一种具备pH响应性的碘酸钙天然高分子微胶囊及其制备方法和用途与流程
本发明涉及微胶囊和饲料添加剂技术领域,尤其涉及一种具备ph响应性的碘酸钙天然高分子微胶囊及其制备方法和用途。
背景技术:
碘元素是公认的动物必需微量元素之一,是甲状腺素的重要组成部分,参与着动物机体的各项生命代谢,碘元素缺乏会严重危害机体健康。我国是碘元素较为缺乏的国家之一,居民和动物的碘元素摄入水平偏低,因而需要额外补充碘元素。动物补充碘元素的主要途径是食用饲料和饮水。研究表明,不同动物吸收碘元素的主要器官不同,在单胃动物中肠道是吸收碘元素的主要器官,而反刍动物中吸收碘元素的主要器官是瘤胃。瘤胃内容物ph值是食糜中挥发性脂肪酸与唾液中缓冲盐相互作用、以及瘤胃上皮对挥发性脂肪酸吸收及随食糜流出等因素综合作用的结果,导致ph值范围一般为6~7。综上,动物主要吸收碘元素的ph为6~7,即在中性或弱碱性环境中有利于碘元素的吸收。
碘酸钙是一种安全性高的碘营养剂,具有易加工、不易吸潮、对设备腐蚀低的特点,在饲料行业得到较为广泛的应用。但是,碘酸钙具有较强的氧化性。碘酸钙在一定条件下能与饲料中的物质相互作用,例如碘酸钙能与饲料中的维生素c(vc)、硫酸亚铁等发生氧化还原反应,将高价态的碘转化生成i-或i2,导致碘元素在使用过程中流失。在碘酸钙被还原过程中,参与反应的物质会破坏饲料的品质、风味以及动物对饲料的吸收。因而,限制了碘酸钙在饲料行业的广泛应用以及动物对其的高效利用。同时,碘酸钙在使用过程中的流失导致其用量存在不确定性,增加了畜牧成本。由于碘酸钙直接施用于饲料中存在碘元素流失量不可控,导致难以控制禽畜等对碘元素的摄入量,而造成实际应用中往往需添加过量的碘酸钙。人或动物长期摄入过量碘元素将会对机体引发诸多不良影响,例如高碘性甲状腺肿等。为此,提高碘元素摄入量的可控性和吸收效率对人和动物机体健康有着重要意义。
通过微胶囊技术可有效改善碘酸钙的稳定性,同时能促进动物吸收碘元素的效率。如中国专利申请201810055932.4公开的“一种缓慢释放碘酸钙的微球及其制备方法”,该技术可一定程度地改善碘酸钙的稳定性,但仍存在一些缺陷,如:1、该技术单纯地以海藻酸钠作为微球的壁材,存在容易暴释的问题,且微球的ph响应性差;2、该方法制备的微球,部分碘酸钙仍会分布在微球的表面,从而容易发生氧化还原反应。
因此,如何提高碘酸钙微胶囊在胃肠液中的稳定性和响应性是碘元素微胶囊化的研究重点之一。
技术实现要素:
为解决上述现有技术中存在的缺点和不足,本发明的目的在于提供一种具备ph响应性的碘酸钙天然高分子微胶囊及其制备方法和用途。本发明的碘酸钙天然高分子微胶囊具有较大的交联密度,在环境中稳定,对碘酸钙的保护性强,提高了碘酸钙的用量可控性及其有效利用率。本发明的碘酸钙天然高分子微胶囊还具备ph响应性,在弱碱性环境下响应性快速释放,提高了动物对碘元素的吸收效率,也提高了动物对碘元素摄入量的可控性。
为实现其目的,本发明采取的技术方案为:
一种具备ph响应性的碘酸钙天然高分子微胶囊的制备方法,其包括如下步骤:
(1)制备壳聚糖溶液:将壳聚糖溶解于乙酸溶液中,得到壳聚糖溶液;
(2)制备壳聚糖微胶囊:将kio3溶解于步骤(1)制得的壳聚糖溶液中,然后加入cacl2,使壳聚糖分子链上的碘酸根与钙离子原位生成碘酸钙,再加入凝聚剂,使壳聚糖析出,再加入交联剂对壳聚糖进行交联,形成壳聚糖微胶囊,滤出壳聚糖微胶囊后对其进行洗涤;
(3)将步骤(2)制得的壳聚糖微胶囊分散到海藻酸钠溶液中,利用静电自组装法对壳聚糖微胶囊进行包衣,过滤、干燥,制得所述具备ph响应性的碘酸钙天然高分子微胶囊。
本发明以海藻酸钠和壳聚糖对碘酸钙进行双重保护,防止碘酸钙与环境接触,提高其在使用过程中的稳定性,有效降低了碘酸钙的流失。而且,海藻酸钠和壳聚糖均为天然高分子材料,具有良好的生物相容性,无毒无害,能被动物消化吸收。此外,海藻酸钠和壳聚糖具有不同的环境溶胀特性,海藻酸钠具有“酸沉碱溶”的特点,而壳聚糖具有“酸溶碱沉”的特点。本发明利用了海藻酸钠与壳聚糖的不同特性,并通过调节组分和制备方法,制备了具有ph响应性的碘酸钙天然高分子微胶囊。在酸性环境下,壳聚糖在微胶囊的内部溶胀,而海藻酸钠则在微胶囊的外部收缩。同时,在酸性环境下壳聚糖的正电性会增加,进一步增强了海藻酸钠与壳聚糖之间的作用力。由此,在酸性环境下,碘酸钙天然高分子微胶囊的内部较为致密,碘酸钙的释放率较低。在弱碱性环境下,壳聚糖的溶胀受阻,而海藻酸钠的溶胀增大,由此,壳聚糖与海藻酸钠之间的静电作用力变弱,碘酸钙天然高分子微胶囊的结构变疏松,更利于碘酸钙的释放。所以,在弱碱性环境下,碘酸钙的释放率较高。经实验验证,本发明的碘酸钙天然高分子微胶囊在弱碱性环境下的累计释放率可达到96.95%,提高了动物对碘元素的吸收效率,也提高了动物对碘元素摄入量的可控性。
本发明在制备过程中,以碘酸钾(kio3)为碘源,将其溶于壳聚糖溶液中,碘酸根能与壳聚糖中质子化的-nh3+相互之间形成静电作用,从而使碘酸根均匀分布在壳聚糖分子链上。此后再加入cacl2,使壳聚糖分子链上的碘酸根与钙离子原位生成碘酸钙,如此,可减少壳聚糖溶液中碘酸钙沉淀的出现。若直接向壳聚糖溶液中加入碘酸钙,会存在碘酸钙在壳聚糖溶液中分布不均匀,碘酸钙沉淀较多等情况,从而导致制备的碘酸钙天然高分子微胶囊载药率不均匀,也会造成碘酸钙的浪费。
现有方法在静电自组装后一般需使用酸或碱等物质去除模板,这个过程会产生污染环境的废液。而本发明利用相分离法制备负载碘酸钙的壳聚糖微胶囊后,直接以该壳聚糖微胶囊作为静电自组装的模板,用海藻酸钠对其进行包衣,形成碘酸钙天然高分子微胶囊。可见,本发明免去了用酸或碱等物质去除模板的过程,减少了静电自组装过程对环境的污染,也简化了微胶囊的制备工序,免去了特殊仪器的使用。同时,本发明的包衣方法还增强了微胶囊的交联密度,提高了微胶囊在环境中的稳定性,进一步加强了壳聚糖和海藻酸钠对碘酸钙的保护。
优选地,步骤(1)中,所述乙酸溶液的体积百分比浓度为1%~3%;更优选地,所述乙酸溶液的体积百分比浓度为2%。上述浓度的乙酸溶液对壳聚糖的溶解性较好,使用成本也较低。
优选地,步骤(1)中,所述壳聚糖溶液的浓度为10~30mg/ml;更优选地,所述壳聚糖溶液的浓度为20mg/ml。本发明通过研究发现,壳聚糖溶液的浓度越大,其颜色越深,所得产物的颜色也会越深。而且,壳聚糖溶液的浓度过大,还会使其流动性下降,致使加入凝聚剂后容易形成粒径较大的颗粒,甚至结块,不利于后续碘酸钙的原位生成。但若壳聚糖溶液的浓度过小,会导致后续形成的微胶囊的粒径过小,不利于产物收集,容易造成产物流失、浪费,也会影响产物的稳定性。而壳聚糖溶液的浓度为10~30mg/ml时,壳聚糖溶液的颜色不会太深,不至于影响产物的外观,流动性较好,利于原位生成碘酸钙,产物也易于收集,流失率低,且产物的稳定性高。壳聚糖溶液的浓度为20mg/ml时,综合效果最好。
优选地,步骤(2)中,所述凝聚剂为无水乙醇;优选地,按所述壳聚糖溶液的体积计,所述无水乙醇的用量为≥6ml/ml;更优选地,按所述壳聚糖溶液的体积计,所述无水乙醇的用量为6ml/ml。现有技术在采用相分离法制备微胶囊时,一般采用苯、环己烷、甲醇等毒性较大的溶剂作为凝聚剂,对环境产生不良影响。而本发明以无水乙醇作为凝聚剂,可减少相分离过程对环境的不良影响。无水乙醇不会以本发明制备微胶囊的物料发生反应,也不会溶解物料,凝聚效果好。无水乙醇可生物分解,对环境不利的影响较小,回收处理的成本低。此外,无水乙醇的用量会显著影响微胶囊的大小均匀度。无水乙醇的用量过少,会导致不能形成大小均匀的微胶囊,还会存在絮状沉淀。无水乙醇的用量过多,会造成无水乙醇的浪费。无水乙醇的用量为6ml/ml时,能获得大小均匀的微胶囊,也不会造成资源浪费。无水乙醇的加入方式也会对微胶囊产生影响,若加入过快容易使壳聚糖形成块状沉淀。因此,滴加无水乙醇时,需逐滴滴加,滴加速度约为1~2滴/秒,此时壳聚糖的凝聚效果较好。
优选地,步骤(2)中,所述交联剂为三聚磷酸钠。现有技术在采用相分离法制备微胶囊时,一般采用戊二醛等二醛类作为交联剂,但戊二醛等交联剂会因残留而导致产生生物毒性。本发明使用无毒的三聚磷酸钠作为交联剂,可减少交联剂对动物和环境的负面影响。优选地,按所述壳聚糖溶液的体积计,所述三聚磷酸钠的用量为1×10-3~3×10-3mol/ml;更优选地,按所述壳聚糖溶液的体积计,所述三聚磷酸钠的用量为2×10-3mol/ml。当三聚磷酸钠采用上述用量时,壳聚糖的交联效果较好,能形成均匀度高的微胶囊。
优选地,步骤(2)中,按所述壳聚糖溶液的体积计,所述kio3的用量为10~30mg/ml;更优选地,按所述壳聚糖溶液的体积计,所述kio3的用量为20mg/ml。
优选地,步骤(2)中,所述kio3与cacl2的质量比为1:0.3。kio3的占比过大,会导致碘酸钾未能完全反应生产碘酸钙,而未反应的碘酸根可能会在后续的洗涤中流失。若kio3的占比过小,则会造成cacl2的浪费。而且,壳聚糖颗粒中存在过量的钙离子时,过量的钙离子会与三聚磷酸根形成三聚磷酸钙等沉淀物,影响微胶囊的交联密度。kio3与cacl2的质量比为1:0.3时,碘酸钾能完全反应生产碘酸钙,防止碘酸根流失,也不会造成cacl2的浪费,体系中也不会产生其它沉淀物,微胶囊的交联密度较大,稳定性较好。
在壳聚糖微胶囊加入海藻酸钠溶液之前,需先将残留于壳聚糖微胶囊上的钙离子洗脱干净,并使壳聚糖微胶囊的表面较为干燥,呈粉状。否则,制得的碘酸钙天然高分子微胶囊会出现结块、团聚等现象,不易分散。而且,壳聚糖微胶囊加入海藻酸钠溶液的过程中,需缓慢加入,不可一次性加入,否则会结块。用浓度为40%~75%的乙醇水溶液对壳聚糖微胶囊进行洗涤,即可有效洗脱壳聚糖微胶囊上的钙离子,还可使壳聚糖微胶囊的表面较为干燥,呈粉状。
优选地,步骤(3)中,所述海藻酸盐溶液的浓度为1~3g/l;更优选地,所述海藻酸盐溶液的浓度为2g/l。本发明通过研究发现,上述浓度的海藻酸盐溶液对壳聚糖微胶囊的包衣效果较好。
作为本发明所述制备方法的一种实施方式,所述具备ph响应性的碘酸钙天然高分子微胶囊的制备方法,包括如下步骤:
(a)制备壳聚糖溶液:用冰醋酸和超纯水配制乙酸溶液,将壳聚糖加入乙酸溶液中,搅拌过夜,得到壳聚糖溶液;
(b)制备壳聚糖微胶囊:将kio3加入步骤(a)制得的壳聚糖溶液中,搅拌分散40~80min,然后加入cacl2,搅拌40~80min,使壳聚糖分子链上的碘酸根与钙离子原位生成碘酸钙,然后逐滴加入无水乙醇,持续搅拌25~45min,使壳聚糖析出,然后加入三聚磷酸盐,交联25~45min后离心分离,滤出壳聚糖微胶囊,用浓度为40%~75%的乙醇水溶液对其进行洗涤;
(c)在搅拌下,将步骤(b)制得的壳聚糖微胶囊加入海藻酸钠溶液中,加入完成后,继续搅拌25~45min,离心分离,滤出产物,干燥,制得所述具备ph响应性的碘酸钙天然高分子微胶囊。
优选地,步骤(a)中:所述乙酸溶液的体积百分比浓度为1%~3%,以所述乙酸溶液的体积计,所述壳聚糖的用量为10~30mg/ml。更优选地,步骤(a)中,所述乙酸溶液的体积百分比浓度为2%,以所述乙酸溶液的体积计,所述壳聚糖的用量为20mg/ml。
优选地,步骤(b)中:以所述壳聚糖溶液的体积计,所述kio3的用量为10~30mg/ml,所述无水乙醇的用量为≥6ml/ml,所述三聚磷酸钠的用量为1×10-3~3×10-3mol/ml;所述kio3与cacl2的质量比为1:0.3。更优选地,步骤(b)中:以所述壳聚糖溶液的体积计,所述kio3的用量为20mg/ml,所述无水乙醇的用量为6ml/ml,所述三聚磷酸钠的用量为2×10-3mol/ml。
优选地,步骤(c)中:海藻酸钠溶液的浓度为1~3g/l,所述干燥为先在40~60℃下鼓风干燥20~30h,然后在25~35℃下真空干燥12h以上。更优选地,步骤(c)中:所述海藻酸钠溶液的浓度为2g/l,所述干燥为先在50℃下鼓风干燥24h,然后在30℃下真空干燥12h。干燥的条件会对微胶囊的颜色及性能产生影响,以本发明的条件进行干燥,不会使微胶囊的颜色变焦黄,也不会影响微胶囊的性能,且干燥效果较好。
本发明还提供了一种具备ph响应性的碘酸钙天然高分子微胶囊,其由上述具备ph响应性的碘酸钙天然高分子微胶囊的制备方法制得。
本发明还提供了所述具备ph响应性的碘酸钙天然高分子微胶囊在饲料制备中的用途。优选地,所述饲料为家畜饲料。
本发明还提供了一种饲料添加剂,其含有所述具备ph响应性的碘酸钙天然高分子微胶囊。
本发明还提供了一种饲料,其含有所述具备ph响应性的碘酸钙天然高分子微胶囊。
与现有技术相比,本发明的有益效果为:
1、本发明以两种不同特性的天然高分子材料对碘酸钙进行双重保护,防止碘酸钙与环境接触,提高其在使用过程中的稳定性,有效降低了碘酸钙的流失,还赋予了碘酸钙天然高分子微胶囊ph响应性,使其在弱碱性环境下的累计释放率高达96.95%,提高了动物对碘元素的吸收效率,也提高了动物对碘元素摄入量的可控性。
2、本发明利用相分离法制备负载碘酸钙的壳聚糖微胶囊,并利用静电自组装法以海藻酸钠包衣壳聚糖微胶囊,制得的碘酸钙天然高分子微胶囊具有较大的交联密度,稳定性好,对碘酸钙的保护性强,制备过程无污染。
3、本发明的碘酸钙天然高分子微胶囊易于保存和使用,具有较好的稳定性,能有效防止其中的碘酸钙流失,实现了碘酸钙的用量可控可调,最大限度地降低了动物碘元素摄入不足或过量的风险,提高了养殖户经济收益。
附图说明
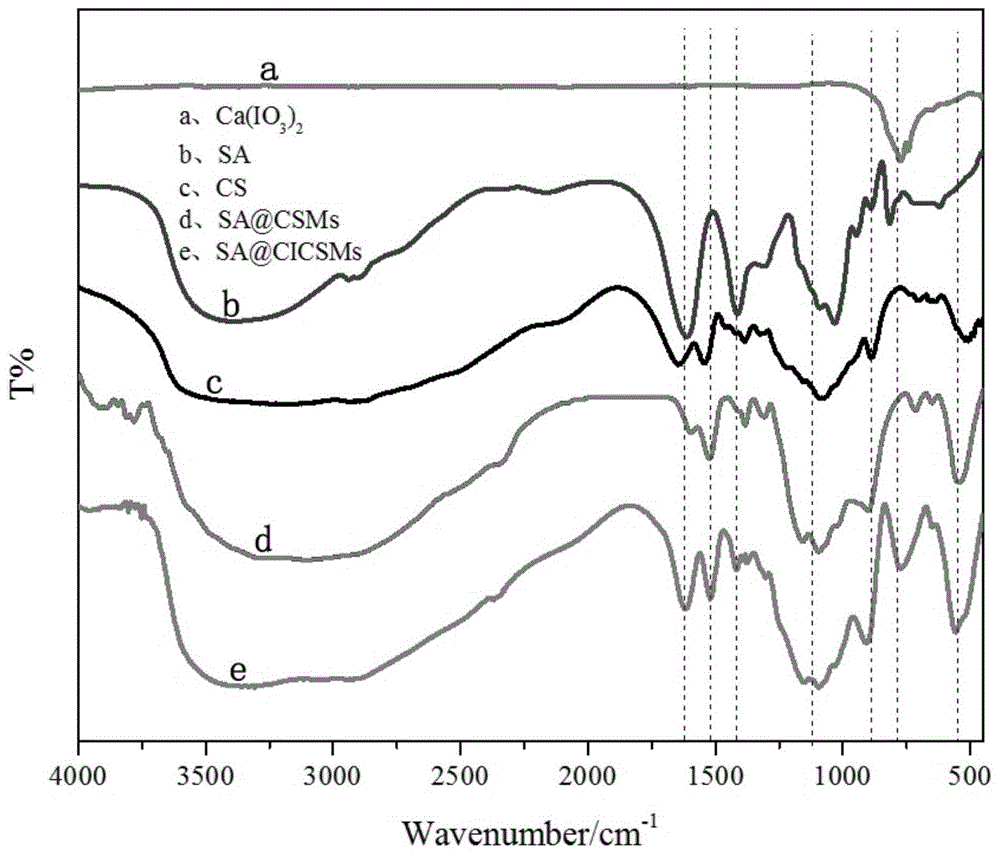
图1为ca(io3)2(a)、sa(b)、cs(c)、sa@csms(d)和sa@cicsms(e)的傅里叶红外图;
图2为sa@csms(a)、csms(b)、sa@cicsms(c)和ca(io3)2·6h2o(d)的xrd图;
图3为sa@cicsms在无酶模拟胃液、超纯水和无酶模拟肠液中的碘累计释放率曲线图;
图4为维生素c的标准曲线图;
图5为sa@cicsms的稳定性图;
图6为sa@cicsms在酸性和碱性环境下的结构示意图。
具体实施方式
为更好的说明本发明的目的、技术方案和优点,本发明通过下列实施例进一步说明。显然,下列实施例仅是本发明的一部分实施例,而不是全部的实施例。应理解,本发明实施例仅用于说明本发明的技术效果,而非用于限制本发明的保护范围。
实施例1
一种具备ph响应性的碘酸钙天然高分子微胶囊的制备方法,其包括如下步骤:
(1)制备壳聚糖溶液:量取49ml超纯水和1ml冰醋酸于容器中,加入1g壳聚糖,搅拌过夜,得到浓度为20mg/ml的壳聚糖溶液;
(2)采用相分离法制备壳聚糖微胶囊:将1gkio3加入步骤(1)制得的壳聚糖溶液中,搅拌分散60min,然后加入0.3gcacl2,搅拌60min,然后逐滴加入300ml无水乙醇,持续搅拌30min,然后加入1×10-3mol三聚磷酸钠,交联30min后以3000r·min-1的转速离心10min,滤出固体产物(即壳聚糖微胶囊),用浓度为50%的乙醇水溶液对壳聚糖微胶囊进行洗涤,至滤液中无钙离子;
(3)在搅拌下,将步骤(2)制得的壳聚糖微胶囊缓慢加入100ml2g/l的海藻酸钠溶液中,加入完成后,继续温和搅拌30min,然后以3000r·min-1的转速离心10min,滤出固体产物,在50℃下鼓风干燥24h,然后在30℃下真空干燥12h,制得所述具备ph响应性的碘酸钙天然高分子微胶囊。
实施例2
一种具备ph响应性的碘酸钙天然高分子微胶囊的制备方法,其包括如下步骤:
(1)制备壳聚糖溶液:量取99ml超纯水和1ml冰醋酸于容器中,加入1g壳聚糖,搅拌过夜,得到浓度为10mg/ml的壳聚糖溶液;
(2)采用相分离法制备壳聚糖微胶囊:将1gkio3加入步骤(1)制得的壳聚糖溶液中,搅拌分散40min,然后加入0.3gcacl2,搅拌40min,然后逐滴加入600ml无水乙醇,持续搅拌25min,然后加入1×10-3mol三聚磷酸钠,交联25min后以3000r·min-1的转速离心10min,滤出固体产物(即壳聚糖微胶囊),用浓度为40%的乙醇水溶液对壳聚糖微胶囊进行洗涤,至滤液中无钙离子;
(3)在搅拌下,将步骤(2)制得的壳聚糖微胶囊缓慢加入100ml1g/l的海藻酸钠溶液中,加入完成后,继续温和搅拌25min,然后以3000r·min-1的转速离心10min,滤出固体产物,在40℃下鼓风干燥20h,然后在25℃下真空干燥14h,制得所述具备ph响应性的碘酸钙天然高分子微胶囊。
实施例3
一种具备ph响应性的碘酸钙天然高分子微胶囊的制备方法,其包括如下步骤:
(1)制备壳聚糖溶液:量取97ml超纯水和3ml冰醋酸于容器中,加入3g壳聚糖,搅拌过夜,得到浓度为30mg/ml的壳聚糖溶液;
(2)采用相分离法制备壳聚糖微胶囊:将3gkio3加入步骤(1)制得的壳聚糖溶液中,搅拌分散80min,然后加入0.9gcacl2,搅拌80min,然后逐滴加入600ml无水乙醇,持续搅拌45min,然后加入3×10-3mol三聚磷酸钠,交联45min后以3000r·min-1的转速离心10min,滤出固体产物(即壳聚糖微胶囊),用浓度为75%的乙醇水溶液对壳聚糖微胶囊进行洗涤,至滤液中无钙离子;
(3)在搅拌下,将步骤(2)制得的壳聚糖微胶囊缓慢加入100ml3g/l的海藻酸钠溶液中,加入完成后,继续温和搅拌45min,然后以3000r·min-1的转速离心10min,滤出固体产物,在60℃下鼓风干燥30h,然后在35℃下真空干燥12h,制得所述具备ph响应性的碘酸钙天然高分子微胶囊。
实验例
制备空白样品:除不加kio3外,其余步骤依照实施例1的制备方法,得到sa@csms。
词语解释:sa为海藻酸钠;cs为壳聚糖;sa@cicsms为本发明实施例制得的碘酸钙天然高分子微胶囊;sa@csms为空白样品,即不含碘的天然高分子微胶囊;csms为不含碘的壳聚糖微胶囊;cicsms为含碘的壳聚糖微胶囊。
一、红外分析
图1为ca(io3)2(a)、sa(b)、cs(c)、sa@csms(d)和sa@cicsms(e)的傅里叶红外图。
结果分析:图1中,红外谱线a、b、c、d、e分别表示碘酸钙、sa、cs、sa@csms和sa@cicsms的傅里叶红外谱线。谱线a中,779cm-1处有强吸收峰出现,此为io3-伸缩振动峰。谱线b中,在3400cm-1附近出现sa的—oh伸缩振动峰峰,1600cm-1附近为sa的c=o伸缩振动峰,在1430cm-1和1022cm-1附近为sa的羧基伸缩带(—coo和—c—o)伸缩振动峰。谱线c中,在3450cm-1附近是o—h、n—h的伸缩振动峰,在1646cm-1附近是cs的酰胺ⅰ峰即c=o伸缩振动峰、酰胺ⅱ峰即n—h弯曲振动;在1388cm-1附近是ch3的弯曲振动峰;在1088cm-1附近是多糖的c—oh的伸缩振动峰;在891cm-1附近是β-构型糖苷键的特征吸收峰。谱线d中,cs与sa反应后,吸收峰从1523cm-1偏移到1542cm-1,吸收峰从1383cm-1偏移到1387cm-1处,—oh和—nh2伸缩振动偏移到3470cm-1,同时峰型变宽;表明,sa中的—cooh与cs中—nh2间存在静电作用力。谱线e中,io3-的特征峰出现在773cm-1处,说明碘酸钙嵌入sa@cicsms中。谱线e与谱线d中对应吸收峰对比,峰值发生偏移,说明碘酸钙在sa@cicsms内与sa和cs存在相互作用。
二、碘酸根存在形式分析
图2为sa@csms(a)、csms(b)、sa@cicsms(c)和ca(io3)2·6h2o(d)的xrd图。
结果分析:图2中,曲线a、b、c和d分别表示sa@csms、csms、sa@cicsms和ca(io3)2·6h2o标准卡的衍射曲线。衍射曲线a中,衍射峰2θ=18.5°消失,在2θ=15至30°之间出现一个宽的衍射峰中,而cs的衍射峰几近湮灭。衍射曲线b中,在2θ=18.5°和24.9°处出现较为宽泛的衍射峰,说明cs是结晶性的天然高分子,其中—oh和—nh2在cs内形成较强的分子间和分子内作用力。sa的加入破坏了cs的结晶结构,导致cs的结晶度降低。衍射曲线c中,特征衍射峰2θ=19.2°、25.6°、28.7°和34.3°,与ca(io3)2·6h2o标准卡的特征衍射峰相似。但衍射峰位置和强度有差异,表明sa包裹cicsms的过程中,进一步破坏了ca(io3)2·6h2o的晶型。
三、释放特性分析
图3为sa@cicsms在无酶模拟胃液、超纯水和无酶模拟肠液中的碘累计释放率曲线图。
结果分析:图3中,曲线a、b、c分别表示sa@cicsms在无酶模拟胃液、超纯水以及无酶模拟肠液中的碘累计释放率曲线。sa@cicsms在无酶模拟胃液、超纯水以及无酶模拟肠液中的最大累计释放率分别为:79.78%、89.37%和96.95%,达到最大累计释放率所用时间为1740min。sa@cicsms在无酶模拟肠液中释放碘酸钙的累计释放率明显大于其在水环境或无酶模拟胃液环境中的累计释放率。同时,sa@cicsms在无酶模拟肠液中释放ca(io3)2的速率高于其在超纯水和无酶模拟胃液中的释放速率。表明,sa@cicsms具有较好的肠道响应性释放特性。sa@cicsms在模拟胃肠液中释放碘酸钙的行为与cicsms在模拟胃肠液中释放碘酸钙的行为截然相反,该现象主要与sa@cicsms的结构、成分以及药物释放环境相关。sa@cicsms形成过程中主要通过sa与cicsms之间的静电作用力进行结合。此外,sa与cicsms存在少量的离子间作用力。sa和cicsms之间相互作用力相对较弱,易在环境中受到破坏。其次,在碱性或者中性的环境中,sa中的—cooh与cs中的—nh2发生去质子化。因而增强了sa和cs分子内与分子间的静电斥力,使得sa@cicsms内部结构疏松,加快了碘酸钙的释放。此外,由于无酶模拟肠液中存在磷酸二氢根、磷酸氢根和磷酸根等带负电的离子,在渗透压的作用下渗透至sa@cicsms内部,增大了sa和cicsms的分子内与分子间的静电斥力。因而,sa@cicsms在ph=6.8的无酶模拟肠液中显示出更高的碘酸钙累计释放率和更快的碘酸钙释放速度。
四、稳定性分析
根据下表式1和式2计算并称取ca(io3)23.7mg、sa@cicsms57.0mg以及sa@csms57.0mg(其中,所称取质量的sa@cicsms中含有的碘酸钙的质量为3.7mg),将称取的样品分别用透析袋包好后分别置于装有100ml0.5mol/l的nacl溶液的棕色锥形瓶内。将样品放进30℃恒温振荡箱,避光震荡,每隔一定时间分别移取1.00ml试液于10ml棕色容量瓶中,同时补充1.00ml0.5mol/l的nacl溶液于棕色锥形瓶中。用0.5mol/l的nacl溶液定容后摇匀,并测定维生素c的吸光度。同时做空白实验,除不加sa@csms、ca(io3)2和sa@cicsms外,其余操作保持不变。碘酸根与维生素c的离子反应式及维生素c的分解率计算公式如下表所示。
式1中,6.25为转换系数,c为紫外光谱测试中样品的浓度,mol/l。
维生素c在此分析条件下浓度与吸光度的关系为c=0.08389a+0.00891,r2=0.9994。维生素c的浓度在0~10μg/ml范围内其浓度与吸光度间呈线性关系,图4为维生素c的标准曲线图。
图5为sa@cicsms的稳定性图,图中,曲线a、b、c、d分别是对比维生素c、sa@csms、ca(io3)2以及sa@cicsms在实验条件下维生素c的分解规律。考察sa@cicsms中碘酸钙的稳定性。如图所示,试验条件下维生素c盐溶液中维生素c的降解几乎可以忽略,说明维生素c在测试条件下内具有较高的稳定性,可满足探究sa@cicsms在还原性物质中稳定性测试的需求。此外,在将sa@csms加入维生素c盐溶液后,样液中维生素c的分解率与维生素c盐溶液的分解率相近,表明sa@csms不会促进维生素c盐溶液中维生素c的分解。然而,在维生素c盐溶液中分别加入碘酸钙和sa@cicsms后,维生素c的分解率明显加快。在体系中,碘酸钙和维生素c发生氧化还原反应,导致维生素c浓度迅速下降。其二者加入维生素c盐溶液后,维生素c分解50%(t50%)用时分别为177.1和225.8min,说明sa@cicsms能够有效改善碘酸钙在还原性物质中的化学稳定性。
图6为sa@cicsms在酸性和碱性环境下的结构示意图。从图中可看出,在酸性环境下,壳聚糖在微胶囊的内部溶胀,而海藻酸钠则在微胶囊的外部收缩,同时,海藻酸钠与壳聚糖之间的作用力增强。所以,在酸性环境下,sa@cicsms的内部较为致密,碘酸钙的释放率较低。在弱碱性环境下,壳聚糖的溶胀受阻,而海藻酸钠的溶胀增大,此时壳聚糖与海藻酸钠之间的静电作用力变弱,sa@cicsms的结构变疏松,更利于碘酸钙的释放。所以,在弱碱性环境下,碘酸钙的释放率较高。
最后应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
- 还没有人留言评论。精彩留言会获得点赞!