示例视为示例性的,本发明的真实范围和实质由以下权利要求书指示。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1