降低排尿频率的药物制剂及其使用方法与流程
本发明总体涉及用于抑制膀胱平滑肌的方法和组合物,特别是用于降低排尿频率的方法和组合物。
背景技术:
逼尿肌(detrusor muscle)是一层由排列成螺旋束、纵向束和环形束的平滑肌纤维组成的膀胱壁。当膀胱牵张时,这指示副交感神经系统使逼尿肌收缩。这促使膀胱经由尿道排出尿液。
为使尿液离开膀胱,自动控制的内括约肌和自主控制的外括约肌均必须打开。这些肌肉出现问题可导致失禁。若尿液量达到膀胱绝对容量的100%,则自主括约肌将变得不自主且尿液将即刻排出。
成人膀胱通常容纳约300至350毫升尿液(工作容积),但充盈的成人膀胱可容纳高达约1000毫升(绝对容积)的尿液,个体间存在差异。尿液积聚时,由膀胱壁折叠产生的褶皱(rugae)变平且膀胱壁随其牵张而变薄,使得膀胱可储存较大量的尿液,而不会使内部压力显著上升。
在大多数个体中,通常在膀胱中的尿液体积达到约200毫升时开始产生尿意。在此阶段,如果需要,个体易于抗拒排尿冲动。随着膀胱继续填充,尿意变强且变得更难忽略。最终,膀胱将填充至排尿冲动无法抗拒的点,此时个体将无法再置之不理。
在一些个体中,当膀胱中的尿液达到小于其工作容积的100%时即开始产生此尿意。这种增强的尿意可干扰正常活动,包括充分不间断的休息期间的睡眠能力。在一些情形下,这种增强的尿意可能与医学疾病相关,如男性的良性前列腺增生或前列腺癌或女性的妊娠。然而,在未受其他医学疾病影响的男性和女性个体中亦会发生尿意增强现象。
在一些个体中,如儿童,可能由于对膀胱肌缺乏控制而发生非自主排尿(例如,尿床)。在其他个体中,可能由于潜在医学疾病而发生非自主排尿(例如,尿失禁)。
因此,存在对用于治疗罹患非期望排尿频率的男性和女性个体的组合物和方法的需求。
技术实现要素:
本发明的一方面涉及一种降低个体的排尿频率的方法。此方法包括对患有导致非期望排尿频率的疾病的个体施用有效量的药学组合物,该药学组合物包含一种或多种前列腺素途径(prostaglandin pathway)抑制剂。
本发明的另一方面涉及一种用于治疗导致非期望排尿频率的疾病的药学组合物。该药学组合物包含一种或多种前列腺素途径抑制剂和药学上可接受的载体。
附图说明
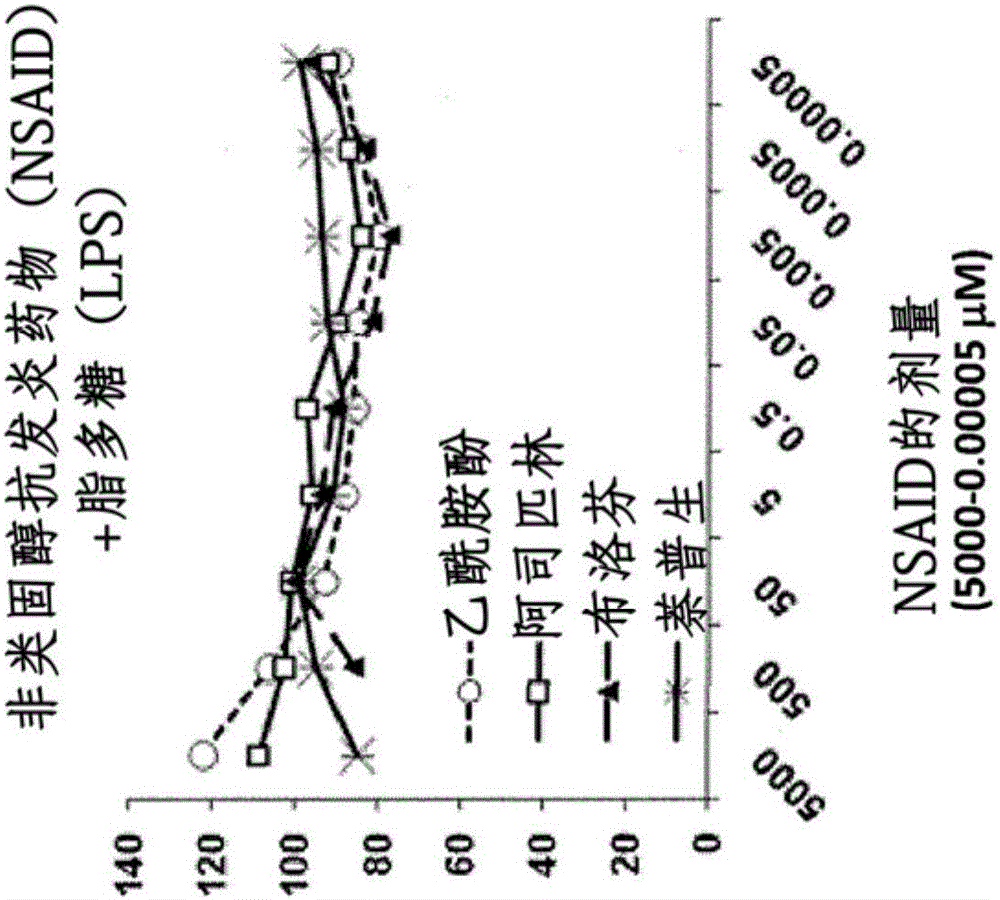
图1A和图1B是显示了在脂多糖(LPS)不存在(图1A)或存在(图1B)的情况下止痛剂调节Raw 264巨噬细胞对协同刺激分子的表达的图表。在止痛剂单独存在或与鼠伤寒沙门氏菌(Salmonela typhimurium)LPS(0.05微克/毫升(μg/ml))一起存在的情况下,将细胞培养24小时(hr)。结果为CD40+CD80+细胞的平均相对%。
具体实施方式
呈现以下详细说明以使得本领域技术人员可实施和利用本发明。为解释目的,陈述特定术语以提供对本发明的完全理解。然而,本领域技术人员将显而易见的是,实践本发明并不需要这些特定细节。关于特定应用的描述仅提供作为代表性实施例。本发明旨不在受限于所示的实施例,而在具有与本文公开的原理和特征一致的可能最宽泛的范畴。
本文所用的术语“前列腺素(prostaglandin,PG)”指经酶促作用(enzymatically)由脂肪酸衍生出的在动物体内具有各种生理学作用(如调节平滑肌组织的收缩和舒张)的一组脂质化合物。每个前列腺素含有20个碳原子,包括一个5-碳环。前列腺素的实例包括前列腺素E1(PGE1)、前列腺素E2(PGE2)、前列腺素D2、前列腺素I2(PGI2,前列腺环素)和前列腺素F2α(PGF2α)。
本文所用的术语“前列腺素(PG)途径抑制剂”指与在PG的合成或PG对靶组织的作用中所涉及的一种或多种组分直接或间接相互作用、且干扰前列腺素的水平或其对靶组织最终作用的试剂。PG途径抑制剂包括,但不限于PG抑制剂、前列腺素转运子(prostaglandin transporter,PGT)抑制剂和前列腺素受体(prostaglandin receptor,PGR)抑制剂。然而,术语“PG途径抑制剂”不包括下文定义的止痛剂。
本文所用的术语“PG抑制剂”包括,但不限于PG合成抑制剂和PG活性抑制剂。本文所用的术语“PG合成抑制剂”指抑制前列腺素产生的试剂,如抑制磷脂酶A2、前列腺素合成酶和组织特异性异构酶以及合成酶如凝血脂素(thromboxane)合成酶、PGF合成酶、细胞溶质PG合成酶(cytosolic PG synthase,cPGES)、前列腺素I合成酶(prostaglandin I synthase,PGIS)和微粒体PGES酶(microsomal PGES enzymes,mPGES))的表达或活性的试剂。PG合成抑制剂的实例包括氟尼辛葡甲胺(flunixin meglumine)。本文所用的术语“PG合成的抑制剂(inhibitors of PG synthesis)”或“PG合成抑制剂(PG synthesis inhibitors)”不包括下文定义的止痛剂。
本文所用的术语“PG活性抑制剂”指以任何方式拮抗前列腺素本身的作用的试剂。仅干扰前列腺素的合成,如通过干扰前列腺素合成酶的作用,但不干扰前列腺素的作用的试剂不包括在本说明书中所用的PG活性抑制剂的定义内。
本文所用的术语“PGT抑制剂”指抑制PG转运子如ATP依赖型多药抗药性(MDR)转运子-4(ATP dependent multi-drug resistance transporter-4)或其他MDR通道(MDR channels)如ABCC1、ABCC2、ABCC3、ABCC6、ABCG2和ABCB11的表达或活性的试剂。抑制PGT活性的PGT抑制剂的实例包括但不限于,抑制MDR膜泵的化合物,如三嗪化合物(triazine compounds)、维拉帕米(verapamil)和钙通道阻断剂;通道包括奎尼丁(quinidine)、酮康唑(ketoconazole)、伊曲康唑(itraconazole)、阿奇霉素(azithromycin)、伐司扑达(valspodar)、环孢素(cyclosporine)、依克立达(elacridar)、烟曲霉毒素C(fumitremorgin-C)、吉非替尼(gefitinib)和红霉素(erythromycin)。抑制PGT表达的PGT抑制剂的实例包括,但不限于通过靶向启动子区和/或与启动子或其他基因控制区结合的转录因子来控制MDR基因转录的试剂。本文所用的术语“PGR抑制剂”指抑制PGR的活性或表达的试剂。在一些实施例中,PGR包含PGE受体的E前列腺素类受体(E prostanoid receptor,EP)1、EP2、EP3和EP4亚型;PGD受体(DP1);PGF受体(FP);PGI受体(IP)和凝血脂素受体(TP)。人类TP的两种其他异形体(TPα与TPβ)和FP的两种其他异型体(FPA与FPB)和八种EP3变体均经由选择性剪切而产生,其仅在C端尾部不同。在一些实施例中,PGR还包含G蛋白偶联受体(G protein-coupled receptor),其被称为趋化因子受体同源分子(chemo-attractant receptor-homologous molecule,CRHME)。在其他实施例中,PGR包括使类视紫质7次跨膜G蛋白偶联受体(rhodopsin-like 7-transmembrane-spanning G protein-coupled receptor)活化的所有受体。
PGR活性抑制剂的实例包括,但不限于抗PGR抗体和抑制G蛋白偶联受体(G-protein coupled receptor)信号传导途径的任何试剂。PGR表达抑制剂包括在转录水平、翻译水平或转录后水平抑制PGR表达的试剂。PGR表达抑制剂的实例包括,但不限于抗-PGR siRNA和miRNA。
本文所用的术语“有效量”意指达成所选结果所必要的量。
本文所用的术语“止痛剂”指用于缓解疼痛且包括抗发炎化合物的试剂、化合物或药物。例示性止痛和/或抗发炎的试剂、化合物或药物包括,但不限于非类固醇抗发炎药物(NSAID)、水杨酸酯(salicylate)、阿司匹林(aspirin)、水杨酸(salicylic acid)、水杨酸甲酯(methyl salicylate)、二氟尼柳(diflunisal)、双水杨酸酯(salsalate)、奥沙拉嗪(olsalazine)、柳氮磺胺吡啶(sulfasalazine)、对氨基苯酚(para-aminophenol)衍生物、乙酰苯胺(acetanilide)、乙酰氨基酚(acetaminophen)、非那西汀(phenacetin)、芬那酸(fenamate)、甲芬那酸(mefenamic acid)、甲氯芬那酸(meclofenamate)、甲氯芬那酸钠(sodium meclofenamate)、杂芳基乙酸(heteroayl acetic acid)衍生物、托美汀(tolmetin)、酮咯酸(ketorolac)、双氯芬酸(diclofenac)、丙酸(propionic acid)衍生物、布洛芬(ibuprofen)、萘普生钠(naproxen sodium)、萘普生(naproxen)、非诺洛芬(fenoprofen)、酮洛芬(ketoprofen)、氟比洛芬(flurbiprofen)、奥沙普嗪(oxaprozin);烯醇酸(enolic acids)、昔康(oxicam)衍生物、吡罗昔康(piroxicam)、美洛昔康(meloxicam)、替诺昔康(tenoxicam)、安吡昔康(ampiroxicam)、屈昔康(droxicam)、吡沃昔康(pivoxicam)、吡唑啉酮(pyrazolon)衍生物、苯基丁氮酮(phenylbutazone)、羟基保泰松(oxyphenbutazone)、安替吡啉(antipyrine)、氨基吡啉(aminopyrine)、安乃近(dipyrone)、昔布类(coxibs)、塞来昔布(celecoxib)、罗非昔布(rofecoxib)、萘丁美酮(nabumetone)、阿扎丙宗(apazone)、吲哚美辛(indomethacin)、舒林酸(sulindac)、依托度酸(etodolac)、异丁基苯基丙酸(isobutylphenyl propionic acid)、罗美昔布(lumiracoxib)、依托昔布(etoricoxib)、帕瑞昔布(parecoxib)、伐地昔布(valdecoxib)、替拉昔布(tiracoxib)、依托度酸(etodolac)、达布非龙(darbufelone)、右旋酮洛芬(dexketoprofen)、醋氯芬酸(aceclofenac)、利克飞龙(licofelone)、溴芬酸(bromfenac)、氯索洛芬(loxoprofen)、普拉洛芬(pranoprofen)、吡罗昔康(piroxicam)、尼美舒利(nimesulide)、西唑利啉(cizolirine)、3-甲酰胺基-7-甲磺酰胺基-6-苯氧基-4H-1-苯并哌喃-4-酮(3-formylamino-7-methylsulfonylamino-6-phenoxy-4H-1-benzopyran-4-one)、美洛昔康(meloxicam)、氯诺昔康(lornoxicam)、d-吲哚布芬(d-indobufen)、莫苯唑酸(mofezolac)、哌氨托美汀(amtolmetin)、普拉洛芬(pranoprofen)、托芬那酸(tolfenamic acid)、氟比洛芬(flurbiprofen)、舒洛芬(suprofen)、奥沙普嗪(oxaprozin)、扎托洛芬(zaltoprofen)、阿米洛芬(alminoprofen)、噻洛芬酸(tiaprofenic acid)、其药理学盐、其水合物和其溶剂化物。
本文所用的术语“昔布类(coxib)”指能抑制COX1和COX2酶的活性或表达的化合物或多种化合物的组合物。
本文所用的术语“衍生物”指经化学改性的化合物,其中该改性是具有一般技能的化学家所常规认为的,如酸的酯或酰胺,或保护基,如用于醇或硫醇的苄基或用于胺的叔丁氧羰基(tert-butoxycarbonyl group)。
本文所用的术语“类似物”指包含特定化合物或其种类的化学改性形式且保持该化合物或种类的医药学和/或药理学活性特征的化合物。
本文所用的术语“药学上可接受的盐”指所公开化合物的衍生物,其中母化合物(parent compound)通过制成其酸盐或碱盐来改性。药学上可接受的盐的实例包括,但不限于碱性残基的无机或有机酸盐如胺、酸性残基的碱盐或有机盐如羧酸、和其类似物。药学上可接受的盐包括由例如无毒无机酸或有机酸形成的母化合物的习知无毒盐或季铵盐。举例而言,这些习知无毒盐包括由无机酸如盐酸、氢溴酸、硫酸、氨基磺酸、磷酸、硝酸和其类似物衍生的那些盐,和由有机酸如乙酸、丙酸、丁二酸、乙醇酸、硬脂酸(stearic acid)、乳酸、苹果酸(malic acid)、酒石酸、柠檬酸、抗坏血酸、帕莫酸(pamoic acid)、顺丁烯二酸(maleic acid)、羟基顺丁烯二酸(hydroxymaleic acid)、苯乙酸、谷氨酸、苯甲酸、水杨酸、对氨基苯磺酸(sulfanilic acid)、2-乙酰氧基苯甲酸(2-acetoxybenzoic acid)、反丁烯二酸(fumaric acid)、甲苯磺酸、甲烷磺酸、乙烷二磺酸(ethane disulfonic acid)、草酸(oxalic acid)、羟乙磺酸(isethionic acid)和其类似物所制备的盐。
本文所用的词组“医药学上可接受”涉及化合物、材料、组合物和/或剂型的使用,它们在合理医学判断的范畴内适合与人类和动物的组织接触使用而按照合理的益处/风险比为无过度毒性、刺激、过敏反应或其他问题或并发症。
本文使用术语“即刻释放(immediate-release)”指不含溶解速度控制材料的药物制剂。在施用即刻释放的制剂后,活性剂的释放基本上不延迟。即刻释放涂层可包括在施用后即刻溶解以释放其中的药物内容物的合适材料。在一些实施例中,使用术语“即刻释放”意指在对患者施用后小于10分钟(min)、20分钟、30分钟、40分钟、50分钟、60分钟、90分钟或120分钟内释放活性成分的药物制剂。
本文所用的术语“延长释放(extended-release)”(亦称为缓释(sustained-release;SR)、持续作用(sustained-action,SA)、定时释放(time-release,TR)、受控释放(controlled-release,CR)、改性释放(modified release,MR)或连续释放(continuous-release,CR))指用于医学片剂或胶囊的随时间缓慢溶解并释放活性成分的机制。延长释放片剂或胶囊的优势在于,其常可比相同药物的即刻释放制剂以较少频率服用且其保持药物在血流中更稳定的水平,由此延长药物作用的持续时间且降低药物在血流中的峰值量。在一些实施例中,术语“延长释放”指片剂或胶囊中的活性成分在施用于患者后在2、3、4、5、6、7、8、9、10、12、14、16、18、20、22或24小时的时间段(period)内连续或脉冲式地释放的释放概况。
本文所用的术语“延迟释放(delayed-release)”指在施用药学组合物后,药学组合物的活性成分的释放被延迟或延后一个给定的时间段(例如,1、2、3、4或5个小时,或在胃部之后)的药物释放概况。
本文所用的术语“延迟延长释放(delayed-extended-release)”指药学组合物的活性成分的释放在施用药学组合物后被延迟或延后一个给定的时间段(例如,1、2、3、4或5个小时的迟滞期(lag period),或在胃部之后)的药物释放概况。一旦开始释放,活性成分即随时间(例如,在2、3、4、5、6、7、8、9、10、12、14、16、18、20、22或24个小时的时间段内)连续或脉冲式地缓慢释放。
降低排尿频率的方法
本发明的一方面涉及一种通过对患有导致非期望排尿频率的疾病的个体施用有效量的药学组合物来降低排尿频率的方法。该药学组合物包含一种或多种PG途径抑制剂和药学上可接受的载体。导致非期望排尿频率的疾病包括,但不限于夜尿症(nocturia)、膀胱过动症(overactive bladder)、尿失禁(urinary incontinence)和尿床(bed wetting)。
在一些实施例中,PG抑制剂是PG合成酶的抑制剂。在其他实施例中,PG抑制剂是PG活性抑制剂。PG活性抑制剂的实例包括,但不限于阻断PG与其任何受体(EP1、EP2、EP3、EP4、DP1、DP2、FP2、IP和TP)结合的试剂。这些类型抑制剂的实例包括,但不限于由罗氏(Roche)研发的IP受体抑制剂RO3244019、EP1受体拮抗剂ONO-85-39、EP1与EP2双重受体拮抗剂AH 6809和EP4拮抗剂RQ-15986。在某些实施例中,该一种或多种PG途径抑制剂包含PGT抑制剂。在一些实施例中,PGT抑制剂为PGT活性抑制剂。PGT活性抑制剂的实例包括,但不限于抗PGT的抗体、和任何已知可抑制显示转运PG的ATP依赖型多药抗药性转运子-4或相关MDR泵的化合物。在其他实施例中,PGT抑制剂为PGT表达抑制剂。PGT表达抑制剂的实例包括,但不限于抗-PGT的siRNA、靶向PGT mRNA的反义RNA、和通过影响DNA甲基化和/或染色质修饰(chromatin modification)来控制基因转录的试剂。
在一些实施例中,该一种或多种PG途径抑制剂包含靶向在COX1与COX2两者中所含的COX活性位点与POX活性位点的抑制剂。在其他实施例中,该一种或多种PG途径抑制剂包含抑制PGE2途径的抑制剂。
在某些实施例中,该一种或多种PG途径抑制剂包含PGR抑制剂。PGR是含有七个跨膜域的G蛋白偶联受体(G-protein-coupled receptors)。PGR的实例包括EP1、EP2、EP3、EP4、DP1、DP2、FP、IP1、IP2、CRTH2和TP受体。在一些实施例中,该一种或多种PG途径抑制剂包含抑制上文所列任何PG受体的抑制剂。在一些实施例中,PGR抑制剂为PGR活性抑制剂。PGR活性抑制剂的实例包括,但不限于抗PGR的抗体。在一些实施例中,PGR抑制剂为PGE2受体活性抑制剂,如EP1活性抑制剂、EP2活性抑制剂、EP3活性抑制剂或EP4活性抑制剂。
在其他实施例中,PGR抑制剂为PGR表达抑制剂。PGR表达抑制剂的实例包括,但不限于抗-PGR的siRNA、靶向PGR mRNA的反义RNA、或通过影响DNA甲基化和/或染色质修饰来控制基因转录的试剂。在一些实施例中,PGR表达抑制剂为PGE2受体表达抑制剂,如EP1表达抑制剂、EP2表达抑制剂、EP3表达抑制剂或EP4表达抑制剂。在一些实施例中,该一种或多种PG途径抑制剂包含小分子抑制剂。本文所用的术语“小分子抑制剂”指具有1000道尔顿(dalton)或1000道尔顿以下的分子量的抑制剂。
在一些实施例中,PG途径抑制剂包含短干扰RNA(siRNA)。siRNA为可经工程设计以诱发对应于PG途径组分的mRNA的序列特异性转录后基因沉默(gene silencing)的双链RNA。siRNA采用RNA干扰(RNAi)机制,用于使基因例如被靶向的PGE2受体基因的表达“沉默”的目的。最初在将双链RNA(dsRNA)转染至细胞中的情形下观测到这种“沉默”。在进入其中时,发现dsRNA经类Rnase-III酶Dicer切割成长度为21至23个核苷酸的在其3’端含有2个核苷酸突出的双链小干扰RNA(siRNA)。在ATP依赖性步骤中,siRNA被整合成多次单元RNAi诱导型沉默复合体(RNAi induced silencing complex,RISC),该复合体给出用于AGO2介导的mRNA互补序列切割的信号,继而导致随后被细胞核酸外切酶(cellular exonucleases)降解。
在一些实施例中,PG途径抑制剂包含合成的siRNA或靶向靶细胞/组织中的PG合成酶RNA、PGT RNA或PGR RNA的其他种类的小RNA。合成产生的siRNA在结构上仿真通常在细胞中由酶Dicer加工的siRNA类型。合成产生的siRNA可向RNA结构引入已知可增强siRNA稳定性和功能性的任何化学修饰。举例而言,在一些情形下,siRNA可合成为锁核酸(locked nucleic acid,LNA)修饰的siRNA。LNA为含有连接核糖的2’-氧与4’碳的亚甲基(methylene)桥的核苷酸类似物。双环结构将LNA分子的呋喃糖环(furanose ring)锁定为3’-内构型(3'-endo conformation),由此在结构上仿真标准RNA单体。
在其他实施例中,PG途径抑制剂包含表达载体,其被工程设计成转录在细胞内经加工成被靶向的siRNA的短双链类发夹RNA(short double-stranded hairpin-like RNA,shRNA)。可使用如Ambion的siRNA构建试剂盒、Imgenex的GENESUPPRESSORTM构建试剂盒和Invitrogen的BLOCK-ITTM可诱导型RNAi质体与慢病毒载体(lentivirus vectors)在合适的表达载体中克隆shRNA。合成的siRNA与shRNA可使用熟知的算法来设计且使用习知的DNA/RNA合成仪来合成。
在一些实施例中,次级活性剂(secondary active agent)包含能够抑制PG途径组分的表达的反义寡核苷酸或多核苷酸。反义寡核苷酸或多核苷酸可包含DNA主链、RNA主链或其化学衍生物。在一个实施例中,反义寡核苷酸或多核苷酸包含靶向降解的单链反义寡核苷酸或多核苷酸。在某些实施例中,抗发炎剂包含与PG途径组分的mRNA序列互补的单链反义寡核苷酸。单链反义寡核苷酸或多核苷酸可被合成产生或可由合适的表达载体来表达。反义核酸被设计为经由互补结合来与mRNA正义链(sense strand)结合以促进RNase H活性(其导致mRNA降解)。反义寡核苷酸优选在化学或结构上经修饰以提高核酸酶稳定性和/或增强结合。
在一些实施例中,反义寡核苷酸经修饰以产生具有非常规化学或主链添加或取代的寡核苷酸,包括,但不限于肽核酸(PNA)、锁核酸(LNA)、吗啉基(morpholino)主链核酸、甲基膦酸酯(methylphosphonates)、双重稳定芪或芘基封端(duplex stabilizing stilbene or pyrenyl caps)、硫代磷酸酯(phosphorothioates)、氨基磷酸盐(phosphoroamidates)、磷酸三酯(phosphotriesters)和其类似物。举例而言,经修饰的寡核苷酸可将一种或多种天然产生的核苷酸与类似物合并或以类似物取代一种或多种天然产生的核苷酸;核苷酸间的修饰引入有例如不带电荷键联(例如,磷酸甲酯(methyl phosphonate)、磷酸三酯(phosphotriester)、磷酰胺酯(phosphoamidate)、氨基甲酸酯(carbamates)等)或带电荷键联(例如,硫代磷酸酯(phosphorothioate)、二硫代磷酸酯(phosphorodithioates)等);修饰引入有嵌入剂(例如,吖啶(acridine)、补骨脂素(psoralen)等)、螯合剂(例如,金属、放射性金属、硼、氧化性金属等)或烷化剂和/或经修饰的键联(例如,α变旋异构核酸(alpha anomeric nucleic acids)等)。
在一些实施例中,单链寡核苷酸经内部修饰以在其主链中包括至少一个中性电荷。举例而言,寡核苷酸可包括与特异性靶序列互补的甲基磷酸酯主链或肽核酸(PNA)。已发现这些修饰预防或减少解旋酶介导的解旋。使用不带电荷的探针还可通过减轻典型杂交中的负电荷核酸链的排斥来增加试样中与多核苷酸靶的杂交速率。
PNA寡核苷酸是磷酸二酯主链经聚酰胺置换的不带电荷的核酸类似物,使得PNA成为由酰胺键将2-氨乙基-甘氨酸(2-aminoethyl-glycine)单元结合在一起的聚合物。使用与标准肽合成中使用的相同Boc或Fmoc化学方法来合成PNA。碱基(腺嘌呤、鸟嘌呤、胞嘧啶和胸腺嘧碇)通过亚甲基羧基键联(methylene carboxyl linkage)与主链连接。因此,PNA为非环状(acyclic)、非手性(achiral)且中性的。PNA的其他特性为相比于核酸提高的特异性和解链温度,形成三链螺旋的能力,在酸性pH下的稳定性,无法被如核酸酶、聚合酶等细胞酶识别。
含甲基磷酸酯的寡核苷酸为中性DNA类似物,含有取代了其中一个非键合磷酰基氧(phosphoryl oxygens)的甲基。具有甲基磷酸酯键联的寡核苷酸首先被报导经由翻译的反义阻断来抑制蛋白质合成。
在一些实施例中,该寡核苷酸中的磷酸酯主链可含有硫代磷酸酯键联或磷酰胺酯。这些寡核苷酸键联的组合亦在本发明的范畴内。
在其他实施例中,该寡核苷酸可含有由磷酸二酯核苷酸间键联接合的经修饰糖的主链。该经修饰的糖可包括呋喃糖(furanose)类似物,包括,但不限于2-脱氧核呋喃糖苷(2-deoxyribofuranosides)、α-D-阿拉伯呋喃糖苷(α-D-arabinofuranosides)、α-2'-脱氧核呋喃糖苷(α-2'-deoxyribofuranosides)和2',3'-二脱氧-3'-氨基核呋喃糖苷(2',3'-dideoxy-3'-aminoribofuranosides)。在替代性实施例中,2-脱氧-β-D-核呋喃糖基(2-deoxy-β-D-ribofuranose groups)可由其他糖(例如β-D-核呋喃糖)置换。另外,可存在β-D-核呋喃糖,其中核糖部分的2-OH经C1-6烷基(2-(O--C1-6烷基)核糖)或经C2-6烯基(2-(O--C2-6烯基)核糖)烷基化或经氟基(2-氟核糖(2-fluororibose))置换。
相关寡聚物形成糖(oligomer-forming sugar)包括在如上文所述的锁核酸(LNA)中所用的糖。例示性LNA寡核苷酸包括具有2'-O-4'-C亚甲基桥的经修饰双环单体单元,如美国专利第6,268,490号中所述。
经化学修饰的寡核苷酸亦可包括单独或任何组合的2'-位糖修饰、5-位嘧啶修饰(例如,5-(N-苄基羧酰胺)-2'-脱氧尿苷(5-(N-benzylcarboxyamide)-2'-deoxyuridine)、5-(N-异丁基羧酰胺)-2'-脱氧尿苷(5-(N-isobutylcarboxyamide)-2'-deoxyuridine)、5-(N-[2-(1H-吲哚-3-基)乙基]羧酰胺)-2'-脱氧尿苷(5-(N-[2-(1H-indole-3yl)ethyl]carboxyamide)-2'-deoxyuridine)、5-(N-[1-(3-三甲基铵)丙基]羧酰胺)-2'-脱氧尿苷氯化物(5-(N-[1-(3-trimethylammonium)propyl]carboxyamide)-2'-deoxyuridine chloride)、5-(N-萘基羧酰胺)-2'-脱氧尿苷(5-(N-napthylcarboxyamide)-2'-deoxyuridine)、5-(N-[1-(2,3-二羟丙基)]羧酰胺)-2'-脱氧尿苷)(5-(N-[1-(2,3-dihydroxypropyl)]carboxyamide)-2'-deoxyuridine))、8-位嘌呤修饰、环外胺(exocyclic amine)的修饰、4-硫尿苷(4-thiouridine)的取代、5-溴-或5-碘-尿嘧啶的取代、甲基化、不寻常的碱基配对组合如异碱基异胞苷(isocytidine)与异胍(isoguanidine)和其类似物。
在一些实施例中,该一种或多种PG途径抑制剂包含能抑制PG途径组分的表达的核糖酶(ribozyme)。核糖酶为能催化分子内或分子间的化学反应的核酸分子。因此,核糖酶为催化性核酸。核糖酶优选催化分子间反应。存在多种不同类型的核糖酶,其催化核酸酶或核酸聚合酶类型的反应,其是基于在天然系统中发现的核糖酶,如锤头核糖酶(hammerhead ribozymes)、发夹核糖酶(hairpin ribozymes)和四膜虫核糖酶(tetrahymena ribozymes)。亦存在多种在天然系统中未发现,但经重新(de novo)工程设计以催化特异性反应的核糖酶。优选的核糖酶切割RNA或DNA底物且更优选切割RNA底物,如PG途径组分的mRNA。核糖酶通常经由识别并与靶底物结合然后切割以切割核酸底物。这种识别通常主要基于典型或非典型的碱基对相互作用。这种特性使得核糖酶成为用于核酸靶向特异性切割的尤其良好的候选物,因为对靶底物的识别是基于靶底物序列。
在一些实施例中,该一种或多种PG途径抑制剂包含能抑制PG途径组分的表达的三螺旋形成寡核苷酸。三螺旋形成寡核苷酸(triplex forming oligonucleotides,TFO)为可与双链和/或单链核酸相互作用的分子,包括基因组DNA靶中的编码区和非编码区。当TFO与标靶区相互作用时,形成所谓三螺旋的结构,其中有三链DNA形成依赖Watson-Crick与Hoogsteen碱基配对的复合物。TFO可以高亲合力和特异性结合标靶区。在一些优选的实施例中,三螺旋形成分子以小于10-6、10-8、10-10或10-12的Kd结合标靶分子。用于本发明中的例示性TFO包括PNA、LNA和经LNA修饰的PNA,如Zorro-LNA。
在一些实施例中,该一种或多种PG途径抑制剂包含外部引导序列(external guide sequences,EGS)。外部引导序列(EGS)为结合靶核酸分子形成复合物的分子。这种复合物由RNase P识别,其切割标靶分子。EGS可经设计以特异性地靶向所选的mRNA分子。RNAse P有助于在细胞内加工转运RNA(transfer RNA,tRNA)。可通过使用导致标靶RNA:EGS复合物模拟天然tRNA底物的EGS,募集细菌RNAse P以实际上切割任何RNA序列。类似地,可利用真核EGS/RNAse P引导的RNA切割来切割真核细胞中的所需标靶。
在其他实施例中,该一种或多种PG途径抑制剂包含生物分子(biomolecule)。本文所用的术语“生物分子”为由活生物体产生的任何分子,包括如蛋白质、多糖、脂质和核酸的大型巨分子以及如初级代谢物、二级代谢物和天然产物的小分子。
在其他实施例中,该一种或多种PG途径抑制剂包含标靶中和剂。本文所用的术语“标靶中和剂”指能直接或间接地特异性地结合PG途径的组分以干扰前列腺素对靶组织的最终作用的抗体、抗体片段或任何其他非抗体肽或合成的结合分子,如适体(aptamer)或合成抗体(synbody)。
标靶中和剂可通过用于产生高亲合力结合配体的任何习知方法来产生,所述方法包括SELEX、噬菌体展示(phase display)和其他方法,包括本领域技术人员已知的组合化学和/或高通量方法。
适体为包含一类寡核苷酸的抗体的核酸变化形式,该寡核苷酸可形成与多种细胞表面分子、蛋白质和/或巨分子结构展现高亲合力结合的特异性三维结构。适体通常由体外(in vitro)选择方法来识别,这种方法有时称作“指数富集配体系统进化(Systematic Evolution of Ligands by EXponential enrichment)”或“SELEX”。SELEX通常由非常大量的随机多核苷酸的集合开始,一般将其限制为每个分子靶向一个适体配体。适体通常为长度在15至50个碱基范围内的小核酸,其折叠成所界定的二级和三级结构,如茎环(stem loop)或G-四分体(G-quartets)。
适体可与上述核酸抑制剂化学连接或接合以形成经标靶的核酸抑制剂,如适体-siRNA嵌合体。适体-siRNA嵌合体含有与siRNA连接的适体形式的标靶部分。当使用适体-siRNA嵌合体时,优选使用细胞内化适体。在与特异性细胞表面分子结合时,适体可促使内化至核酸抑制剂作用的细胞中。在一个实施例中,适体与siRNA均包含RNA。适体与siRNA可包含如本文进一步描述的任何的核苷酸修饰。适体优选包含特异性指导结合表达趋化因子-(chemokine-)、细胞因子-(cytokine-)和/或受体标靶基因的细胞(如类淋巴细胞、上皮细胞和/或内皮细胞)的标靶部分。
合成抗体(synbody)由包含经筛选用于与所关注的标靶蛋白质结合的随机肽串组成的文库中所产生的合成的抗体(synthetic antibody)。
标靶中和剂(包括适体和合成抗体)可经工程设计以10-10至10-12摩尔浓度(M)的Kd非常紧密地结合标靶分子。在一些实施例中,标靶中和剂以小于10-6、小于10-8、小于10-9、小于10-10或小于10-12M的Kd结合标靶分子。
在某些实施例中,该一种或多种PG途径抑制剂为包含编码PGT抑制剂和/或PGR抑制剂且适于表达PGT抑制剂和/或PGR抑制剂的多核苷酸。在其他实施例中,该一种或多种PG途径抑制剂为包含编码PGT抑制剂和/或PGR抑制剂且适于表达PGT抑制剂和/或PGR抑制剂的表达载体。
在一些实施例中,PG途径抑制剂为工程化蛋白,其含有针对编码PG途径的任何组分的基因的TALE序列或工程化锌指(Zinc Finger)。这种TALE或锌指可设计为直接与基因结合,并通过切割基因、改变其核苷酸序列或将抑制蛋白质与基因拴在一起使该基因沉默来抑制其表达。
在一些实施例中,使用CRISPR/CAS系统产生PG途径抑制剂。在此策略中,对每一PG途径基因的基因序列设计有特异性的引导分子(guide molecule),并使用上文所述的递送系统(病毒、质体等)将其引入细胞或组织中。CRISPR/CAS系统的运行将修饰基因的DNA序列,以使得PG途径基因得以删除或抑制表达RNA的能力。
在一些实施例中,PG途径抑制剂能通过靶向在染色质中翻译后修饰组织蛋白的染色质相关酶来关闭对一种或多种PG途径基因的转录。该酶的实例为(但不限于)组织蛋白去乙酰酶(histone deacetylases)、组织蛋白去甲基酶(histone demethylases)、组织蛋白乙酰基移转酶(histone acetyltransferases)、组织蛋白甲基转移酶(histone methyltransferases)和解旋酶(helicases)。
在一些实施例中,PG途径抑制剂通过改变基因的DNA甲基化状态靶向编码每种组分的基因。靶向DNA去甲基酶(demethylases)与DNA甲基转移酶(DNMT1、DNMTa和DNMTb)的TET家族的化合物可改变来自PG途径中的任何基因的RNA的表达。
本发明的表达载体包含编码PG途径抑制剂或其一部分的多核苷酸。表达载体亦包括一个或多个与所表达的多核苷酸操作性连接的调节序列。该调节序列基于宿主细胞的类型来选择。本领域技术人员将了解,表达载体的设计取决于如宿主细胞的选择以及所期望的表达水平等因素。
在一些实施例中,表达载体为一质体载体。在其他实施例中,表达载体为病毒载体。病毒载体的例子包括,但不限于逆转录病毒(retrovirus)、慢病毒(lentivirus)、腺病毒(adenovirus)、腺相关病毒(adeno-associated virus,AAV)、疱疹病毒(herpes virus)或α病毒(alphavirus)载体。病毒载体亦可为星状病毒(astrovirus)、冠状病毒(coronavirus)、正粘液病毒(orthomyxovirus)、乳多泡病毒(papovavirus)、副粘病毒(paramyxovirus)、小病毒(parvovirus)、小核糖核酸病毒(picornavirus)、痘病毒(poxvirus)或披衣病毒(togavirus)载体。在用于哺乳动物细胞中时,表达载体的控制功能常由病毒调节元件来提供。举例而言,常用的启动子来自于多瘤病毒(polyoma)、腺病毒2型(adenovirus 2)、巨细胞病毒(cytomegalovirus)和猿猴病毒40(Simian Virus 40)。在一些实施例中,表达载体含有组织特异性调节元件。表达载体的递送包括,但不限于使用病毒载体直接感染、使靶组织暴露于与被杀死的病毒连接或未连接的聚阳离子缩合DNA(polycationic condensed DNA)、配体连接的DNA、基因枪、离子化辐射(ionizing radiation)、核电荷(nucleic acid)中和或与细胞膜融合。亦可采用裸质体(naked plasmid)或病毒DNA。可使用生物可降解的乳胶珠粒来改良吸收效率。这种方法可通过对珠粒进行处理以增加其疏水性而进一步得到改良。亦可使用基于脂质体(liposome)的方法将质体或病毒载体引入靶组织中。
在一些实施例中,所述药学组合物还包含一种或多种选自由以下活性组分组成的组中的一种或者多种活性组分:止痛剂、抗毒蕈碱剂(antimuscarinic agent)、抗利尿剂(antidiuretic)、解痉剂(spasmolytic)、磷酸二酯酶第5型(phosphodiesterase type 5)抑制剂(PDE 5抑制剂)和唑吡坦(zolpedim)。
抗毒蕈碱剂的实例包括,但不限于奥昔布宁(oxybutynin)、索利那新(solifenacin)、达非那新(darifenacin)、弗斯特罗定(fesoterodine)、托特罗定(tolterodine)、曲司氯胺(trospium)、阿托品(atropine)和三环类抗抑郁剂(tricyclic antidepressant)。抗利尿剂的实例包括,但不限于抗利尿激素(ADH)、血管收缩素II(angiotensin II)、醛固酮(aldosterone)、血管加压素(vasopressin)、血管加压素类似物(例如,去氨加压素(desmopressin)、精氨酸加压素(argipressin)、赖氨酸加压素(lypressin)、苯赖加压素(felypressin)、鸟氨酸加压素(ornipressin)、特利加压素(terlipressin))、血管加压素受体促效剂、心房利钠肽(atrial natriuretic peptide,ANP)和C型利钠肽(C-type natriuretic peptide,CNP)受体(亦即,NPR1、NPR2和NPR3)拮抗剂(例如,HS-142-1、靛红(isatin)、[Asu7,23']b-ANP-(7-28)]、安南汀(anantin)(一种来自天蓝色链霉菌(Streptomyces coerulescens)的环状肽)和3G12单克隆抗体)、生长抑制素第2型(somatostatin type 2)受体拮抗剂(例如,生长抑制素)、药学上可接受的衍生物和其类似物、盐、水合物和溶剂化物。解痉剂的例子包括,但不限于肌安宁(carisoprodol)、苯二氮类药物(benzodiazepines)、巴氯芬(baclofen)、环苯扎林(cyclobenzaprine)、美他沙酮(metaxalone)、美索巴莫(methocarbamol)、可乐宁(clonidine)、可乐宁类似物和丹曲林(dantrolene)。PDE 5抑制剂的实例包括,但不限于他达那非(tadalafil)、西地那非(sildenafil)和伐地那非(vardenafil)。
药学组合物可被配制成用于即刻释放、延长释放、延迟释放、或其组合。
在一些实施例中,所述药学组合物被配制成用于即刻释放。
在其他实施例中,所述药学组合物被配制成通过将活性成分嵌入如丙烯酸树脂(acrylics)或甲壳素(chitin)等非可溶性物质的基质中而用于延长释放。延长释放形式经设计从而通过在特定时间段内维持恒定药物水平来以预定速率释放活性成分。这可经由各种制剂来实现,该制剂包括,但不限于脂质体和药物-聚合物结合物,如水凝胶(hydrogels)。
延长释放制剂可经设计从而以预定速率释放活性成分,以恒定药物水平维持指定的持续时间段,如在施用后或在与活性成分的延迟释放相关的迟滞期后长达约24小时、约22小时、约20小时、约18小时、约16小时、约14小时、约12小时、约10小时、约9小时、约8小时、约7小时、约6小时、约5小时、约4小时、约3小时或约2小时。可通过连续释放活性成分或脉冲式地释放活性成分来维持恒定的活性成分水平。
在某些实施例中,延长释放制剂中的活性成分在约2小时至约12小时间的时间间隔内释放。或者,活性成分可在约3、约4、约5、约6、约7、约8、约9、约10小时、约11小时或约12小时内释放。又在其他实施例中,延长释放制剂中的活性成分在施用后在约5至约8小时间的时间段内释放。
在一些实施例中,延长释放制剂包含由一个或多个惰性粒子组成的活性核(active core),该粒子各自为珠粒、丸剂、药片、颗粒、微胶囊(microcapsule)、微球体(microsphere)、微粒剂(microgranule)、纳米胶囊(nanocapsule)或纳米球体(nanosphere)的形式,其表面上使用例如流体床技术(fluid bed techniques)或本领域技术人员已知的其他方法涂布有含药涂层或成膜组合物的形式的药物。惰性粒子可具有各种尺寸,只要其足够大以维持不易溶解即可。作为另一选择,可通过粒化(granulating)并研磨和/或通过挤出并滚圆(spheronization)含有药物物质的聚合物组合物来制备活性核。本文所用的术语“药物”指药学组合物的活性成分。
可通过本领域技术人员已知的技术将活性成分引入惰性载体,该技术包括如药物层化(drug layering)、粉末涂布、挤出/滚圆、碾压(roller compaction)或粒化。核中活性成分的量将取决于所需的剂量且通常在约1至100重量%、约5至100重量%、约10至100重量%、约20至100重量%、约30至100重量%、约40至100重量%、约50至100重量%、约60至100重量%,、约70至100重量%、或约80至100重量%间变化。
一般而言,视所需的迟滞时间(lag time)和/或所选的聚合物和涂层溶剂而定,活性核上的聚合物涂层基于经涂布的粒子的重量计为约1至50%。本领域技术人员将能够选择适当量的药物用于涂布至核上或并入核中来达成所期望的剂量。在一个实施例中,非活性核可为糖球体或缓冲剂晶体或经囊封的缓冲剂晶体,如碳酸钙、碳酸氢钠、反丁烯二酸、酒石酸等,其使药物的微环境发生改变以促使其释放。
延长释放制剂可利用促使活性剂随时间逐渐释放的各种延长释放涂层或机制。在一些实施例中,延长释放剂包含通过溶解控制的释放来控制释放的聚合物。在一个特定实施例中,将活性剂并入基质中,该基质包含非可溶性聚合物和涂布有不同厚度的聚合材料的药物粒子或颗粒。聚合材料可包含脂质障壁(lipid barrier),脂质障壁包含蜡状材料(如棕榈蜡(carnauba wax)、蜂蜡(beewax)、鲸蜡(spermaceti wax)、小烛树蜡(candellila wax)、虫胶蜡(shallac wax)、可可脂(cocoa butter)、鲸蜡硬脂醇(cetostearyl alcohol)、部分氢化植物油、地蜡(ceresin)、石蜡(paraffin wax)、纯地蜡(ceresine)、肉豆蔻醇(myristyl alcohol)、硬脂醇(stearyl alcohol)、鲸蜡醇(cetyl alcohol)和硬脂酸(stearic acid))以及表面活性剂(如聚氧乙烯去水山梨醇单油酸酯(polyoxyethylene sorbitan monooleate))。在与例如生物流体(biological fluid)等水性介质接触时,聚合物涂层在预定的迟滞时间后乳化或腐蚀,视聚合物涂层的厚度而定。该迟滞时间与胃肠活动力(motility)、pH或胃滞留(gastric residence)无关。
在其他实施例中,延长释放剂包含实现扩散控制的释放的聚合基质。所述基质可包含一种或多种亲水性和/或水溶胀性基质形成聚合物、pH依赖性聚合物和/或非pH依赖性聚合物。
在一个实施例中,延长释放制剂包含水溶性或水溶胀性基质形成聚合物,视情况可选地含有一种或多种溶解增强剂和/或释放促进剂。在水溶性聚合物溶解时,活性剂溶解(若可溶)并经基质的水合部分逐渐扩散。随着更多水渗透至基质的核中,凝胶层随时间而增长,使凝胶层的厚度增加且提供阻碍药物释放的扩散障壁。当外层完全水合时,聚合物链完全松弛且不能再维持凝胶层的完整性,导致在基质表面上的外部水合聚合物解缠(disentanglement)以及浸蚀(erosion)。水穿过凝胶层继续向核渗透,直至其已被完全浸蚀。尽管可溶性药物通过扩散与浸蚀机制的组合来释放,浸蚀是非可溶性药物的主要机制,与剂量无关。
类似地,水溶胀性聚合物通常在生物流体中水合且溶胀而形成均质基质结构,该均质基质结构在药物释放期间保持其形状且作为药物、溶解性增强剂和/或释放促进剂的载体。初始的基质聚合物水合阶段导致药物的缓慢释放(迟滞期)。一旦水溶胀性聚合物完全水合且溶胀,则基质中的水可同样溶解药物物质且使其可穿过基质涂层向外扩散。
另外,基质的孔隙率(porosity)可因pH-依赖性释放促进剂的滤出(leaching out)而增加,从而以较快速率释放药物。药物释放的速率随后变为恒定,且为透过水合聚合物凝胶的药物扩散的函数。自基质释放的速率视各种因素而定,所述因素包括聚合物的类型和水平、药物的溶解度和剂量、聚合物与药物的比率、填充剂的类型和水平、聚合物与填充剂的比率、药物与聚合物的颗粒尺寸以及基质的孔隙率和形状。
例示性亲水性和/或水溶胀性基质形成聚合物包括,但不限于纤维素聚合物,包括羟烷基纤维素(hydroxyalkyl cellulose)和羧烷基纤维素(carboxyalkyl cellulose),如羟丙基甲基纤维素(hydroxypropylmethylcellulose,HPMC)、羟丙基纤维素(hydroxypropylcellulose,HPC)、羟乙基纤维素(hydroxyethylcellulose,HEC)、甲基纤维素(methylcellulose,MC)、羧甲基纤维素(carboxymethylcellulose,CMC);粉末状纤维素,如微晶纤维素(microcrystalline cellulose)、乙酸纤维素(cellulose acetate)、乙基纤维素(ethylcellulose)、其盐、及其组合;海藻酸盐(alginate);胶类,包括异源多糖胶(heteropolysaccharide gums)和同源多糖胶(homopolysaccharide gums),如黄原胶(xanthan)、黄蓍胶(tragacanth)、果胶、阿拉伯胶(acacia)、刺梧桐胶(karaya)、海藻酸盐、琼脂、瓜尔胶(guar)、羟丙基瓜尔胶(hydroxypropyl guar)、硅酸镁铝(veegum)、角叉菜胶(carrageenan)、刺槐豆胶(locust bean gum)、结兰胶(gellan gum)、及其衍生物;丙烯酸系树脂(acrylic resin),包括丙烯酸(acrylic acid)、甲基丙烯酸(methacrylic acid)、丙烯酸甲酯(methyl acrylate)、和甲基丙烯酸甲酯(methyl methacrylate)的聚合物和共聚物;和交联聚丙烯酸(cross-linked polyacrylic acid)衍生物,如卡波姆(Carbomer)(例如,包括自俄亥俄州辛辛那提市诺誉有限公司(Noveon,Inc.,Cincinnati,OH)可购得的各种分子量等级的71G NF)、角叉菜胶;聚乙酸乙烯酯(polyvinylacetate)(例如,SR);和聚乙烯吡咯烷酮(polyvinyl pyrrolidone)和其衍生物,如交联聚维酮(crospovidone)、聚氧化乙烯(polyethylene oxide)和聚乙烯醇(polyvinyl alcohol)。优选的亲水性和水溶胀性聚合物包括纤维素聚合物,尤其为HPMC。
延长释放制剂可进一步包含至少一种能使亲水性化合物在水性介质(包括生物流体)中交联以形成亲水性聚合物基质(亦即,凝胶基质)的粘合剂。
例示性粘合剂包括同源多糖,如半乳甘露聚糖胶
(galactomannan gums)、瓜尔胶、羟丙基瓜尔胶、羟丙基纤维素(HPC;例如Klucel EXF)和刺槐豆胶。在其他实施例中,粘合剂为海藻酸(alginic acid)衍生物、HPC或微晶化纤维素(microcrystallized cellulose,MCC)。其他粘合剂包括,但不限于淀粉、微晶纤维素、羟丙基纤维素、羟乙基纤维素、羟丙基甲基纤维素和聚乙烯吡咯烷酮。
在一实施例中,引入方法为通过向惰性载体上喷洒活性剂悬浮液和粘合剂的药物层化法。
粘合剂在珠粒制剂中的存在量按重量计可为约0.1%至约15%,且优选为按重量计为约0.2%至约10%。
在一些实施例中,亲水性聚合物基质更可包括离子型聚合物、非离子型聚合物或非水溶性疏水性聚合物,以提供更结实的凝胶层和/或减小基质中的孔的数量和尺寸,从而减缓活性剂的扩散速率与浸蚀速率以及伴随的活性剂的释放。另外这还可抑制初始的突释效应(burst effect),并产生更稳定的活性剂的“零级释放(zero order release)”。
用于减缓溶解速率的例示性离子型聚合物包括阴离子型和阳离子型聚合物。例示性阴离子型聚合物包括,例如羧甲基纤维素钠(Na CMC);海藻酸钠、丙烯酸或卡波姆(例如,934、940、974P NF)的聚合物;肠溶性(enteric)聚合物,如聚醋酸乙烯邻苯二甲酸酯(polyvinyl acetate phthalate,PVAP)、甲基丙烯酸共聚物(methacrylic acid copolymer)(例如,L100、L 30D 55、A和FS 30D)和羟丙甲纤维素醋酸琥珀酸酯(hypromellose acetate succinate)(AQUAT HPMCAS);和黄原胶。例示性阳离子型聚合物包括,例如甲基丙烯酸二甲氨基乙酯共聚物(dimethylaminoethyl methacrylate copolymer)(例如,E 100)。阴离子型聚合物(尤其为肠溶性聚合物)的引入有助于研发相对于单独亲水性聚合物的弱碱性药物的非pH依赖性释放概况。
用于减缓溶解速率的例示性非离子型聚合物包括,例如羟丙基纤维素(HPC)和聚氧化乙烯(PEO)(例如,POLYOXTM)。
例示性疏水性聚合物包括乙基纤维素(例如,ETHOCELTM、)、乙酸纤维素、甲基丙烯酸共聚物(例如,NE 30D)、铵基-甲基丙烯酸酯(ammonio-methacrylate)共聚物(例如,RL 100或PO RS 100)、聚乙酸乙烯酯、单硬脂酸甘油酯(glyceryl monostearate)、脂肪酸(如柠檬酸乙酰基三丁酯(acetyl tributyl citrate))、及其组合和衍生物。
膨胀性聚合物可以按重量计为1%至50%、优选按重量计为5%至40%、最佳按重量计为5%至20%的比例并入制剂中。膨胀性聚合物和粘合剂可在粒化之前或之后并入制剂中。聚合物亦可分散于有机溶剂或水-醇中并在粒化期间喷洒。
例示性释放促进剂包括在低于约4.0的pH值下保持完整,并且在高于4.0、优选高于5.0、最佳约6.0的pH值下溶解的pH依赖性肠溶性聚合物,且其作为释放促进剂被认为有助于本发明。例示性pH依赖性聚合物包括,但不限于甲基丙烯酸共聚物;甲基丙烯酸-甲基丙烯酸甲酯共聚物(methacrylic acid-methyl methacrylate copolymer)(例如,L100(A型)、S100(B型),德国罗姆股份有限公司(Rohm GmbH,Germany))、甲基丙烯酸-丙烯酸乙酯共聚物(methacrylic acid-ethyl acrylate copolymer)(例如,L100-55(C型)和L30D-55共聚物分散液,德国罗姆股份有限公司(Rohm GmbH,Germany));甲基丙烯酸-甲基丙烯酸甲酯与甲基丙烯酸甲酯的共聚物(FS);甲基丙烯酸、甲基丙烯酸酯和丙烯酸乙酯的三元共聚物(terpolymer)、邻苯二甲酸乙酸纤维素(cellulose acetate phthalates,CAP);邻苯二甲酸羟丙基甲基纤维素(hydroxypropyl methylcellulose phthalate,HPMCP)(例如,HP-55、HP-50、HP-55S,日本信越化学公司(Shinetsu Chemical,Japan));聚醋酸乙烯邻苯二甲酸酯(PVAP)(例如,肠溶性白色OY-P-7171);聚乙酸丁酸乙烯酯(polyvinylbutyrate acetate)、丁二酸乙酸纤维素(cellulose acetate succinates,CAS);羟丙基甲基纤维素乙酸琥珀酸酯(hydroxypropyl methylcellulose acetate succinate,HPMCAS)(例如,HPMCAS LF等级、MF等级和HF等级,包括LF和MF,日本信越化学公司(Shin-Etsu Chemical,Japan))、虫胶(shellac)(例如,MARCOATTM 125和MARCOATTM 125N);乙酸乙烯酯-顺丁烯二酸酐共聚物(vinyl acetate-maleic anhydride copolymer)、苯乙烯-顺丁烯二酸单酯共聚物(styrene-maleic monoester copolymer)、羧甲基乙基纤维素(carboxymethyl ethylcellulose,CMEC,日本弗罗因德公司(Freund Corporation,Japan));邻苯二甲酸乙酸纤维素(CAP)(例如,)、偏苯三酸乙酸纤维素(cellulose acetate trimellitates,CAT)、及其中两者或两者以上的重量比在约2:1至约5:1之间的混合物,如重量比为约3:1至约2:1的L 100-55与S 100的混合物,或重量比为约3:1至约5:1的L 30D-55与FS的混合物。
这些聚合物可单独或组合使用,或与除上文提及聚合物以外的聚合物一起使用。优选的肠溶性pH依赖性聚合物为药学上可接受的甲基丙烯酸共聚物。这些共聚物为基于甲基丙烯酸和甲基丙烯酸甲酯的阴离子型聚合物,且优选具有约50,000至200,000,优选约135,000的平均分子量。该共聚物中的游离羧基与甲基酯化羧基的比率可在,例如1:1至1:3的范围内,例如约1:1或1:2。释放促进剂不限于pH依赖性聚合物。快速溶解且从剂型快速滤出而留下多孔结构的其他亲水性分子亦可用于同一目的。
在一些实施例中,基质可包括释放促进剂与溶解度增强剂的组合。溶解度增强剂可为离子型和非离子型表面活性剂、络合剂、亲水性聚合物、和pH改性剂(如酸化剂与碱化剂)以及经由分子截留(entrapment)增加可溶性不佳药物的溶解度的分子。可同时使用若干种溶解度增强剂。
溶解度增强剂可包括表面活性剂,如多库酯钠(sodium docusate);月桂基硫酸钠(sodium lauryl sulfate);硬脂酰反丁烯二酸钠(sodium stearyl fumarate);和Spans(PEO改性的去水山梨醇单酯(sorbitan monoester)和脂肪酸去水山梨醇酯(fatty acid sorbitan ester));聚(氧化乙烯)-聚氧化丙烯-聚(氧化乙烯)嵌段共聚物(poly(ethylene oxide)-polypropylene oxide-poly(ethylene oxide)block copolymer)(亦称PLURONICSTM);络合剂,如低分子量聚乙烯吡咯烷酮和低分子量羟丙基甲基纤维素;经由分子截留辅助溶解度的分子,如环糊精;和pH改性剂,包括酸化剂(如柠檬酸、反丁烯二酸、酒石酸、和盐酸)和碱化剂(如葡甲胺(meglumine)和氢氧化钠)。
溶解度增强剂通常占剂型的按重量计的1%至80%、按重量计的1%至60%、按重量计的1%至50%、按重量计的1%至40%和按重量计的1%至30%,且可以各种方式并入。它们可在粒化之前以干或湿的形式并入制剂中。其亦可在剩余材料经粒化或以其他方式加工后加入制剂中。粒化期间,溶解度增强剂可以含或不含粘合剂的溶液形式喷洒。
在一个实施例中,延长释放制剂包含在活性核上形成有包含一种或多种非水溶性的透水性成膜聚合物的非水溶性的透水性聚合物涂层或基质。涂层可另外包括一种或多种水溶性聚合物和/或一种或多种增塑剂(plasticizer)。非水溶性聚合物涂层包含针对核中活性剂的释放的障壁涂层,其中较低分子量(粘度)等级与较高粘度等级相比展示较快的释放速率。
在一些实施例中,非水溶性成膜聚合物包括一种或多种烷基纤维素醚,如乙基纤维素及其混合物(例如,乙基纤维素等级PR100、PR45、PR20、PR10和PR7;Dow)。
在一些实施例中,非水溶性聚合物无需增塑剂即可提供合适特性(例如,延长释放特征、机械特性和涂层特性)。举例而言,包含聚乙酸乙烯酯(PVA)、丙烯酸酯/甲基丙烯酸酯的中性共聚物(如可购自赢创工业集团(Evonik Industries)的Eudragit NE30D)、乙基纤维素与羟丙基纤维素组合、蜡等的涂层可在无增塑剂的情形下应用。
在又一实施例中,非水溶性聚合物基质还可包括增塑剂。所需增塑剂的量取决于增塑剂、非水溶性聚合物的特性和涂层的最终所需特性。增塑剂的合适含量相对于涂层的总重量而言,按重量计在约1%至约20%、约3%至约20%、约3%至约5%、约7%至约10%、约12%至约15%、约17%至约20%的范围内,或按重量计为约1%、约2%、约3%、约4%、约5%、约6%、约7%、约8%、约9%、约10%、约15%或约20%,包括其间的所有范围和子范围。
例示性增塑剂包括,但不限于三乙酸甘油(triacetin)、乙酰化单甘油酯(acetylated monoglyceride)、油类(蓖麻油、氢化蓖麻油、葡萄籽油、芝麻油、橄榄油等)、柠檬酸酯、柠檬酸三乙酯、乙酰基柠檬酸三乙酯(acetyltriethyl citrate)、乙酰基柠檬酸三丁酯、柠檬酸三丁酯、乙酰基柠檬酸三正丁酯、邻苯二甲酸二乙酯、邻苯二甲酸二丁酯、邻苯二甲酸二辛酯、对羟苯甲酸甲酯、对羟苯甲酸丙酯、对羟苯甲酸丁酯、癸二酸二乙酯、癸二酸二丁酯、三丁酸甘油酯(glyceroltributyrate)、经取代的三酸甘油酯和甘油酯、单乙酰化(monoacetylated)和二乙酰化(diacetylated)甘油酯(例如,9-45)、单硬脂酸甘油酯、三丁酸甘油酯、聚山梨醇酯80、聚乙二醇(如PEG-4000和PEG-400)、丙二醇、1,2-丙二醇、甘油、山梨糖醇(sorbitol)、草酸二乙酯、苹果酸二乙酯、反丁烯二酸二乙酯(diethyl fumarate)、丙二酸二乙酯、丁二酸二丁酯、脂肪酸、顺丁烯二酸二乙酯、丁二酸二乙酯、邻苯二甲酸二辛酯、癸二酸二丁酯和其混合物。增塑剂可具有表面活性剂特性以使其可充当释放改性剂(release modifier)。举例而言,可使用非离子型清洁剂,如Brij 58(聚氧乙烯(20)鲸蜡醚(polyoxyethylene(20)cetyl ether))和其类似物。
增塑剂可为用于对硬质或脆性(brittle)聚合材料赋予挠性(flexibility)的高沸点有机溶剂,且可影响活性剂的释放概况。增塑剂一般导致沿聚合物链的分子间内聚力减小,从而导致聚合物特性发生各种改变。该改变包括,但不限于抗拉强度(tensile strength)减小与伸长率增加和聚合物的玻璃转变温度(trnasition)或软化温度降低。增塑剂的量和选择可影响,例如片剂的硬度,甚至可影响其溶解或崩解(disintegration)特征和其物理与化学稳定性。某些增塑剂可增加涂层的弹性和/或可挠性(pliability),由此减小涂层的脆性。
在另一实施例中,延长释放制剂包含至少两种凝胶形成聚合物的组合,包括至少一种非离子型凝胶形成聚合物和/或至少一种阴离子型凝胶形成聚合物。由凝胶形成聚合物的组合形成的凝胶提供受控制的释放,使得当制剂被摄入并且与胃肠道流体接触时,最接近表面的聚合物水合以形成粘性凝胶层。由于粘度高,同一过程中粘性层仅逐渐溶解掉使下方材料暴露。团块因此缓慢溶解掉,由此缓慢释放活性成分至胃肠道流体中。至少两种凝胶形成聚合物的组合使得所得凝胶的特性,如粘度可被操控,从而提供所需的释放概况。
在一个特定实施例中,制剂包含至少一种非离子型凝胶形成聚合物和至少一种阴离子型凝胶形成聚合物。在另一实施例中,制剂包含两种不同的非离子型凝胶形成聚合物。在又一实施例中,制剂包含具有相同化学性,但溶解度、粘度和/或分子量不同的非离子型凝胶形成聚合物的组合(例如,具有不同粘度等级的羟丙基甲基纤维素的组合,如HPMC K100与HPMC K15M或HPMC K100M)。
例示性阴离子型凝胶形成聚合物包括,但不限于羧甲基纤维素钠(Na CMC)、羧甲基纤维素(CMC)、阴离子型多糖,如海藻酸钠、海藻酸、果胶、聚葡萄糖醛酸(polyglucuronic acid)(聚-α-和-β-1,4-葡萄糖醛酸(poly-α-and-β-1,4-glucuronic acid))、聚半乳糖醛酸(polygalacturonic acid)(果胶酸)、硫酸软骨素、角叉菜胶(carrageenan)、富塞兰藻胶(furcellaran)、阴离子型胶(如黄原胶(xanthan gum))、丙烯酸或卡波姆(934、940、974P NF)的聚合物、共聚物、聚合物、聚卡波非(polycarbophil)及其他。
例示性非离子型凝胶形成聚合物包括,但不限于聚维酮(Povidone)(PVP:聚乙烯吡咯烷酮)、聚乙烯醇、PVP与聚乙酸乙烯酯的共聚物、羟丙基纤维素(HPC)、羟丙基甲基纤维素(HPMC)、羟乙基纤维素、羟甲基纤维素、明胶、聚氧化乙烯、阿拉伯胶、糊精、淀粉、聚甲基丙烯酸羟乙酯(polyhydroxyethylmethacrylate,PHEMA)、水溶性非离子型聚甲基丙烯酸酯(polymethacrylates)和其共聚物、经改性的纤维素、经改性的多糖、非离子型胶、非离子型多糖和/或其混合物。
制剂可选择性包含如上所述的肠溶性聚合物和/或至少一种赋形剂,如填充剂、粘合剂(如上所述)、崩解剂和/或流动助剂或助流剂(glidant)。
例示性填充剂包括,但不限于乳糖、葡萄糖、果糖、蔗糖、磷酸氢钙、糖醇(亦称为“糖多元醇”)(如山梨糖醇、甘露糖醇、乳糖醇(lactitol)、木糖醇、异麦芽酮糖醇(isomalt)、赤藻糖醇和氢化淀粉水解产物(若干种糖醇的共混物(blend)))、玉米淀粉、马铃薯淀粉、羧甲基纤维素钠、乙基纤维素和乙酸纤维素、肠溶性聚合物或其混合物。
例示性粘合剂包括,但不限于水溶性亲水性聚合物,如聚维酮(PVP:聚乙烯吡咯烷酮)、共聚维酮(copovidone)(聚乙烯吡咯烷酮与聚乙酸乙烯酯的共聚物)、低分子量羟丙基纤维素(HPC)、低分子量羟丙基甲基纤维素(HPMC)、低分子量羧甲基纤维素、乙基纤维素、明胶、聚氧化乙烯、阿拉伯胶、糊精、硅酸镁铝(magnesium aluminum silicate)和淀粉,以及聚甲基丙烯酸酯如Eudragit NE 30D、Eudragit RL、Eudragit RS、Eudragit E,聚乙酸乙烯酯、肠溶性聚合物或其混合物)。
例示性崩解剂包括,但不限于低取代的羧甲基纤维素钠、交联聚维酮(crospovidone)(交联聚乙烯吡咯烷酮)、羧甲基淀粉钠(sodium carboxymethyl starch)(乙醇酸淀粉钠(sodium starch glycolate))、交联羧甲基纤维素钠(交联羧甲纤维素(Croscarmellose))、预胶化淀粉(淀粉1500)、微晶纤维素、非水溶性淀粉、羧甲基纤维素钙、低取代的羟丙基纤维素和硅酸镁或硅酸铝。
例示性助流剂包括,但不限于镁、二氧化硅、滑石、淀粉、二氧化钛和其类似物。
在又一实施例中,延长释放制剂通过以涂布材料和可选择的造孔剂和其他赋形剂来涂布水溶性/分散性含药粒子(如其中的珠粒或珠粒群体(如上文所述))而形成。涂布材料优选选自包含以下的组:纤维素聚合物,如乙基纤维素(例如,)、甲基纤维素、羟丙基纤维素、羟丙基甲基纤维素、乙酸纤维素和邻苯二甲酸乙酸纤维素;聚乙烯醇;丙烯酸聚合物,如聚丙烯酸酯、聚甲基丙烯酸酯和其共聚物;和其他基于水或基于溶剂的涂布材料。用于给定的珠粒群体的控释涂层(release-controlling coating)可由该控释涂层的至少一种参数来控制,如涂层的性质、涂布水平、造孔剂的类型和浓度、加工参数和其组合。因此,改变参数(如造孔剂浓度或固化条件)使得自任何指定珠粒群体的活性剂释放发生改变,由此可选择性地将制剂调整至预定的释放概况。
适用于本文的控释涂层中的造孔剂可为有机剂或无机剂且包括在使用的环境中可自涂层中溶解出、萃取(extract)出或滤出的材料。例示性造孔剂包括,但不限于有机化合物,如单糖、寡糖和多糖(包括蔗糖、葡萄糖、果糖、甘露糖醇、甘露糖、半乳糖、山梨糖醇、普鲁兰糖(pullulan)和聚葡糖(dextran));在使用的环境中可溶的聚合物,如水溶性亲水性聚合物、羟烷基纤维素、羧烷基纤维素、羟丙基甲基纤维素、纤维素醚、丙烯酸树脂、聚乙烯吡咯烷酮、交联聚乙烯吡咯烷酮、聚氧化乙烯、卡波蜡(Carbowax)、卡波普(Carbopol)和其类似物、二醇、多元醇、聚羟基醇(polyhydric alcohol)、聚亚烷基二醇(polyalkylene glycol)、聚乙二醇(polyethylene glycol)、聚丙二醇或其嵌段聚合物、聚二醇(polyglycol)和聚(α-Ω)烷二醇(poly(α-Ω)alkylenediols);和无机化合物,如碱金属盐、碳酸锂、氯化钠、溴化钠、氯化钾、硫酸钾、磷酸钾、乙酸钠、柠檬酸钠、合适的钙盐、其组合和其类似物。
控释涂层可进一步包含本领域已知的其他添加剂,如增塑剂、抗粘剂、助流剂(或流动助剂)和消泡剂。在一些实施例中,经涂布的粒子或珠粒可额外包括“外涂层(overcoat)”,以对珠粒提供例如防潮保护、减少静电荷、掩味、调味、着色和/或抛光或其他美化。用于这种外涂层的合适涂布材料在本领域中已知且包括,但不限于纤维素聚合物,如羟丙基甲基纤维素、羟丙基纤维素和微晶纤维素或其组合(例如,各种涂布材料)。
经涂布的粒子或珠粒可额外含有增强剂,其可例示为,但不限于溶解度增强剂、溶解增强剂、吸收增强剂、渗透性增强剂、稳定剂、络合剂、酶抑制剂、p-糖蛋白抑制剂和多药抗性蛋白抑制剂。作为另一选择,该制剂亦可含有与涂布粒子分离的增强剂,例如在单独的珠粒群体中或呈粉末形式。在又一实施例中,在控释涂层下方或上方的经涂布的粒子上的隔离层(separate layer)中可含有增强剂。
在其他实施例中,延长释放制剂被配制成通过渗透机制释放活性剂。举例而言,胶囊可被配制成具有单渗透单元或其可引入囊封于硬明胶胶囊中的2、3、4、5或6个推拉式单元(push-pull unit),由此每个双层推拉式单元含有渗透推动层(osmatic push layer)和药物层(drug layer),二者均由半渗透膜所围绕。对与药物层相邻的膜钻出一个或多个孔。这种膜可额外以pH依赖性肠溶衣覆盖,使直至胃排空之后才进行释放。明胶胶囊在摄入后即刻溶解。在推拉式单元进入小肠时,肠溶衣分解,随后可使流体经由半渗透膜流出,使渗透压推动隔室溶胀,以驱使药物以经半渗透膜输送水的速率精确控制的速率经小孔推出。药物的释放可在恒定速率下进行长达24小时或更长时间。
渗透推动层包含一种或多种渗透剂,从而产生驱动力用于将水经半渗透膜输送至递送媒剂(delivery vehicle)的核中。一类渗透剂包括水溶胀性亲水性聚合物,亦称为“渗透聚合物(osmopolymer)”和“水凝胶(hydrogel)”,其包括,但不限于亲水性乙烯和丙烯酸聚合物、多糖(如海藻酸钙)、聚氧化乙烯(PEO)、聚乙二醇(PEG)、聚丙二醇(PPG)、聚(2-羟乙基甲基丙烯酸酯)(poly(2-hydroxyethyl methacrylate))、聚(丙烯)酸、聚(甲基丙烯)酸、聚乙烯吡咯烷酮(PVP)、交联PVP、聚乙烯醇(PVA)、PVA/PVP共聚物、含有疏水性单体如甲基丙烯酸甲酯和乙酸乙烯酯的PVA/PVP共聚物、含有大PEO嵌段的亲水性聚氨酯、交联羧甲纤维素钠、角叉菜胶、羟乙基纤维素(HEC)、羟丙基纤维素(HPC)、羟丙基甲基纤维素(HPMC)、羧甲基纤维素(CMC)和羧乙基纤维素(CEC)、海藻酸钠、聚卡波非、明胶、黄原胶和乙醇酸淀粉钠。
另一类渗透剂包括渗透原(osmogen),其能够吸水以影响跨越半渗透膜的渗透压梯度。例示性渗透原包括,但不限于无机盐,如硫酸镁、氯化镁、氯化钙、氯化钠、氯化锂、硫酸钾、磷酸钾、碳酸钠、亚硫酸钠、硫酸锂、氯化钾和硫酸钠;糖类,如右旋糖(dextrose)、果糖、葡萄糖、肌醇(inositol)、乳糖、麦芽糖、甘露糖醇、棉籽糖(raffinose)、山梨糖醇、蔗糖、海藻糖和木糖醇;有机酸,如抗坏血酸、苯甲酸、反丁烯二酸、柠檬酸、顺丁烯二酸、癸二酸、山梨酸、己二酸、四乙酸乙二胺(edetic acid)、谷氨酸、对甲苯磺酸、丁二酸和酒石酸;尿素;和其混合物。
可用于形成半渗透膜的材料包括在生理相关pH下具有透水性和非水溶性或易于通过化学改变(如交联)而具备非水溶性的各种等级的丙烯酸类、乙烯类、醚类、聚酰胺类、聚酯类和纤维素衍生物。
在一些实施例中,延长释放制剂包含在胃和肠中均可抗浸蚀的多糖涂层。该聚合物仅可在结肠中降解,结肠中含有大微生物群,其含有的含生物降解酶降解例如多糖涂层从而以受控制的时间依赖性的方式来释放药物内容物。例示性多糖涂层可包括,例如直链淀粉、阿拉伯半乳聚糖(arabinogalactan)、几丁聚糖(chitosan)、硫酸软骨素、环糊精、聚葡糖、瓜尔胶、果胶、木胶(xylan)和其组合或衍生物。
在一些实施例中,药学组合物被配制成用于延迟释放或延迟-延长释放。在一些实施例中,延迟延长释放制剂包括涂布有肠溶衣的延长释放制剂,肠溶衣为涂覆于口服药物上以防止药物在到达小肠之前释放的障壁。延迟释放制剂(如肠溶衣)防止对胃具有刺激作用的药物(如阿司匹林)在胃中溶解。本文所用的术语“肠溶衣(enteric coating)”包含一种或多种具有pH依赖性或非pH依赖性释放概况的聚合物的涂层。经肠溶性涂布的药丸在胃的酸性液(pH约为3)中不溶解,但其将在小肠或结肠的碱性(pH为7至9)环境中溶解。肠溶性聚合物涂层通常抗拒活性剂释放直至施用后约3至4小时的胃排空迟滞期后一个时间段。
这些涂层亦用于保护酸不稳定药物免于暴露于胃酸中,而是将其递送至其不发生降解且产生其所期望作用的碱性pH环境(肠的pH 5.5和以上)。术语“脉冲式释放(pulsatile-release)”为一种延迟释放,其在本文中的使用涉和药物制剂,该药物制剂在预定迟滞期后即刻在短时间内提供快速和短暂的药物释放,由此在药物施用后产生药物的“脉冲式”血浆分布。制剂可被设计为在施用后以预定的时间间隔提供单次脉冲式释放或多次脉冲式释放,或以脉冲式释放(例如,20%至60%的活性成分)而随后进行一个时间段的延长释放(例如,连续释放剩余的活性成分)。延迟释放或脉冲式释放制剂一般包含一种或多种包覆有障壁涂层的成分,其在指定迟滞期后溶解、浸蚀或破裂。
用于延迟释放的障壁涂层视目标而定可由各种不同材料组成。此外,制剂可包含复数种障壁涂层以促使以短暂的(temporal)方式释放。涂层可为糖涂层、膜涂层(例如,基于羟丙基甲基纤维素、甲基纤维素、甲基羟乙基纤维素、羟丙基纤维素、羧甲基纤维素、丙烯酸酯共聚物、聚乙二醇和/或聚乙烯吡咯烷酮)或基于甲基丙烯酸共聚物、邻苯二甲酸乙酸纤维素、邻苯二甲酸羟丙基甲基纤维素、乙酸丁二酸羟丙基甲基纤维素(hydroxypropyl methylcellulose acetate succinate)、聚醋酸乙烯邻苯二甲酸酯(polyvinyl acetate phthalate)、虫胶(shellac)和/或乙基纤维素的涂层。此外,制剂可额外包括延迟时间的材料,如单硬脂酸甘油酯或二硬脂酸甘油酯(glyceryl distearate)。
在一些实施例中,延迟-延长释放制剂包括肠溶衣,其包含一种或多种促使活性剂在胃肠道的近端区或远程区中释放的聚合物。pH依赖性肠溶衣包含一种或多种pH依赖性或pH敏感性聚合物,其在低pH下(如在胃中)维持其结构完整性但在胃肠道更远程区(如小肠)的较高pH环境中溶解并释放药物内容物。为本发明的目的,“pH依赖性(pH dependent)”定义为具有根据环境pH而变化的特征(例如,溶解)。例示性的pH依赖性聚合物已于前文描述。pH依赖性聚合物通常展示对于溶解而言的特征性pH最佳值。在一些实施例中,pH依赖性聚合物展示在约5.0与5.5之间、约5.5与6.0之间、约6.0与6.5之间或约6.5与7.0之间的pH最佳值。在其他实施例中,pH依赖性聚合物展示≥5.0、≥5.5、≥6.0、≥6.5或≥7.0的pH最佳值。
在某些实施例中,涂布方法采用一种或多种pH依赖性聚合物与一种或多种非pH依赖性聚合物的共混。当可溶性聚合物达到其溶解的最佳pH时,pH依赖性聚合物与非pH依赖性聚合物的共混可降低活性成分的释放速率。
在一些实施例中,“延迟释放”或“延迟-延长释放”的概况可使用含有一种或多种活性剂的非水溶性胶囊体来获得,其中该胶囊体的一端以不溶性、但具渗透性和溶胀性的水凝胶插塞(plug)来封闭。在与胃肠道流体或溶解介质接触时,插塞溶胀、将其自身推出胶囊且在预定的迟滞时间后释放药物,这可由例如插塞的位置和尺寸来控制。胶囊体可进一步涂布有外部pH依赖性肠溶衣,使胶囊在到达小肠前保持完整。合适的插塞材料包括,例如聚甲基丙烯酸酯、易受浸蚀的压缩聚合物(例如,HPMC、聚乙烯醇)、经凝结(congealed)的熔融聚合物(例如,单油酸甘油酯(glycerl mono oleate))和酶促控制的可受浸蚀聚合物(例如,多糖,如直链淀粉、阿拉伯半乳聚糖、几丁聚糖、硫酸软骨素、环糊精、聚葡糖、瓜尔胶、果胶和木糖胶)。
在其他实施例中,胶囊或双层片剂可被配制成含有含药核(drug-containing core),其被溶胀层和外部不溶性、但具半渗透性的聚合物涂层或膜所包覆。破裂前的迟滞时间可由聚合物涂层的渗透性与机械特性和溶胀层的溶胀行为来控制。通常,溶胀层包含一种或多种溶胀剂,如溶胀且在其结构中保留水的溶胀性亲水性聚合物。
用于延迟释放涂层中的例示性水溶胀性材料包括,但不限于聚氧化乙烯(具有例如1,000,000与7,000,000之间的平均分子量,如);甲基纤维素;羟丙基纤维素;羟丙基甲基纤维素;具有100,000至6,000,000的重量平均分子量的聚环氧烷(polyalkylene oxides),包括,但不限于聚(亚甲基氧化物)(poly(methylene oxide))、聚(环氧丁烷)(poly(butylene oxide))、具有25,000至5,000,000的分子量的聚(羟烷基甲基丙烯酸酯)(poly(hydro alkyl mrthacrylate))、具有低缩醛残基(low acetal residue)且与乙二醛、甲醛或戊二醛交联且具有200至30,000间的聚合度的聚(乙烯)醇(poly(vinyl)alcohol);甲基纤维素、经交联的琼脂和羧甲基纤维素的混合物;通过形成顺丁烯二酸酐(maleic anhydride)与苯乙烯、乙烯、丙烯、丁烯或异丁烯的细碎共聚物(finely divided copolymer)的分散体系所产生的水凝胶形成共聚物,其以共聚物中每摩尔顺丁烯二酸酐用0.001至0.5摩尔的饱和交联剂交联;具有450,000至4,000,000的分子量的酸性羧基聚合物;聚丙烯酰胺;经交联的水溶胀性茚顺丁烯二酸酐(indenemaleicanhydride)聚合物;具有80,000至200,000的分子量的聚丙烯酸;淀粉接枝共聚物(starch graft copolymers);由缩合葡萄糖单元组成的丙烯酸酯聚合物多糖,如二酯交联聚葡萄糖(diester cross-linked polyglucan);具有3,000至60,000毫帕˙秒(mPa˙s)的粘度,且呈质量体积比(w/v)为0.5%至1%的水溶液形式的卡波姆(carbomer);纤维素醚,如具有约1000至7000毫帕·秒的粘度,且呈重量/重量比为1%的水溶液形式(25℃)的羟丙基纤维素;具有约1000或更高、优选2,500或更高至最大25,000毫帕·秒的粘度,且呈重量体积比为2%的水溶液形式的羟丙基甲基纤维素;在20℃下具有约300至700毫帕·秒的粘度,且呈重量体积比为10%的水溶液形式的聚乙烯吡咯烷酮;和其组合。
作为另一选择,可由崩解迟滞时间(disintegration lag time)来控制药物的释放时间,这取决于在主体底部含有预定微孔的非水溶性聚合物膜(如乙基纤维素,EC)的耐受度和厚度与溶胀性赋形剂(如经低取代的羟丙基纤维素(L-HPC)和乙醇酸钠)的量之间的平衡。口服之后,GI流体经微孔渗透,致使溶胀性赋形剂溶胀,这产生使胶囊组件脱离的内压,该组件包括含有溶胀性材料的第一胶囊主体、含有药物的第二胶囊主体和附接至第一胶囊主体的外盖。
延迟释放涂层可进一步包含抗粘剂,如滑石和单硬脂酸甘油酯。延迟释放涂层可进一步包含一种或多种增塑剂,包括,但不限于柠檬酸三乙酯、乙酰基柠檬酸三乙酯、乙酰基柠檬酸三丁酯、聚乙二醇乙酰化单甘油酯(polyethylene glycol acetylated monoglycerides)、甘油、三乙酸甘油酯、丙二醇、邻苯二甲酸酯(例如,邻苯二甲酸二乙酯、邻苯二甲酸二丁酯)、二氧化钛、氧化铁、蓖麻油、山梨糖醇和癸二酸二丁酯。
在另一个实施例中,延迟释放制剂采用透水性但非可溶性的膜涂层来封闭活性成分和渗透剂。当来自肠道的水经此膜缓慢扩散至核中时,核溶胀直至膜胀裂,由此释放活性成分。膜涂层可被调整成实现各种水渗透速率或释放时间。
在另一个体实施例中,延迟释放制剂采用不透水的片剂涂层,借此水经受控制的孔隙进入涂层中直至核胀裂。当片剂胀裂时,药物内容物即刻释放或经较长时间段释放。该技术和其他技术可经改进以允许在开始释放药物之前存在预定的迟滞时间。
在另一个实施例中,活性剂在制剂中递送以提供延迟释放和延长释放(延迟-延长释放)。本文中所使用的术语“延迟-延长释放”涉及药物制剂,提供在施用后以预定的时间或迟滞期活性剂的脉冲式释放,其后接续着活性剂的延长释放。
在一些实施例中,即刻释放、延长释放、延迟释放或延迟-延长释放制剂包含由一个或多个惰性粒子组成的活性核,该惰性粒子各自为珠粒、丸剂、药片、颗粒、微胶囊、微球体、微颗粒、纳米胶囊或纳米球体的形式,其表面使用例如流体床技术或本领域技术人员已知的其他方法涂布有呈例如含药膜形成组合物(drug-containing film-forming composition)形式的药物。惰性粒子可具有各种尺寸,只要其足够大以保持不易溶解即可。作为另一选择,可通过粒化并研磨和/或通过挤压并滚圆含有药物物质的聚合物组合物来制备活性核。
核中的药物的量将取决于所需的剂量且通常在约1至100重量%、约5至100重量%、约10至100重量%、约20至100重量%、约30至100重量%、约40至100重量%、约50至100重量%、约60至100重量%、约70至100重量%或约80至100重量%的范围内变化。一般而言,视所需释放概况的迟滞时间与类型和/或所选的聚合物与涂料溶剂而定,活性核上的聚合物涂层基于被涂布粒子的重量计将为约1至50%。本领域技术人员将能够选择适当量的药物用于涂布至核上或并入核中以达成所期望的剂量。在一个实施例中,非活性核可为糖球体或缓冲剂晶体或经囊封的缓冲剂晶体,如碳酸钙、碳酸氢钠、反丁烯二酸、酒石酸等,其使药物的微环境发生改变以促进其释放。
在一些实施例中,举例而言,延迟释放或延迟-延长释放组合物可通过以非水溶性聚合物与肠溶性聚合物的混合物涂布水溶性/分散性含药粒子(如珠粒)来形成,其中非水溶性聚合物与肠溶性聚合物可以4:1至1:1的重量比存在,且涂层的总重量以所涂布珠粒的总重量计为10至60重量%。药物层积珠粒可视情况包括控制内部溶解速率的乙基纤维素膜。聚合膜外层的组成以及内层与外层各自的重量经优化,基于体外/体内的相关性进行预测,以对于给定的活性而言达成所期望的生理节律(circadian rhythm)释放概况。
在其他实施例中,制剂可包含即刻释放含药粒子(无溶解速率控制聚合物膜)与在例如口服后展示2至4小时的迟滞时间的延迟-延长释放珠粒的混合物,由此提供双脉冲释放(two-pulse profile)概况。
在一些实施例中,活性核涂布有一层或多层溶解速率受控制的聚合物,以获得具有或不具有迟滞时间的所期望的释放概况。内层膜在水或体液吸入核中后可在很大程度上控制药物的释放速率,而外层膜可提供所期望的迟滞时间(在水或体液吸入核中后无药物释放或药物释放极少的时期)。内层膜可包含非水溶性聚合物或非水溶性聚合物与水溶性聚合物的混合物。
适用于在很大程度上控制长达6小时的迟滞时间的外膜的聚合物可包含如上所述的肠溶性聚合物和10至50重量%的非水溶性聚合物。非水溶性聚合物与肠溶性聚合物的比率可在4:1至1:2的范围内变化,这些聚合物优选以约1:1的比率存在。通常所用的非水溶性聚合物为乙基纤维素。
例示性非水溶性聚合物包括乙基纤维素、聚乙酸乙烯酯(来自BASF的Kollicoat SR#0D)、基于丙烯酸乙酯和甲基丙烯酸甲酯的中性共聚物、带季铵基的丙烯酸酯和甲基丙烯酸酯的共聚物,如NE、RS和RS30D、RL或RL30D和其类似物。例示性水溶性聚合物包括低分子量HPMC、HPC、甲基纤维素、聚乙二醇(分子量>3000的PEG),其厚度视活性剂在水中的溶解度和所用的基于溶剂或乳胶悬浮液的涂料制剂而定,在1重量%至高达10重量%的范围内。非水溶性聚合物与水溶性聚合物的比率通常可在95:5至60:40、优选80:20至65:35的范围内变化。在一些实施例中,使用AMBERLITETM IRP69树脂作为延长释放载体。AMBERLITETM IRP69为不溶、强酸性、钠形式阳离子交换树脂,其适合作为阳离子性(碱性)物质的载体。在其他实施例中,使用DUOLITETM AP143/1093树脂作为延长释放载体。DUOLITETMAP143/1093为不溶、强碱性、阴离子交换树脂,其适合作为阴离子性(酸性)物质的载体。当用作药物载体时,AMBERLITETM IRP69或/和DUOLITETM AP143/1093树脂提供一种使试剂与不溶性聚合基质结合的方法。经由形成树脂-药物复合物(药物树脂酸盐(drug resinates))来实现延长释放。当药物与高电解质浓度(此为胃肠道的典型特征)达到平衡时,药物在体内自树脂释放。由于与阳离子交换系统的芳香族结构的疏水性相互作用,更多疏水性药物通常将以较低速率自树脂溶离。
在一些实施例中,药学组合物被配制成用于口服。口服剂型包括例如片剂、胶囊和囊片且亦可包含复数个可经囊封或未经囊封的颗粒、珠粒、粉末或丸剂。片剂和胶囊代表最便利的口服剂型,在此情形下采用固体医药载体。
在延迟释放制剂中,可向丸剂、片剂或胶囊涂覆一或更多层障壁涂层以促使药物缓慢溶解同时释放至肠道中。障壁涂层通常含有一种或多种在治疗性组合物或活性核周围围绕、包围或形成层或膜的聚合物。在一些实施例中,活性剂在制剂中递送从而在施用后以预定时间提供延迟释放。延迟可长达约10分钟、约20分钟、约30分钟、约1小时、约2小时、约3小时、约4小时、约5小时、约6小时或更长时间。
可对含有活性剂的颗粒、珠粒、粉末或丸剂、片剂、胶囊或其组合应用各种涂布技术以产生不同且独特的释放概况。在一些实施例中,药学组合物为含有单层涂层的片剂或胶囊形式。在其他实施例中,药学组合物为含有多层涂层的片剂或胶囊形式。在一些实施例中,本发明的药学组合物被配制成用于延长释放或延迟延长释放100%的活性成分。
在其他实施例中,本发明的药学组合物被配制成用于双阶段延长释放(two-phase extended-release)或延迟双阶段延长释放(delayed two-phase extended-release),其特征在于“即刻释放”组分在施用两小时内释放,且“延长释放”组分在2至12小时的时间段内释放。在一些实施例中,“即刻释放”组分提供欲由药物制剂递送的活性剂总剂量的约1%至90%,而“延长释放”组分提供欲由药物制剂递送的活性剂总剂量的10%至99%。举例而言,即刻释放组分可提供欲由药物制剂递送的活性剂总剂量的约10%至90%、或约10%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%或90%。延长释放组分提供欲由制剂递送的活性剂总剂量的约10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%或99%。在一些实施例中,即刻释放组分与延长释放组分含有相同的活性成分。在其他实施例中,即刻释放组分与延长释放组分含有不同的活性成分(例如,一种组分中为一种PG途径抑制剂,而另一种组分中为另一种PG途径抑制剂)。在一些实施例中,即刻释放组分与延长释放组分各自含有PG途径抑制剂和选自由阿司匹林、布洛芬、萘普生钠、吲哚美辛、萘丁美酮和乙酰氨基酚所组成的组的止痛剂。在其他实施例中,即刻释放组分和/或延长释放组分进一步包含一种或多种选自由抗毒蕈碱剂、抗利尿剂、解痉剂、磷酸二酯酶(phosphodiesterase)型抑制剂(PDE 5抑制剂)和唑吡坦所组成的组的额外活性剂。
在一些实施例中,药学组合物包含复数种选自由PG途径抑制剂、止痛剂、抗毒蕈碱剂、抗利尿剂、解痉剂、PDE 5抑制剂和唑吡坦所组成的组的活性成分。抗毒蕈碱剂的实例包括,但不限于奥昔布宁、索利那新、达非那新、弗斯特罗定、托特罗定、曲司氯胺、阿托品和三环状抗抑郁剂。抗利尿剂的例子包括,但不限于抗利尿剂激素(ADH)、血管收缩素II、醛固酮、血管加压素、血管加压素类似物(例如,去胺加压素、精氨酸加压素、赖氨酸加压素、苯赖加压素、鸟氨酸加压素、特利加压素)、血管加压素受体促效剂、心房利钠肽(ANP)和C型利钠肽(CNP)受体(亦即,NPR1、NPR2和NPR3)拮抗剂(例如,HS-142-1、靛红、[Asu7,23']b-ANP-(7-28)]、安南汀(一种来自天蓝色链霉菌的环状肽)、和3G12单克隆抗体)、生长抑制素2型受体拮抗剂(例如,生长抑制素)、药学上可接受的衍生物和其类似物、盐、水合物和溶剂化物。解痉剂的例子包括,但不限于肌安宁、苯二氮类药物、巴氯芬、环苯扎林、美他沙酮、美索巴莫、可乐宁、可乐宁类似物和丹曲林。PDE 5抑制剂的例子包括,但不限于他达那非、西地那非和伐地那非。
在一些实施例中,药学组合物包含复数种活性成分,其包含(1)一种或多种PG途径抑制剂和(2)一种或多种选自由止痛剂、抗毒蕈碱剂、抗利尿剂、解痉剂、PDE 5抑制剂和唑吡坦所组成的组的其他活性成分。在一些实施例中,复数种活性成分被配制成用于即刻释放。在其他实施例中,复数种活性成分被配制成用于延长释放。在其他实施例中,复数种活性成分被配制成用于延迟释放。在其他实施例中,复数种活性成分被配制成用于即刻释放与延长释放(例如,每种活性成分的第一部分被配制成用于即刻释放而每种活性成分的第二部分被配制成用于延长释放)。在又一些其他实施例中,该活性成分中的一些被配制成用于即刻释放且该活性成分中的一些被配制成用于延长释放(例如,活性成分A、B、C被配制成用于即刻释放而活性成分C和D被配制成用于延长释放)。在一些其他实施例中,复数种活性成分被配制成用于延迟-延长释放。
在某些实施例中,药学组合物包含即刻释放组分与延长释放组分。即刻释放组分可包含一种或多种选自由PG途径抑制剂、止痛剂、抗毒蕈碱剂、抗利尿剂、解痉剂、PDE 5抑制剂和唑吡坦所组成的组的活性成分。延长释放组分可包含一种或多种选自由PG途径抑制剂、止痛剂、抗毒蕈碱剂、抗利尿剂、解痉剂、PDE 5抑制剂和唑吡坦所组成的组的活性成分。在一些实施例中,即刻释放组分与延长释放组分具有完全相同的活性成分。在其他实施例中,即刻释放组分与延长释放组分具有不同的活性成分。在又一些其他实施例中,即刻释放组分与延长释放组分具有一种或多种共同的活性成分。在一些其他实施例中,即刻释放组分和/或延长释放组分进一步涂布有延迟释放涂层(如肠溶衣)。在其他实施例中,药学组合物包含二种或更多种被配制成二种延长释放组分的活性成分,所述二种延长释放组分各自提供不同的延长释放概况。举例而言,第一延长释放组分以第一释放速率释放第一活性成分而第二延长释放组分以第二释放速率释放第二活性成分。
在一些实施例中,药学组合物包含即刻释放组分和延迟释放组分。在其他实施例中,药学组合物包含二种或更多种被配制成二种延迟释放组分的活性成分,所述延迟释放组分各自提供不同的延迟释放概况。举例而言,第一延迟释放组分在第一时间点释放第一活性成分而第二延迟释放组分在第二时间点释放第二活性成分。
组合释放概况制剂中的组分(例如,具有即刻释放组分与延长释放组分的组合;即刻释放组分与延迟释放组分的组合;即刻释放组分、延迟释放组分与延长释放组分的组合;二种或更多种延迟释放组分的组合;或二种或更多种延长释放组分的组合的制剂)可含有相同活性成分或不同活性成分。在一些实施例中,即刻释放组分可提供欲由药物制剂递送的活性剂总剂量的约1%至约80%。在一些实施例中,组合释放概况制剂含有即刻释放组分且该即刻释放组分提供欲由制剂递送的各种活性成分的总剂量的约1%至约90%、约10%至约90%、约20%至约90%、约30%至约90%、约40%至约90%、约50%至约90%、约60%至约90%、约70%至约90%或约80%至约90%。在替代实施例中,即刻释放组分提供欲由制剂递送各种成分的总剂量的高达约10%、20%、30%、40%、50%、60%、70%、80%或90%。
在一些实施例中,药物制剂包含由一个或多个惰性粒子所组成的活性核,所述粒子各自为珠粒、丸剂、药片、颗粒、微胶囊、微球体、微粒剂、纳米胶囊、或纳米球体的形式,其表面使用例如流体床技术或本领域技术人员已知的其他方法涂布有呈例如含药膜形成组合物的形式的药物。惰性粒子可具有各种尺寸,只要其足够大以维持不易溶解即可。作为另一选择,可通过粒化并研磨和/或通过挤出并滚圆含有药物物质的聚合物组合物来制备活性核。
核中的药物的量将取决于所需的剂量且通常在约5至90重量%的范围内变化。一般而言,视所需释放概况的迟滞时间与类型和/或所选的聚合物与涂料溶剂而定,活性核上的聚合物涂层基于经涂布粒子的重量计将为约1至50%。本领域技术人员将能够选择适当量的药物用于涂布至核上或并入核中以达成所期望的剂量。在一个实施例中,非活性核可为糖球体或缓冲剂晶体或囊封缓冲剂晶体,如碳酸钙、碳酸氢钠、反丁烯二酸、酒石酸等,其使药物的微环境发生改变以促进其释放。
在一些实施例中,该药学组合物包含通过以非水溶性聚合物与肠溶性聚合物的混合物涂布水溶性/分散性含药粒子(如一珠粒)所形成的延迟释放组分,其中非水溶性聚合物与肠溶性聚合物可以4:1至1:1的重量比存在,且涂层的总重量以所涂布珠粒的总重量计为10至60重量%。药物层化珠粒可视情况包括控制内部溶解速率的乙基纤维素膜。聚合膜外层的组成以及内层与外层各自的重量经优化,基于体外/体内相关性进行预测,以对于给定活性而言达成所期望的生理节律释放概况。
在其他实施例中,制剂包含即刻释放含药粒子(无控制溶解速率的聚合物膜)与在例如口服后展示2至4小时的迟滞时间的延迟释放珠粒的混合物,由此提供双脉冲释放概况。在又一些其他实施例中,制剂包含两种类型的延迟释放珠粒的混合物:第一种类型展示1至3小时的迟滞时间而第二种类型展示4至6小时的迟滞时间。在又一些其他实施例中,制剂包含两种类型的释放珠粒的混合物:第一种类型展示即刻释放,而第二种类型展示1至4小时的迟滞时间,其后为延长释放。
在一些实施例中,制剂被设计为具有某种释放概况以使得一部分活性成分(例如,10%至80%)在施用后即刻释放或在两小时内释放,而剩余部分在一个延长的时间段内(例如,在2至24小时的时间段内)释放。在其他实施例中,制剂被设计为具有某种释放概况以使得一种活性成分(例如,止痛剂)在施用后即刻释放或在两小时内释放,而一种或多种其他活性成分(例如,PG途径抑制剂)在一个延长的时间段内(例如,在2至24小时的时间段内)释放。
药学组合物可每日施用或基于需要进行施用。药学组合物可经口、静脉、或肌肉施用。在优选实施例中,药学组合物为经口施用。在其他实施例中,药学组合物通过经泌尿道的逆行灌注(retrograde perfusion)来施用。在其他实施例中,药学组合物通过直接注射至膀胱肌中来施用。
在一些实施例中,药学组合物以一天两次或一天三次的方式来每天施用。在其他实施例中,药学组合物以每隔一天、每3天、每4天、每5天、每6天、每周、每2周、每3周、每月、每2个月或每3个月的方式来施用。
在一些实施例中,药学组合物在就寝时间施用。在一些实施例中,药学组合物在就寝时间前约两小时内、优选在就寝时间前约一小时内施用。在另一实施例中,药学组合物在就寝时间前约2至4小时施用。在又一实施例中,药学组合物在就寝时间前至少4小时施用。
即刻释放组分、延长释放组分、延迟释放组分或延迟-延长释放组分中的活性成分的适当剂量(“治疗有效量”)将取决于例如疾病的严重性和过程、施用模式、特定成分的生物利用度(bioavailability)、患者的年龄和体重、患者的临床病史和对活性剂的反应、医生的判断等。
作为一般主张,即刻释放组分、延迟释放组分、延长释放组分或延迟-延长释放组分中的PG途径抑制剂的治疗有效量以每天每公斤体重约1微克至每天每公斤体重约100毫克的范围施用,而无论通过一次或多次施用。在一些实施例中,每种活性剂每日单剂量或多剂量施用的范围为每天每公斤体重约1微克至每天每公斤体重约100毫克、每天每公斤体重1微克至每天每公斤体重约30毫克、每天每公斤体重1微克至每天每公斤体重约10毫克、每天每公斤体重1微克至每天每公斤体重约3毫克、每天每公斤体重1微克至每天每公斤体重约1毫克、每天每公斤体重1微克至每天每公斤体重约300微克、每天每公斤体重1微克至每天每公斤体重约100微克、每天每公斤体重1微克至每天每公斤体重约30微克、每天每公斤体重1微克至每天每公斤体重约10微克、每天每公斤体重l微克至每天每公斤体重约3微克、每天每公斤体重10微克至每天每公斤体重约100毫克、每天每公斤体重10微克至每天每公斤体重约30毫克、每天每公斤体重10微克至每天每公斤体重约10毫克、每天每公斤体重10微克至每天每公斤体重约3毫克、每天每公斤体重10微克至每天每公斤体重约1毫克、每天每公斤体重10微克至每天每公斤体重约300微克、每天每公斤体重10微克至每天每公斤体重约100微克、每天每公斤体重10微克至每天每公斤体重约30微克、每天每公斤体重30微克至每天每公斤体重约100毫克、每天每公斤体重30微克至每天每公斤体重约30毫克、每天每公斤体重30微克至每天每公斤体重约10毫克、每天每公斤体重30微克至每天每公斤体重约3毫克、每天每公斤体重30微克至每天每公斤体重约1毫克、每天每公斤体重30微克至每天每公斤体重约300微克、每天每公斤体重30微克至每天每公斤体重约100微克、每天每公斤体重100微克至每天每公斤体重约100毫克、每天每公斤体重100微克至每天每公斤体重约30毫克、每天每公斤体重100微克至每天每公斤体重约10毫克、每天每公斤体重100微克至每天每公斤体重约3毫克、每天每公斤体重100微克至每天每公斤体重约1毫克、每天每公斤体重100微克至每天每公斤体重约300微克、每天每公斤体重300微克至每天每公斤体重约100毫克、每天每公斤体重300微克至每天每公斤体重约30毫克、每天每公斤体重300微克至每天每公斤体重约10毫克、每天每公斤体重300微克至每天每公斤体重约3毫克、每天每公斤体重300微克至每天每公斤体重约1毫克、每天每公斤体重1毫克至每天每公斤体重约100毫克、每天每公斤体重1毫克至每天每公斤体重约30毫克、每天每公斤体重1毫克至每天每公斤体重约10毫克、每天每公斤体重1毫克至每天每公斤体重约3毫克、每天每公斤体重3毫克至每天每公斤体重约100毫克、每天每公斤体重3毫克至每天每公斤体重约30毫克、每天每公斤体重3毫克至每天每公斤体重约10毫克、每天每公斤体重10毫克至每天每公斤体重约100毫克、每天每公斤体重10毫克至每天每公斤体重约30毫克或每天每公斤体重30毫克至每天每公斤体重约100毫克。
作为一般主张,即刻释放组分、延迟释放组分、延长释放组分或延迟-延长释放组分中的止痛剂的治疗有效量以每天每公斤体重约10微克至每天每公斤体重约100毫克的范围施用,而无论通过一次或多次施用。在一些实施例中,每种活性剂每日单剂量或多剂量施用的范围为每天每公斤体重约10微克至每天每公斤体重约100毫克、每天每公斤体重10微克至每天每公斤体重约30毫克、每天每公斤体重10微克至每天每公斤体重约10毫克、每天每公斤体重10微克至每天每公斤体重约3毫克、每天每公斤体重10微克至每天每公斤体重约1毫克、每天每公斤体重10微克至每天每公斤体重约300微克、每天每公斤体重10微克至每天每公斤体重约100微克、每天每公斤体重10微克至每天每公斤体重约30微克、每天每公斤体重30微克至每天每公斤体重约100毫克、每天每公斤体重30微克至每天每公斤体重约30毫克、每天每公斤体重30微克至每天每公斤体重约10毫克、每天每公斤体重30微克至每天每公斤体重约3毫克、每天每公斤体重30微克至每天每公斤体重约1毫克、每天每公斤体重30微克至每天每公斤体重约300微克、每天每公斤体重30微克至每天每公斤体重约100微克、每天每公斤体重100微克至每天每公斤体重约100毫克、每天每公斤体重100微克至每天每公斤体重约30毫克、每天每公斤体重100微克至每天每公斤体重约10毫克、每天每公斤体重100微克至每天每公斤体重约3毫克、每天每公斤体重100微克至每天每公斤体重约1毫克、每天每公斤体重100微克至每天每公斤体重约300微克、每天每公斤体重300微克至每天每公斤体重约100毫克、每天每公斤体重300微克至每天每公斤体重约30毫克、每天每公斤体重300微克至每天每公斤体重约10毫克、每天每公斤体重300微克至每天每公斤体重约3毫克、每天每公斤体重300微克至每天每公斤体重约1毫克、每天每公斤体重1毫克至每天每公斤体重约100毫克、每天每公斤体重1毫克至每天每公斤体重约30毫克、每天每公斤体重1毫克至每天每公斤体重约10毫克、每天每公斤体重1毫克至每天每公斤体重约3毫克、每天每公斤体重3毫克至每天每公斤体重约100毫克、每天每公斤体重3毫克至每天每公斤体重约30毫克、每天每公斤体重3毫克至每天每公斤体重约10毫克、每天每公斤体重10毫克至每天每公斤体重约100毫克、每天每公斤体重10毫克至每天每公斤体重约30毫克或每天每公斤体重30毫克至每天每公斤体重约100毫克。
本文所述的止痛剂可包括于即刻释放组分或延长释放组分、延迟释放组分、延迟-延长释放组分或其组合中以用于每日口服施用,其单次剂量或组合剂量范围为1毫克至2000毫克、1毫克至1000毫克、1毫克至300毫克、1毫克至100毫克、1毫克至30毫克、1毫克至10毫克、1毫克至3毫克、3毫克至2000毫克、3毫克至1000毫克、3毫克至300毫克、3毫克至100毫克、3毫克至30毫克、3毫克至10毫克、10毫克至2000毫克、10毫克至1000毫克、10毫克至300毫克、10毫克至100毫克、10毫克至30毫克、30毫克至2000毫克、30毫克至1000毫克、30毫克至300毫克、30毫克至100毫克、100毫克至2000毫克、100毫克至1000毫克、100毫克至300毫克、300毫克至2000毫克、300毫克至1000毫克或1000毫克至2000毫克。如所预期,剂量将取决于患者的疾病、尺寸、年龄和情况。
在一些实施例中,药学组合物包含单一止痛剂。在一个实施例中,单一止痛剂为阿司匹林。在另一实施例中,单一止痛剂为布洛芬。在另一实施例中,单一止痛剂为萘普生或萘普生钠。在另一实施例中,单一止痛剂为吲哚美辛。在另一实施例中,单一止痛剂为萘丁美酮。在另一实施例中,单一止痛剂为乙酰氨基酚。
在其他实施例中,药学组合物包含一对止痛剂。这种成对止痛剂的实例包括,但不限于乙酰基水杨酸(acetylsalicylic acid)与布洛芬、乙酰基水杨酸与萘普生钠、乙酰基水杨酸与萘丁美酮、乙酰基水杨酸与乙酰氨基酚、乙酰基水杨酸与吲哚美辛(indomethancin)、布洛芬与萘普生钠、布洛芬与萘丁美酮、布洛芬与乙酰氨基酚、布洛芬与吲哚美辛、萘普生、萘普生钠与萘丁美酮、萘普生钠与乙酰氨基酚、萘普生钠与吲哚美辛、萘丁美酮与乙酰氨基酚、萘丁美酮与吲哚美辛、和乙酰氨基酚与吲哚美辛。成对止痛剂以在0.1:1至10:1、0.2:1至5:1或0.3:1至3:1范围内的重量比混合。在一个实施例中,成对止痛剂以1:1的重量比混合。
在一些其他实施例中,本申请的药学组合物进一步包含一种或多种抗毒蕈碱剂。抗毒蕈碱剂的实例包括,但不限于奥昔布宁、索利那新、达非那新、弗斯特罗定、托特罗定、曲司氯胺、阿托品、和三环状抗抑郁剂。抗毒蕈碱剂的每日剂量在1微克至300毫克、1微克至100毫克、1微克至30毫克;1微克至10毫克、1微克至3毫克、1微克至1毫克、1微克至300微克、1微克至100微克、1微克至30微克、1微克至10微克、1微克至3微克、3微克至100毫克、3微克至30毫克;3微克至10毫克、3微克至3毫克、3微克至1毫克、3微克至300微克、3微克至100微克、3微克至30微克、3微克至10微克、10微克至300毫克、10微克至100毫克、10微克至30毫克;10微克至10毫克、10微克至3毫克、10微克至1毫克、10微克至300微克、10微克至100微克、10微克至30微克、30微克至300毫克、30微克至100毫克、30微克至30毫克;30微克至10毫克、30微克至3毫克、30微克至1毫克、30微克至300微克、30微克至100微克、100微克至300毫克、100微克至100毫克、100微克至30毫克;100微克至10毫克、100微克至3毫克、100微克至1毫克、100微克至300微克、300微克至300毫克、300微克至100毫克、300微克至30毫克;300微克至10毫克、300微克至3毫克、300微克至1毫克、1毫克至300毫克、1毫克至100毫克、1毫克至30毫克、1毫克至3毫克、3毫克至300毫克、3毫克至100毫克、3毫克至30毫克、3毫克至10毫克、10毫克至300毫克、10毫克至100毫克、10毫克至30毫克、30毫克至300毫克、30毫克至100毫克或100毫克至300毫克的范围内。
在一些其他实施例中,本申请的药学组合物进一步包含一种或多种抗利尿剂。抗利尿剂的实例包括,但不限于抗利尿剂激素(ADH)、血管收缩素II、醛固酮、血管加压素、血管加压素类似物(例如,去氨加压素、精氨酸加压素、赖氨酸加压素、苯赖加压素、鸟氨酸加压素和特利加压素)、血管加压素受体促效剂、心房利钠肽(ANP)和C型利钠肽(CNP)受体(亦即,NPR1、NPR2和NPR3)拮抗剂(例如,HS-142-1、靛红、[Asu7,23']b-ANP-(7-28)]、安南汀(一种来自天蓝色链霉菌的环状肽)、和3G12单克隆抗体)、生长抑制素2型受体拮抗剂(例如,生长抑制素)、药学上可接受的衍生物和其类似物、盐、水合物和溶剂化物。在一些实施例中,该一种或多种抗利尿剂包含去氨加压素。在其他实施例中,该一种或多种抗利尿剂为去氨加压素。抗利尿剂的每日剂量在1微克至300毫克、1微克至100毫克、1微克至30毫克;1微克至10毫克、1微克至3毫克、1微克至1毫克、1微克至300微克、1微克至100微克、1微克至30微克、1微克至10微克、1微克至3微克、3微克至100毫克、3微克至100毫克、3微克至30毫克;3微克至10毫克、3微克至3毫克、3微克至1毫克、3微克至300微克、3微克至100微克、3微克至30微克、3微克至10微克、10微克至300毫克、10微克至100毫克、10微克至30毫克;10微克至10毫克、10微克至3毫克、10微克至1毫克、10微克至300微克、10微克至100微克、10微克至30微克、30微克至300毫克、30微克至100毫克、30微克至30毫克;30微克至10毫克、30微克至3毫克、30微克至1毫克、30微克至300微克、30微克至100微克、100微克至300毫克、100微克至100毫克、100微克至30毫克;100微克至10毫克、100微克至3毫克、100微克至1毫克、100微克至300微克、300微克至300毫克、300微克至100毫克、300微克至30毫克;300微克至10毫克、300微克至3毫克、300微克至1毫克、1毫克至300毫克、1毫克至100毫克、1毫克至30毫克、1毫克至3毫克、3毫克至300毫克、3毫克至100毫克、3毫克至30毫克、3毫克至10毫克、10毫克至300毫克、10毫克至100毫克、10毫克至30毫克、30毫克至300毫克、30毫克至100毫克或100毫克至300毫克的范围内。
在其他实施例中,本申请的药学组合物进一步包含一种或多种解痉剂。解痉剂的实例包括,但不限于肌安宁、苯二氮类药物、巴氯芬、环苯扎林、美他沙酮、美索巴莫、可乐宁、可乐宁类似物、和丹曲林。在一些实施例中,解痉剂以0.1毫克至1000毫克、0.1毫克至300毫克、0.1毫克至100毫克、0.1毫克至30毫克、0.1毫克至10毫克、0.1毫克至3毫克、0.1毫克至1毫克、0.1毫克至0.3毫克、0.3毫克至1000毫克、0.3毫克至300毫克、0.3毫克至100毫克、0.3毫克至30毫克、0.3毫克至10毫克、0.3毫克至3毫克、0.3毫克至1毫克、1毫克至1000毫克、1毫克至300毫克、1毫克至100毫克、1毫克至30毫克、1毫克至10毫克、1毫克至3毫克、3毫克至1000毫克、3毫克至300毫克、3毫克至100毫克、3毫克至30毫克、3毫克至10毫克、10毫克至1000毫克、10毫克至300毫克、10毫克至100毫克、10毫克至30毫克、30毫克至1000毫克、30毫克至300毫克、30毫克至100毫克、100毫克至1000毫克、100毫克至300毫克或300毫克至1000毫克的每日剂量使用。
在其他实施例中,本申请的药学组合物进一步包含一种或多种PDE 5抑制剂。PDE 5抑制剂的实例包括,但不限于他达那非、西地那非和伐地那非。在一些实施例中,一种或多种PDE 5抑制剂包含他达那非。在其他实施例中,一种或多种PDE 5抑制剂为他达那非。在一些实施例中,PDE 5抑制剂以0.1毫克至1000毫克、0.1毫克至300毫克、0.1毫克至100毫克、0.1毫克至30毫克、0.1毫克至10毫克、0.1毫克至3毫克、0.1毫克至1毫克、0.1毫克至0.3毫克、0.3毫克至1000毫克、0.3毫克至300毫克、0.3毫克至100毫克、0.3毫克至30毫克、0.3毫克至10毫克、0.3毫克至3毫克、0.3毫克至1毫克、1毫克至1000毫克、1毫克至300毫克、1毫克至100毫克、1毫克至30毫克、1毫克至10毫克、1毫克至3毫克、3毫克至1000毫克、3毫克至300毫克、3毫克至100毫克、3毫克至30毫克、3毫克至10毫克、10毫克至1000毫克、10毫克至300毫克、10毫克至100毫克、10毫克至30毫克、30毫克至1000毫克、30毫克至300毫克、30毫克至100毫克、100毫克至1000毫克、100毫克至300毫克或300毫克至1000毫克的每日剂量使用。
在一些其他实施例中,本申请的药学组合物进一步包含唑吡坦。唑吡坦的每日剂量在100微克至100毫克、100微克至30毫克、100微克至10毫克、100微克至3毫克、100微克至1毫克、100微克至300微克、300微克至100毫克、300微克至30毫克、300微克至10毫克、300微克至3毫克、300微克至1毫克、1毫克至100毫克、1毫克至30毫克、1毫克至10毫克、1毫克至3毫克、10毫克至100毫克、10毫克至30毫克或30毫克至100毫克的范围内。
抗毒蕈碱剂、抗利尿剂、解痉剂、唑吡坦和/或PDE 5抑制剂可单独或与其他活性成分一起配制在药学组合物中以用于即刻释放、延长释放、延迟释放、延迟-延长释放或其组合。
在某些实施例中,药学组合物被配制成用于延长释放且包含(1)选自由乙酰基水杨酸、布洛芬、萘普生、萘普生钠、萘丁美酮、乙酰氨基酚和吲哚美辛所组成的组的止痛剂和(2)PDE 5抑制剂,如他达那非。
药学组合物可被配制成片剂、胶囊、糖衣锭(dragee)、散剂、颗粒剂、液体、凝胶或乳液形式。该液体、凝胶或乳液可以裸形式(naked form)或包含于胶囊中由个体摄入。
在一些实施例中,药学组合物包含单一止痛剂与单一PDE 5抑制剂。在一个实施例中,单一止痛剂为阿司匹林。在另一实施例中,单一止痛剂为布洛芬。在另一实施例中,单一止痛剂为萘普生或萘普生钠。在另一实施例中,单一止痛剂为吲哚美辛。在另一实施例中,单一止痛剂为萘丁美酮。在另一实施例中,单一止痛剂为乙酰氨基酚。在另一实施例中,单一PDE 5抑制剂为他达那非。止痛剂与PDE 5抑制剂可以前文所述范围内的剂量施用。
在一些实施例中,药学组合物个别地或组合地包含一种或多种止痛剂,其量在10至1000毫克、10至800毫克、10至600毫克、10至500毫克、10至400毫克、10至300毫克、10至250毫克、10至200毫克、10至150毫克、10至100毫克、30至1000毫克、30至800毫克、30至600毫克、30至500毫克、30至400毫克、30至300毫克、30至250毫克、30至200毫克、30至150毫克、30至100毫克、100至1000毫克、100至800毫克、100至600毫克、100至400毫克、100至250毫克、300至1000毫克、300至800毫克、300至600毫克、300至400毫克、400至1000毫克、400至800毫克、400至600毫克、600至1000毫克、600至800毫克或800至1000毫克之间,其中该组合物被配制成用于延长释放,其释放概况为其中一种或多种止痛剂在2至12小时或5至8小时的时间段内连续释放。
在一些实施例中,组合物被配制成用于延长释放,其释放概况为其中一种或多种止痛剂的至少90%在2至12小时或5至8小时的时间段内连续释放。
在一些实施例中,组合物被配制成用于延长释放,其释放概况为其中一种或多种止痛剂在5、6、7、8、10或12小时的时间段内连续释放。在一些实施例中,药学组合物进一步包含抗毒蕈碱剂、抗利尿剂、解痉剂、唑吡坦或PDE 5抑制剂。
在其他实施例中,组合物被配制成用于延长释放,其释放概况为其止痛剂在2至12小时或5至8小时的时间段内以稳定速率释放。在其他实施例中,组合物被配制成用于延长释放,其释放概况为其止痛剂在5、6、7、8、10或12小时的时间段内以稳定速率释放。本文所用的“在一时间段内的稳定速率”被定义为其中在给定时间段期间的任意点的释放速率为在此给定时间段内的平均释放速率的30%至300%以内的释放概况。举例而言,若80毫克阿司匹林在8小时的时间段内以稳定速率释放,则在此时间段期间的平均释放速率为10毫克/小时,且此时间段期间任意时间的实际释放速率在3毫克/小时至30毫克/小时的范围内(亦即,在8小时期间10毫克/小时的平均释放速率的30%至300%以内)。在一些实施例中,药学组合物进一步包含抗毒蕈碱剂、抗利尿剂、解痉剂、唑吡坦或PDE 5抑制剂。
在一些实施例中,止痛剂选自由阿司匹林、布洛芬、萘普生钠、萘普生、吲哚美辛、萘丁美酮和乙酰氨基酚所组成的组。在一个实施例中,止痛剂为乙酰氨基酚。药学组合物被配制成提供小量止痛剂的稳定释放来维持血液中的有效药物浓度,从而使单一剂量中的药物总量相较于即刻释放制剂减少。
在一些其他实施例中,药学组合物个别地或组合地包含一种或多种止痛剂,其量在10至1000毫克、10至800毫克、10至600毫克、10至500毫克、10至400毫克、10至300毫克、10至250毫克、10至200毫克、10至150毫克、10至100毫克、30至1000毫克、30至800毫克、30至600毫克、30至500毫克、30至400毫克、30至300毫克、30至250毫克、30至200毫克、30至150毫克、30至100毫克、100至1000毫克、100至800毫克、100至600毫克、100至400毫克、100至250毫克、300至1000毫克、300至800毫克、300至600毫克、300至400毫克、400至1000毫克、400至800毫克、400至600毫克、600至1000毫克、600至800毫克或800至1000毫克之间,其中止痛剂被配制成用于延长释放,其特征为双阶段释放概况,其中止痛剂的20%至60%在施用2小时内释放,且剩余部分在2至12小时或5至8小时的时间段内连续释放或以稳定速率释放。在又一个实施例中,止痛剂被配制成用于具有双阶段释放概况的延长释放,其中止痛剂的20%、30%、40%、50%或60%在施用2小时内释放,且剩余部分在2至12小时或5至8小时的时间段内连续释放或以稳定速率释放。在一个实施例中,止痛剂选自由阿司匹林、布洛芬、萘普生钠、萘普生、吲哚美辛、萘丁美酮、和乙酰氨基酚所组成的组。在一个实施例中,止痛剂为乙酰氨基酚。在另一实施例中,止痛剂为乙酰氨基酚。在一些实施例中,药学组合物进一步包含抗毒蕈碱剂、抗利尿剂、解痉剂、唑吡坦和/或PDE 5抑制剂。在一些实施例中,抗毒蕈碱剂、抗利尿剂、解痉剂、唑吡坦和/或PDE 5抑制剂被配制成用于即刻释放。
本发明的另一方面涉及一种降低排尿频率的方法,其通过对有需要的个体交替施用二种或更多种PG途径抑制剂以防止产生抗药性。在一个实施例中,该方法包含施用第一PG途径抑制剂持续第一时间段且随后施用第二PG途径抑制剂持续第二时间段。在另一个实施例中,该方法进一步包含施用第三PG途径抑制剂持续第三时间段。该第一PG途径抑制剂、第二PG途径抑制剂、和第三PG途径抑制剂彼此不同且可被配制成用于即刻释放、延长释放、延迟释放或其组合。
本发明的另一方面涉及一种治疗夜尿症的方法,其通过对有需要者施用利尿剂,接着施用本申请的药学组合物。利尿剂经定剂量且被配制成在施用6小时内具有利尿效应(diuretic effect),且在就寝时间前至少8或7小时施用。本申请的药学组合物被配制成用于延长释放或延迟延长释放,且在就寝时间前2小时内施用。
利尿剂的例子包括,但不限于酸化盐,如CaCl2和NH4Cl;精氨酸血管加压素受体2拮抗剂,如两性霉素B(amphotericin B)和柠檬酸锂;排水剂(aquaretics),如秋麒麟草(Goldenrod)和桧(Juniper);Na-H交换拮抗剂(Na-H exchanger antagonist),如多巴胺(dopamine);碳酸酐酶(carbonic anhydrase)抑制剂,如乙酰偶氮胺(acetazolamide)和杜塞酰胺(dorzolamide);环利尿剂(loop diuretics),如布美他尼(bumetanide)、依他尼酸(ethacrynic acid)、呋塞米(furosemide)和托拉塞米(torsemide);渗透利尿剂,如葡萄糖和甘露糖醇;保钾利尿剂(potassium-sparing diuretics),如阿米洛利(amiloride)、螺内酯(spironolactone)、氨苯喋啶(triamterene)、坎利酸钾(potassium canrenoate);噻嗪类(thiazides),如苄氟噻嗪(bendroflumethiazide)和氢氯噻嗪(hydrochlorothiazide);和黄嘌呤类(xanthines),如咖啡因(caffeine)、茶碱(theophylline)和可可碱(theobromine)。
本申请的另一方面涉及一种降低排尿频率的方法,其包括对有需要的个体施用有效量的本申请的药学组合物和有效量的肉毒杆菌毒素(botulinum toxin)。
在一些实施例中,肉毒杆菌毒素通过注射至膀胱肌中来施用,且经口对个体施用本申请的药学组合物。在一些实施例中,注射步骤包含在膀胱肌中的5至20个部位以每个部位2至10单位的注射剂量注射10至200单位的肉毒杆菌毒素。在一个实施例中,注射步骤包含在膀胱肌中的5个部位以每个部位2至10单位的注射剂量注射肉毒杆菌毒素。在另一实施例中,注射步骤包含在膀胱肌中的10个部位以每个部位2至10单位的注射剂量注射肉毒杆菌毒素。在另一实施例中,注射步骤包含在膀胱肌中的15个部位以每个部位2至10单位的注射剂量注射肉毒杆菌毒素。在又一实施例中,注射步骤包含在膀胱肌中的20个部位以每个部位2至10单位的注射剂量注射肉毒杆菌毒素。在一些实施例中,每3个月、4个月、6个月、8个月、10个月或12个月重复该注射步骤。
以下实施例进一步说明本发明,该实施例不应理解为限制本发明。本发明中引用的所有参考文件、专利、和公开专利申请的内容均以引用方式并入本文中。
例1:用布洛芬抑制尿意
招募20名志愿者个体(包括男性和女性),其各自均经历过尿意或排尿欲望提早而干扰其无法进行足够时间的睡眠以得到充分休息。每位个体在就寝时间前以单次施用的形式摄入400至800毫克的布洛芬。至少14位个体报告其可得到更佳的休息,因为其并未频繁地被尿意唤醒。
若干个体报告在每夜使用布洛芬数周后,不再感受到尿意频率减少的益处。然而,所有这些个体均进一步报告在停止服用此剂量数天后,益处恢复。最近测试已证实以低许多的剂量可达成类似结果,而益处于后续无任何减少。
例2:止痛剂、肉毒神经毒素、和抗毒蕈碱剂对巨噬细胞对炎性刺激和非炎性刺激反应的影响
实验设计
设计此研究来确定止痛剂和抗毒蕈碱剂在控制巨噬细胞对于由COX2和前列腺素(PGE、PGH等)所介导的炎性与非炎性刺激的反应的剂量和体外功效。建立对膀胱细胞中炎性与非炎性效应物(effector)的反应的基线(剂量与动力学(kinetic))。简而言之,在不存在或存在各种效应物的情形下,使所培养的细胞暴露于止痛剂和/或抗毒蕈碱剂。
效应物包括:脂多糖(LPS),一种致炎剂,以及COX2诱导物,作为炎性刺激;碳酰胆碱(carbachol)或乙酰胆碱,平滑肌收缩的刺激物,作为非炎性刺激;肉毒杆菌神经毒素A,一种已知的乙酰胆碱释放抑制剂,作为阳性对照;和花生油酸(arachidonic acid,AA)、γ次亚麻油酸(linolenic acid,DGLA)、或二十碳五烯酸(eicosapentaenoic acid,EPA),作为前列腺素的前驱体,该前列腺素由环氧合酶(cyclooxygenase)(COX1和COX2)和末端前列腺素合成酶连续氧化细胞内部的AA、DGLA或EPA而产生。
止痛剂包括:水杨酸酯,如阿司匹林;异丁基-丙酸-酚酸(iso-butyl-propanoic-phenolic acid)衍生物(布洛芬),如雅维(Advil)、美林(Motrin)、努普林(Nuprin)、和梅迪普莱恩(Medipren);萘普生钠,如阿莱韦(Aleve)、阿纳普思(Anaprox)、安特经(Antalgin)、超非明那斯(Feminax Ultra)、非拉那斯(Flanax)、因萨(Inza)、米朵延伸舒缓剂(Midol Extended Relief)、那捷新(Nalgesin)、能百镇(Naposin)、那皮兰(Naprelan)、那波捷新(Naprogesic)、那波新(Naprosyn)、那波新悬浮液、EC-那波新、那罗新(Narocin)、普伦森(Proxen)、迅斐利(Synflex)和新诺比(Xenobid);乙酸衍生物,如吲哚美辛(印多新(Indocin));1-萘乙酸(1-naphthaleneacetic acid)衍生物,如萘丁美酮或瑞力芬(relafen);N-乙酰基-对氨基苯酚(N-acetyl-para-aminophenol,APAP)衍生物,如乙酰氨基酚或对乙酰氨基酚(泰勒诺(Tylenol));和塞来昔布(Celecoxib)。
抗毒蕈碱剂包括奥昔布宁、索利那新、达非那新、和阿托品。
巨噬细胞经以下的试剂短期(1至2小时)或长期(24至48小时)刺激:
(1)每种止痛剂单独以各种剂量刺激。
(2)每种止痛剂在LPS存在的情形下以各种剂量刺激。
(3)每种止痛剂在碳酰胆碱或乙酰胆碱存在的情形下以各种剂量刺激。
(4)每种止痛剂在AA、DGLA或EPA存在的情形下以各种剂量刺激。
(5)肉毒杆菌神经毒素A单独以各种剂量刺激。
(6)肉毒杆菌神经毒素A在LPS存在的情形下以各种剂量刺激。
(7)肉毒杆菌神经毒素A在碳酰胆碱或乙酰胆碱存在的情形下以各种剂量刺激。
(8)肉毒杆菌神经毒素A在AA、DGLA或EPA存在的情形下以各种剂量刺激。
(9)每种抗毒蕈碱剂单独以各种剂量刺激。
(10)每种抗毒蕈碱剂在LPS存在的情形下以各种剂量刺激。
(11)每种抗毒蕈碱剂在碳酰胆碱或乙酰胆碱存在的情形下以各种剂量刺激。
(12)每种抗毒蕈碱剂在AA、DGLA或EPA存在的情形下以各种剂量刺激。
随后分析细胞的PGH2的释放;PGE的释放;PGE2的释放;前列腺环素的释放;凝血脂素的释放;IL-1β的释放;IL-6的释放;TNF-α的释放;COX2活性;cAMP和cGMP的产生;IL-1β、IL-6、TNF-α、和COX2mRNA的产生;和CD80、CD86、和MHC II类分子的表面表达。
材料和方法
巨噬细胞
在本研究中使用鼠类RAW264.7或J774巨噬细胞(由ATCC获得)。使细胞维持在含有添加有10%胎牛血清(FBS)、15毫摩尔浓度(mM)的HEPES、2毫摩尔浓度的左旋谷氨酰胺(L-glutamine)、100单位/毫升(U/ml)青霉素、和100微克/毫升(μg/ml)链霉素的RPMI 1640的培养基中。在5%CO2的气氛(atmosphere)中在37℃下培养细胞,细胞每周分裂(继代)一次。
在体外以止痛剂处理巨噬细胞
将RAW264.7巨噬细胞在100微升培养基中以每孔1.5×105个细胞的细胞密度接种(seed)于96孔培养板中。用以下各物处理细胞:(1)各种浓度的止痛剂(乙酰氨基酚、阿司匹林、布洛芬或萘普生)、(2)各种浓度的脂多糖(LPS),其为对巨噬细胞的炎性刺激效应物,(3)各种浓度的碳酰胆碱或乙酰胆碱,其为非炎性刺激的效应物,(4)止痛剂和LPS或(5)止痛剂和碳酰胆碱或乙酰胆碱。简而言之,将止痛剂溶解于无FBS的培养基(亦即,添加有15毫摩尔浓度的HEPES、2毫摩尔浓度的左旋谷氨酰胺、100单位/毫升青霉素和100微克/毫升链霉素的RPMI 1640)中,且使用相同的培养基通过系列稀释(serial dilution)而稀释至所需浓度。对于在不存在LPS的情形下经止痛剂处理的细胞,于每孔中添加50微升的止痛剂溶液以和50微升的无FBS培养基。对于在存在LPS的情形下经止痛剂处理的细胞,于每孔中添加50微升的止痛剂溶液和50微升在无FBS培养基中的LPS(来自鼠伤寒沙门氏菌(Salmonella typhimurium))。所有条件都进行二重复测试。
培养24或48小时后,收集150微升的培养物上清液,在4℃下以每分钟8,000转离心沉降(spin down)2分钟以移除细胞和碎片并储存在-70℃,以备通过ELISA来分析细胞因子反应。收集细胞并通过在500微升的磷酸盐缓冲液(PBS)中离心(4℃,每分钟1,500转,5分钟)来洗涤。随后将一半细胞在液态氮中速冻并储存在-70℃。剩余细胞经荧光单克隆抗体染色且由流式细胞仪进行分析。
对协同刺激分子表达的流式细胞仪分析
对于流式细胞仪分析,将巨噬细胞在100微升FACS缓冲液(具有2%牛血清白蛋白(bovine serum albumin,BSA)和0.01%NaN3的磷酸盐缓冲生理盐水)中稀释,并通过添加经FITC结合的抗CD40的抗体、经PE结合的抗CD80的抗体、经PE结合的抗CD86的抗体、抗MHC II类(I-Ad)PE(BD BioScience)而在4℃下染色30分钟。随后通过在300微升的FACS缓冲液中离心(4℃下,每分钟1,500转,5分钟)洗涤细胞。第二次洗涤后,将细胞再悬浮于200微升的FACS缓冲液中,并藉助于Accuri C6流式细胞仪(BD Biosciences)分析表达给定的标记(单阳性(single positive))或标记的组合(双阳性(double positive))的细胞百分比。
通过ELISA分析细胞因子反应
对培养物上清液进行细胞因子特异性的ELISA,以测定在经止痛剂、单独的LPS或LPS与止痛剂的组合处理的巨噬细胞培养物中的IL-1β、IL-6、和TNF-α反应。对在0.1摩尔浓度碳酸氢钠缓冲液(pH 9.5)中的100微升抗小鼠IL-6(anti-mouse IL-6)、TNF-α单克隆抗体(TNF-αmAb,BD Biosciences)或IL-1β单克隆抗体(IL-1βmAb,R&D Systems)包覆过夜的Nunc MaxiSorp Immunoplates(Nunc)进行分析。在以PBS(每孔200微升)洗涤两次之后,于每孔中添加200微升的PBS(3%BSA)(封闭,blocking)且将培养板在室温下培育2小时。于每孔添加200微升而将培养板再洗涤两次,添加二重复的100微升细胞因子标准物和培养物上清液的系列稀释液,并将培养板置于4℃下培育过夜。最后,将培养板洗涤两次并以100微升经二级生物素化(biotinylated)的抗小鼠IL-6、TNFα单克隆抗体(BD Biosciences)或IL-1β(R&D Systems)培育,接着以经过氧化物酶标记的山羊抗生物素单克隆抗体(peroxidase-labled goat anti-biotin mAb,Vector Laboratories)培育。通过添加2,2’-次偶氮基-双(3)-乙基苄基噻唑林-6-磺酸(2,2’-azino-bis(3)-ethylbenzylthiazoline-6-sulfonic acid,ABTS)底物和H2O2(Sigma)来进行比色反应(colorimetric reaction),且在415纳米下由V多标记培养板读取器(multilabel plate reader,PerkinElmer)来测量吸光度。
测定COX2活性以及cAMP和cGMP的产生
通过连续竞争性ELISA(sequential competitive ELISA,R&D Systems)来测定所培养巨噬细胞中COX2的活性。通过cAMP分析与cGMP分析来测定cAMP与cGMP的产生。该分析以本领域的常规手段进行。
表1总结了以Raw 264巨噬细胞株进行的试验以及关于止痛剂对协同刺激分子CD40和CD80的细胞表面表达的影响的主要发现。该分子的表达由COX2和炎性信号来刺激,且因此经评估而确定COX2的抑制作用的功能性结果。
如表2中所示,除了最高剂量(亦即,5×106纳摩尔浓度(nM)摩尔)以外,乙酰氨基酚、阿司匹林、布洛芬、和萘普生在所有测试剂量(亦即,5×105nM摩尔、5×104nM摩尔、5×103nM摩尔、5×102nM摩尔、50nM摩尔、和5nM摩尔)下均抑制巨噬细胞对协同刺激分子CD40与CD80的基础表达,而最高剂量似乎为增强而非抑制协同刺激分子的表达。如图1A和图1B所示,在低至0.05纳摩尔浓度(亦即,0.00005微摩尔浓度(μM))的止痛剂剂量下观测到对CD40和CD50表达的抑制效果。这个发现支持如下观点,即受控释放的小剂量止痛剂比急性递送的大剂量更佳。这个实验还表明,乙酰氨基酚、阿司匹林、布洛芬、和萘普生对LPS所诱发的CD40与CD80表达有类似的抑制效果。
表1.实验总结
表2.主要发现总结
ND*:未进行(毒性)
表3总结了测量成人口服治疗剂量后的止痛剂血清水平的若干研究的结果。如表3所示,在口服治疗剂量后,止痛剂的最大血清水平在104至105nM摩尔的范围内。因此,表2中的体外(in vitro)测试的止痛剂剂量涵盖在人类体内(in vivo)可达到的浓度范围。
表3.口服治疗剂量后,人类血液中止痛剂的血清水平
例3:止痛剂、肉毒神经毒素、和抗毒蕈碱剂对小鼠膀胱平滑肌细胞对炎性刺激和非炎性刺激反应的影响
实验设计
设计本研究旨在表征实施例2中所确定的止痛剂的最佳剂量如何影响细胞培养物或组织培养物中的膀胱平滑肌细胞,并解决不同种类的止痛剂是否可协同作用以更有效地抑制COX2和PGE2反应。
效应物、止痛剂和抗毒蕈碱剂描述于实施例2中。
小鼠膀胱平滑肌细胞的初级培养物经以下的试剂短期(1至2小时)或长期(24至48小时)刺激:
(1)每种止痛剂单独以各种剂量刺激。
(2)每种止痛剂在LPS存在的情形下以各种剂量刺激。
(3)每种止痛剂在碳酰胆碱或乙酰胆碱存在的情形下以各种剂量刺激。
(4)每种止痛剂在AA、DGLA、或EPA存在的情形下以各种剂量刺激。
(5)肉毒杆菌神经毒素A单独以各种剂量刺激。
(6)肉毒杆菌神经毒素A在LPS存在的情形下以各种剂量刺激。
(7)肉毒杆菌神经毒素A在碳酰胆碱或乙酰胆碱存在的情形下以各种剂量刺激。
(8)肉毒杆菌神经毒素A在AA、DGLA、或EPA存在的情形下以各种剂量刺激。
(9)每种抗毒蕈碱剂单独以各种剂量刺激。
(10)每种抗毒蕈碱剂在LPS存在的情形下以各种剂量刺激。
(11)每种抗毒蕈碱剂在碳酰胆碱或乙酰胆碱存在的情形下以各种剂量刺激。
(12)每种抗毒蕈碱剂在AA、DGLA、或EPA存在的情形下以各种剂量刺激。
随后分析细胞的PGH2的释放;PGE的释放;PGE2的释放;前列腺环素的释放;凝血脂素的释放;IL-1β的释放;IL-6的释放;TNF-α的释放;COX2活性;cAMP和cGMP的产生;IL-1β、IL-6、TNF-α、和COX2mRNA的产生;和CD80、CD86、和MHC II类分子的表面表达。
材料和方法
分离且纯化小鼠膀胱细胞
自安乐死动物C57BL/6小鼠(8至12周大)移除膀胱细胞,并通过酶消化、接着在Percoll梯度上纯化来分离细胞。简而言之,将来自10只小鼠的膀胱在10毫升的消化缓冲液(RPMI 1640,2%胎牛血清、0.5毫克/毫升胶原酶、30微克/毫升脱氧核糖核酸酶(Dnase))中用剪刀剪碎成细浆。使膀胱浆料在37℃下经酶消化30分钟。未消化的片段经细胞训练器(cell-trainer)进一步分散。将细胞悬浮液制成丸剂且添加至不连续的20%、40%和75%Percoll梯度,以供纯化单核细胞。每次实验使用50至60个膀胱。
在RPMI 1640中洗涤之后,将膀胱细胞再悬浮于添加有10%胎牛血清、15毫摩尔浓度(mM)的HEPES、2毫摩尔浓度的左旋谷氨酰胺、100单位/毫升青霉素和100微克/毫升链霉素的RPMI 1640中,并以100微升的每孔3×104个细胞的细胞密度接种于透明底黑色的96孔细胞培养微量培养板中。在37℃下在5%CO2的气氛中培养细胞。
在体外以止痛剂处理细胞
以止痛剂溶液(每孔50微升)单独或与碳酰胆碱(10摩尔浓度(Molar)摩尔,每孔50微升)(作为非炎性刺激的实例)一起处理膀胱细胞,或与鼠伤寒沙门氏菌的脂多糖(LPS)(1微克/毫升,每孔50微升)(作为炎性刺激的实例)一起处理膀胱细胞。当未向细胞添加其他效应物时,于孔中添加50微升的无胎牛血清的RPMI 1640,以将最终体积调整至200微升。
培养24小时后,收集150微升的培养物上清液并在4℃下以每分钟8,000转离心沉降2分钟以移除细胞和碎片,并储存在-70℃,以备通过ELISA来分析前列腺素E2(PGE2)反应。将细胞固定、透性化(permeabilized)、并封闭,以使用荧光底物来检测环氧合酶-2(COX2)。在选定的实验中,在体外刺激细胞12小时以分析COX2的反应。
分析COX2的反应
根据制造商的说明,通过以细胞为基础的ELISA,使用人类/小鼠总COX2免疫分析(human/mouse total COX2immunoassay,R&D Systems)来分析COX2的反应。简而言之,在细胞被固定和透性化后,于透明底黑色的96孔细胞培养微量培养板的孔中添加小鼠抗总COX2(mouse anti-total COX2)和兔抗总GAPDH(rabbit anti-total GAPDH)。在培育和洗涤后,添加经HRP结合的抗小鼠IgG(HRP-conjugated anti-mouse IgG)和经AP结合的抗兔IgG(AP-conjugated anti-rabbit IgG)至孔中。在另一次培育和另一组洗涤后,添加HRP荧光底物和AP荧光底物。最后,使用V多标记培养板读取器(PerkinElmer)来读取在600纳米(COX2荧光)和450纳米(GAPDH荧光)下的发射荧光。通过由相对荧光单位(RFU)测定并以看家蛋白(housekeeping protein)GAPDH为基准的COX2的相对水平表示结果。
分析PGE2的反应
通过连续竞争性ELISA(R&D Systems)来分析前列腺素E2的反应。更具体地说,将培养物上清液或PGE2标准物添加至涂布有山羊抗小鼠多株抗体(goat anti-mouse polyclonal antibody)的96孔聚苯乙烯微量培养板的孔中。在微量培养板震荡器上培育一小时后,添加经HRP结合的PGE2(HRP-conjugated PGE2),并将培养板置于室温下再培育两小时。随后洗涤培养板且于每孔中添加HRP底物溶液。显色30分钟,并在570纳米的波长校正下读取450纳米下的培养板之后,通过添加硫酸来终止反应。结果以PGE2的平均皮克/毫升来表示。
其他分析
如实施例2中所述测定PGH2的释放;PGE的释放、前列腺环素的释放;凝血脂素的释放;IL-1β的释放;IL-6的释放;和TNF-α的释放;cAMP和cGMP的产生;IL-1β、IL-6、TNF-α、和COX2mRNA的产生;和CD80、CD86、和MHC II类分子的表面表达。
结果
止痛剂抑制小鼠膀胱细胞对炎性刺激的COX2反应
在小鼠膀胱细胞上以5微摩尔浓度(μM)或50微摩尔浓度的浓度测试若干种止痛剂(乙酰氨基酚、阿司匹林、布洛芬、和萘普生),以确定止痛剂是否可诱发COX2反应。对24小时培养物的分析显示,所测试的止痛剂在体外对小鼠膀胱细胞均未诱发COX2反应。
亦测试于体外该止痛剂于小鼠膀胱细胞对碳酰胆碱或LPS刺激的COX2反应的影响。如表1所示,所测试的碳酰胆碱的剂量对小鼠膀胱细胞中COX2的水平并无显著效应。另一方面,LPS使总COX2水平显著增加。有趣的是,乙酰氨基酚、阿司匹林、布洛芬、和萘普生均可抑止LPS对COX2水平的影响。在以5微摩尔浓度或50微摩尔浓度测试该药物时(表4),可见止痛剂的抑止效果。
表4.在体外刺激并用止痛剂处理后小鼠膀胱细胞的COX2表达
止痛剂抑制小鼠膀胱细胞对炎性刺激的PGE2反应
测量小鼠膀胱细胞的培养物上清液中PGE2的分泌,以确定由止痛剂改变小鼠膀胱细胞COX2水平的生物学意义。如表5所示,在未受刺激的膀胱细胞或在碳酰胆碱存在的情形下培养的膀胱细胞的培养物上清液中并未检测到PGE2。与上述COX2反应一致,以LPS刺激小鼠膀胱细胞诱发分泌高水平的PGE2。添加止痛剂(乙酰氨基酚、阿司匹林、布洛芬、和萘普生)抑止LPS对PGE2分泌的影响,且在经5或50微摩尔浓度剂量的止痛剂处理的细胞反应之间并未见差异。
表5.在体外刺激并用止痛剂处理后小鼠膀胱细胞所分泌的PGE2
总而言之,这些数据显示,单独5微摩尔浓度(μM)或50微摩尔浓度的止痛剂在小鼠膀胱细胞中并不诱发COX2和PGE2反应。然而,5微摩尔浓度或50微摩尔浓度的止痛剂显著抑制体外的经LPS(1微克/毫升(μg/ml))刺激的小鼠膀胱细胞的COX2和PGE2反应。未观测到止痛剂对经碳酰胆碱(1毫摩尔浓度(mM))刺激的小鼠膀胱细胞的COX2和PGE2反应具有显著影响。
例4:止痛剂、肉毒神经毒素、和抗毒蕈碱剂对小鼠膀胱平滑肌细胞收缩的影响
实验设计
使培养的小鼠或大鼠膀胱平滑肌细胞和小鼠或大鼠膀胱平滑肌组织在各种浓度的止痛剂和/或抗毒蕈碱剂存在的情形下暴露于炎性刺激和非炎性刺激。测量刺激所诱发的肌肉收缩,以评估止痛剂和/或抗毒蕈碱剂的抑制效果。
效应物、止痛剂和抗毒蕈碱剂描述于实施例2中。
小鼠膀胱平滑肌细胞的初级培养物经以下的试剂短期(1至2小时)或长期(24至48小时)刺激:
(1)每种止痛剂单独以各种剂量刺激。
(2)每种止痛剂在LPS存在的情形下以各种剂量刺激。
(3)每种止痛剂在碳酰胆碱或乙酰胆碱存在的情形下以各种剂量刺激。
(4)每种止痛剂在AA、DGLA、或EPA存在的情形下以各种剂量刺激。
(5)肉毒杆菌神经毒素A单独以各种剂量刺激。
(6)肉毒杆菌神经毒素A在LPS存在的情形下以各种剂量刺激。
(7)肉毒杆菌神经毒素A在碳酰胆碱或乙酰胆碱存在的情形下以各种剂量刺激。
(8)肉毒杆菌神经毒素A在AA、DGLA、或EPA存在的情形下以各种剂量刺激。
(9)每种抗毒蕈碱剂单独以各种剂量刺激。
(10)每种抗毒蕈碱剂在LPS存在的情形下以各种剂量刺激。
(11)每种抗毒蕈碱剂在碳酰胆碱或乙酰胆碱存在的情形下以各种剂量刺激。
(12)每种抗毒蕈碱剂在AA、DGLA、或EPA存在的情形下以各种剂量刺激。
材料和方法
如实施例3所述分离初级小鼠膀胱细胞。在选定的实验中,使用膀胱组织培养物。以Grass多种波动描写器(Grass polygraph,Quincy Mass,USA)记录膀胱平滑肌细胞收缩。
例5:口服止痛剂和抗毒蕈碱剂对小鼠膀胱平滑肌细胞COX2和PGE2反应的影响
实验设计
给予正常小鼠和患有膀胱过动症(over active bladder syndrome)的小鼠口服剂量的阿司匹林、萘普生钠、布洛芬、印多新、萘丁美酮、泰勒诺、塞来昔布、奥昔布宁、索利那新、达非那新、阿托品、和其组合。对照组包括未经处理的正常小鼠和未经处理的患有膀胱过动症的OAB小鼠。给予最后剂量的30分钟后,收集膀胱并以碳酰胆碱或乙酰胆碱离体(ex vivo)刺激。在选定的实验中,在以碳酰胆碱刺激之前,先以肉毒杆菌神经毒素A对膀胱进行处理。使动物保持在代谢笼(metabolic cages)中并评估其排尿频率(和体积)。通过监控水摄入量和笼垃圾的重量来测定膀胱输出量。通过ELISA来测定血清PGH2、PGE、PGE2、前列腺环素、凝血脂素、IL-1β、IL-6、TNF-α、cAMP、和cGMP的水平。通过流式细胞仪测定全血细胞中的CD80、CD86、和MHC II类表达。
实验结束时,将动物安乐死,并以Grass多种波动描写器记录离体膀胱收缩。使膀胱部分固定于福尔马林(formalin)中,并通过免疫组织化学来分析COX2反应。
例6:止痛剂、肉毒神经毒素、和抗毒蕈碱剂对人类膀胱平滑肌细胞对炎性刺激和非炎性刺激反应的影响
实验设计
设计本研究旨在表征实施例1至实施例5中确定的止痛剂的最佳剂量如何影响细胞培养物或组织培养物中的人类膀胱平滑肌细胞,并解决不同种类的止痛剂是否可协同作用以更有效地抑制COX2和PGE2反应。
效应物、止痛剂和抗毒蕈碱剂描述于实施例2中。
人类膀胱平滑肌细胞经以下的试剂短期(1至2小时)或长期(24至48小时)刺激:
(1)每种止痛剂单独以各种剂量刺激。
(2)每种止痛剂在LPS存在的情形下以各种剂量刺激。
(3)每种止痛剂在碳酰胆碱或乙酰胆碱存在的情形下以各种剂量刺激。
(4)每种止痛剂在AA、DGLA、或EPA存在的情形下以各种剂量刺激。
(5)肉毒杆菌神经毒素A单独以各种剂量刺激。
(6)肉毒杆菌神经毒素A在LPS存在的情形下以各种剂量刺激。
(7)肉毒杆菌神经毒素A在碳酰胆碱或乙酰胆碱存在的情形下以各种剂量刺激。
(8)肉毒杆菌神经毒素A在AA、DGLA、或EPA存在的情形下以各种剂量刺激。
(9)每种抗毒蕈碱剂单独以各种剂量刺激。
(10)每种抗毒蕈碱剂在LPS存在的情形下以各种剂量刺激。
(11)每种抗毒蕈碱剂在碳酰胆碱或乙酰胆碱存在的情形下以各种剂量刺激。
(12)每种抗毒蕈碱剂在AA、DGLA、或EPA存在的情形下以各种剂量刺激。
随后分析细胞的PGH2的释放;PGE的释放;PGE2的释放;前列腺环素的释放;凝血脂素的释放;IL-1β的释放;IL-6的释放;TNFα的释放;COX2活性;cAMP和cGMP的产生;IL-1β、IL-6、TNFα、和COX2mRNA的产生;和CD80、CD86、和MHC II类分子的表面表达。
例7:止痛剂、肉毒神经毒素、和抗毒蕈碱剂对人类膀胱平滑肌细胞收缩的影响
实验设计
使培养的人类膀胱平滑肌细胞在各种浓度的止痛剂和/或抗毒蕈碱剂存在的情形下暴露于炎性刺激和非炎性刺激。测量刺激所诱发的肌肉收缩,以评估止痛剂和/或抗毒蕈碱剂的抑制效果。
效应物、止痛剂和抗毒蕈碱剂描述于实施例2中。
人类膀胱平滑肌细胞经以下的试剂短期(1至2小时)或长期(24至48小时)刺激:
(1)每种止痛剂单独以各种剂量刺激。
(2)每种止痛剂在LPS存在的情形下以各种剂量刺激。
(3)每种止痛剂在碳酰胆碱或乙酰胆碱存在的情形下以各种剂量刺激。
(4)每种止痛剂在AA、DGLA、或EPA存在的情形下以各种剂量刺激。
(5)肉毒杆菌神经毒素A单独以各种剂量刺激。
(6)肉毒杆菌神经毒素A在LPS存在的情形下以各种剂量刺激。
(7)肉毒杆菌神经毒素A在碳酰胆碱或乙酰胆碱存在的情形下以各种剂量刺激。
(8)肉毒杆菌神经毒素A在AA、DGLA、或EPA存在的情形下以各种剂量刺激。
(9)每种抗毒蕈碱剂单独以各种剂量刺激。
(10)每种抗毒蕈碱剂在LPS存在的情形下以各种剂量刺激。
(11)每种抗毒蕈碱剂在碳酰胆碱或乙酰胆碱存在的情形下以各种剂量刺激。
(12)每种抗毒蕈碱剂在AA、DGLA、或EPA存在的情形下以各种剂量刺激。
以Grass多种波动描写器(Quincy Mass,USA)记录膀胱平滑肌细胞收缩。
例8:止痛剂对正常人类膀胱平滑肌细胞对炎性刺激和非炎性刺激反应的影响
实验设计
培养正常人类膀胱平滑肌细胞
通过酶自目视正常的人类膀胱切片消化分离正常的人类膀胱平滑肌细胞。在体外通过以下扩繁细胞:在5%CO2的气氛中在37℃,在添加有10%胎牛血清、15毫摩尔浓度的HEPES、2毫摩尔浓度的左旋谷氨酰胺、100单位/毫升(U/ml)青霉素和100毫克/毫升链霉素的RPMI 1640中培养细胞,并通过胰蛋白酶处理细胞来分离细胞,接着再接种于新的培养瓶中来每周继代一次。在培养的第一周,对培养基添加0.5纳克/毫升表皮生长因子(epidermal growth factor)、2纳克/毫升纤维母细胞生长因子(fibroblast growth factor)和5微克/毫升胰岛素(insulin)。
在体外以止痛剂处理正常人类膀胱平滑肌细胞
以止痛剂溶液(每孔50微升)单独或与碳酰胆碱(10摩尔浓度(Molar),每孔50微升)(作为非炎性刺激的实例)一起,或与鼠伤寒沙门氏菌的脂多糖(LPS)(1微克/毫升,每孔50微升)(作为炎性刺激的例子)一起,处理经胰蛋白酶处理(trypsinized)且以100微升的每孔3×104个细胞的细胞密度接种于微量培养板中的膀胱平滑肌细胞。当未向细胞中添加其他效应物时,添加50微升的无胎牛血清的RPMI 1640至孔中,以将最终体积调整至200微升。
培养24小时后,收集150微升的培养物上清液,在4℃以每分钟8,000转离心沉降2分钟以移除细胞和碎片,并储存在-70℃,以备通过ELISA来分析前列腺素E2(PGE2)反应。将细胞固定、透性化并封闭,使用荧光底物来检测COX2。在选定的实验中,在体外刺激细胞12小时,以分析COX2、PGE2、和细胞因子反应。
分析COX2、PGE2和细胞因子反应
如实施例3所述分析COX2和PGE2反应。如实施例2所述分析细胞因子反应。
结果
止痛剂抑制正常人类膀胱平滑肌细胞对炎性与非炎性刺激的COX2反应-培养24小时后对细胞和培养物上清液的分析显示,所测试的止痛剂均未单独在正常人类膀胱平滑肌细胞中诱发COX2反应。然而,如表6所总结的,碳酰胆碱在正常人类膀胱平滑肌细胞中诱发低但显著的COX2反应。另一方面,LPS处理导致正常人类膀胱平滑肌细胞中更高水平的COX2反应。乙酰氨基酚、阿司匹林、布洛芬、和萘普生均可抑止碳酰胆碱和LPS对COX2水平的影响。在以5微摩尔浓度或50微摩尔浓度测试该药物时,可见止痛剂对LPS诱发的反应的抑止效果。
表6.在体外以炎性和非炎性刺激物刺激并用止痛剂处理后,正常人类膀胱平滑肌细胞的COX2表达
#数据表示为重复实验的平均值
止痛剂抑制正常人类膀胱平滑肌细胞对炎性与非炎性刺激的PGE2反应-与上述COX2反应的诱发一致,碳酰胆碱与LPS两者均诱发正常人类膀胱平滑肌细胞产生PGE2。亦发现乙酰氨基酚、阿司匹林、布洛芬、和萘普生在5微摩尔浓度或50微摩尔浓度下抑止LPS诱发的PGE2反应(表7)。
表7.在体外以炎性和非炎性刺激物刺激并用止痛剂处理后,正常人类膀胱平滑肌细胞的PGE2表达
#数据表示为重复实验的平均值
止痛剂抑制正常人类膀胱细胞对炎性刺激的细胞因子反应-在培养24小时后对细胞和培养物上清液的分析显示,所测试的止痛剂均未单独在正常人类膀胱平滑肌细胞中诱发IL-6或TNFα分泌。如表8和表9所示,所测试的碳酰胆碱剂量在正常人类膀胱平滑肌细胞中诱发低但显著的TNFα和IL-6反应。另一方面,LPS处理导致大量诱发该促炎性细胞因子。乙酰氨基酚、阿司匹林、布洛芬、和萘普生抑止碳酰胆碱和LPS对TNFα和IL-6反应的影响。在以5微摩尔浓度或50微摩尔浓度测试该药物时,可见止痛剂对LPS诱发的反应的抑止效果。
表8.在体外以炎性和非炎性刺激物刺激并用止痛剂处理后,正常人类膀胱平滑肌细胞的TNFα分泌
#数据表示为重复实验的平均值
表9.在体外以炎性和非炎性刺激物刺激并用止痛剂处理后,正常人类膀胱平滑肌细胞的IL-6分泌
#数据表示为重复实验的平均值
将初级正常人类膀胱平滑肌细胞分离、培养,并评估其在非炎性(碳酰胆碱)与炎性(LPS)刺激存在的情形下对止痛剂的反应。本研究的目标在于确定正常人类膀胱平滑肌细胞是否重现先前由鼠类膀胱细胞所得出的观测结果。
对在延迟释放制剂或延长释放制剂或延迟-延长释放制剂中的止痛剂和/或抗毒蕈碱剂重复上述实验。
以上描述用于教示本领域中具有通常知识者如何实践本发明,且其并非旨在详细描述熟习操作者在阅读此描述后将显而易见的本发明的所有那些明显修改和变化。然而,所有该明显修改和变化皆包括在本发明的范畴内,其由权利要求书来界定。除非上下文有明确相反指示,否则该权利要求旨在涵盖有效满足预期目标的所请求保护的成分和以任何次序的步骤。
- 还没有人留言评论。精彩留言会获得点赞!