癌症治疗剂的组合的制作方法
发明背景
在WO 2012/069146中公开了化合物A、其制备方法及其用于治疗癌症的用途。如在各种基于细胞的测定中证实的,这种化合物是p70S6K和Akt的新型选择性高效双重抑制剂。显示化合物A对广谱(broad panel of)癌细胞系表现出强效抗肿瘤活性。乳腺癌细胞、胶质母细胞瘤细胞、子宫内膜癌细胞和卵巢癌细胞对化合物A特别敏感。
蛋白激酶构成负责控制细胞内的多种信号转导过程的一大族结构相关酶(Hardie, G和Hanks, S. (1995) The Protein Kinase Facts Book, I and II, Academic Press, San Diego, CA)。激酶可通过被它们磷酸化的底物(例如蛋白质-酪氨酸、蛋白质-丝氨酸/苏氨酸、脂质等)分族。已经识别了大致与各激酶族对应的序列基序(例如Hanks, S.K., Hunter, T., FASEB J., 9:576-596 (1995);Knighton等人, Science, 253:407-414 (1991);Hiles等人, Cell, 70:419-429 (1992);Kunz等人, Cell, 73:585-596 (1993);Garcia-Bustos等人, EMBO J., 13:2352-2361 (1994))。
蛋白激酶可通过它们的调节机制表征。这些机制包括例如,自磷酸化、通过其它激酶转磷酸化、蛋白质-蛋白质相互作用、蛋白质-脂质相互作用和蛋白质-多核苷酸相互作用。可能通过多于一种机制调节单一蛋白激酶。
激酶通过将磷酸基团添加到靶蛋白上来调节许多不同的细胞过程,包括但不限于,增殖、分化、凋亡、运动、转录、翻译(translation)和其它信号传导过程。这些磷酸化事件充当可调制或调节靶蛋白生物功能的分子通/断开关(on/off switches)。响应各种细胞外信号(激素、神经递质、生长和分化因子等)、细胞周期事件、环境或营养应激等发生靶蛋白的磷酸化。适当的蛋白激酶在信号通路中用于活化或失活(直接或间接)例如代谢酶、调节蛋白、受体、细胞骨架蛋白、离子通道或泵或转录因子。在许多疾病中已经牵涉由蛋白质磷酸化的控制缺陷引起的不受控信号传导,所述疾病包括例如炎症、癌症、过敏症/哮喘、免疫系统疾病和病症、中枢神经系统疾病和病症和血管生成。
蛋白激酶70S6K——70 kDa核糖体蛋白激酶p70S6K(也称作SK6、p70/p85 S6激酶、p70/p85核糖体S6激酶和pp70S6K)是蛋白激酶的AGC亚族的成员。p70S6K是丝氨酸-苏氨酸激酶,其是磷脂酰肌醇3激酶(PI3K)/AKT通路的组分。p70S6K是PI3K的下游,并通过响应许多有丝分裂原、激素和生长因子在许多位点磷酸化而发生活化。p70S6K活性也受到含mTOR的复合物(TORC1)的控制,因为雷帕霉素起到抑制p70S6K活性的作用。通过PI3K下游靶AKT和PKCζ调节p70S6K。Akt将TSC2直接磷酸化和失活,由此活化mTOR。此外,使用受渥曼青霉素而不受雷帕霉素抑制的p70S6K的突变等位基因的研究表明,Pl3K通路可独立于mTOR活性的调节表现出对p70S6K的作用。
酶p70S6K通过S6核糖体蛋白的磷酸化调节蛋白质合成。S6磷酸化与翻译装置(translational apparatus)的mRNA编码组分的提高的翻译相关联,所述翻译装置包括核糖体蛋白和翻译延伸因子(其表达的提高对细胞生长和增殖是必不可少的)。这些mRNA在它们的5'转录起始点(被称作5'TOP)含有寡嘧啶片段(oligopyrimidime tract),已表明这对它们在翻译水平下的调节是必不可少的。
除其参与翻译外,还已经在细胞周期控制、神经细胞分化、细胞运动性的调节和在肿瘤转移、免疫响应和组织修复中重要的细胞响应中牵涉p70S6K活化。p70S6K的抗体消除促有丝分裂响应驱动的大鼠成纤维细胞进入S期,表明p70S6K功能对细胞周期中从G1进展到S期是必不可少的。此外,通过雷帕霉素在细胞周期的G1至S期的细胞周期增殖的抑制已被确定为是抑制产生过度磷酸化的活化形式的p70S6K的结果。
p70S6K在肿瘤细胞增殖和防止细胞免于细胞凋亡中的作用基于其参与肿瘤组织中的生长因子受体信号转导、过度表达和活化维持。例如,Northern和Western分析揭示,PS6K基因的扩增分别伴随着mRNA和蛋白质表达的相应提高(Cancer Res. (1999) 59: 1408-11-Localization of PS6K to Chromosomal Region 17q23 and Determination of Its Amplification in Breast Cancer)。
染色体17q23在多至20%的原发性乳腺肿瘤、87%的含有BRCA2突变的乳腺肿瘤和50%的含有BRCA1突变的肿瘤以及其它癌症类型,如胰腺癌、膀胱癌和成神经细胞瘤中扩增(参见M. Barlund, O. Monni, J. Kononen, R. Cornelison, J. Torhorst, G. Sauter, O.-P. Kallioniemi和Kallioniemi A., Cancer Res., 2000, 60:5340-5346)。已经表明,乳腺癌中的17q23扩增涉及PAT1、RAD51C、PS6K和SIGMA1B基因(Cancer Res. (2000): 60, 第5371-5375页)。已确定p70S6K基因是这一区域中的扩增和过度表达的靶并已经观察到扩增与不良预后之间的统计上显著的联系。
在用CCI-779(雷帕霉素酯)——上游激酶mTOR的抑制剂——治疗的肾癌患者中观察到p70S6K活化的临床抑制。报道了疾病进程与p70S6K活性的抑制之间的显著线性联系。
在响应能量应激时,肿瘤抑制因子LKB1活化AMPK,AMPK将TSC1/2复合物磷酸化并能够使其使mTOR/p70S6K通路失活。LKB1中的突变造成黑斑息肉综合征(PJS),其中患有PJS的患者产生癌症的可能性是普通人群的15倍。此外,1/3的肺腺癌潜伏有失活的LKB1突变。
在代谢疾病和障碍中已经牵涉p70S6K。据报道,p70S6K的缺失防止年龄和饮食诱发的肥胖症,同时提高胰岛素敏感性。基于这些发现支持了p70S6K在代谢疾病和障碍,如肥胖症、糖尿病、代谢综合征、胰岛素抵抗、高血糖、高氨基酸血症和高脂血症中的作用。
在WO 03/064397、WO 04/092154、WO 05/054237、WO 05/056014、WO 05/033086、WO 05/117909、WO 05/039506、WO 06/120573、WO 06/136821、WO 06/071819、WO 06/131835、WO 08/140947、WO 10/093419、WO 12/013282和WO 12/069146中公开了被描述为适合于p70S6K抑制的化合物。
已经表明,不仅抑制p70S6K还抑制激酶 Akt(在PI3K通路中在p70S6K的上游)的化合物A提供更有效的PI3K通路切断(Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Proc. Natl Acad Sci U S A. 2008年11月11日;105(45):17414-9.)并能够捕获任何Akt反馈回路活化(Tamburini等人Blood 2008;111:379–82)。
本发明的目的是找到进一步推进化合物A的药物效用的方式。在这方面,在体外和体内研究化合物A与HER2抑制剂的组合。
也被称作CD340(分化抗原簇340)或原癌基因Neu的受体酪氨酸-蛋白激酶erbB-2是人体中由ERBB2基因编码的蛋白质。ERBB2基因也常被称作HER2(来自人表皮生长因子受体)。
HER2是表皮生长因子受体(EGFR/ERBB)家族的成员。这种致癌基因的扩增或过度表达已表明在某些侵袭型乳腺癌的发展和进展中起到重要作用。近年来,蛋白质已成为大约30%乳腺癌患者的重要的生物标记和治疗靶。
ErbB家族由四种质膜结合的受体酪氨酸激酶构成。所有四种都含有胞外配体结合域、跨膜域和可与多种信号分子相互作用并表现出配体依赖性和配体不依赖性活性的胞内域。HER2可以与任何其它三种受体异二聚(heterodimerise)并被视为其它ErbB受体的优选二聚搭档。二聚造成受体的胞浆域内的酪氨酸残基的自磷酸化并引发各种信号通路。该家族的其它成员是表皮生长因子受体erbB-3(神经调节蛋白-结合;缺乏激酶域)和erbB-4。
通过HER2活化的信号通路包括:促分裂原活化蛋白激酶(MAPK)和磷脂酰肌醇3-激酶(PI3K/Akt)。
概括而言,通过ErbB受体家族的信号传导促进细胞增殖和抵抗细胞凋亡,因此必须严格调节以防止发生不受控细胞生长。
在大约15-30%的乳腺癌中发生ERBB2基因的扩增或过度表达。其与提高的疾病复发和不良预后强烈相关。过度表达也已知在卵巢癌、胃癌和侵袭形式的子宫癌,如子宫浆液性子宫内膜癌中发生。
此外,已经识别出造成这种受体的配体不依赖性firing的各种结构变化,在不存在受体过度表达的情况下实现。在各种肿瘤中发现HER2且这些肿瘤的一些在指定HER2的跨膜域的序列中带有点突变。用缬氨酸替代跨膜域中的谷氨酸可导致这种蛋白质在不存在配体的情况下的组成性二聚(constitutive dimerization)。
令人惊讶地,本专利申请的发明人已经发现,化合物A在与HER2抑制剂组合时以协同方式发挥作用。
HER2的抑制剂的一个实例是单克隆抗体曲妥珠单抗(作为Herceptin销售)。曲妥珠单抗是以高亲合力和特异性结合到HER2受体的胞外域上的高度纯化的重组DNA衍生的人源化单克隆IgGI κ抗体。其已被批准用于治疗HER2过度表达的癌症。
HER2的小分子抑制剂的一个实例是拉帕替尼(作为Tyverb销售)。
附图
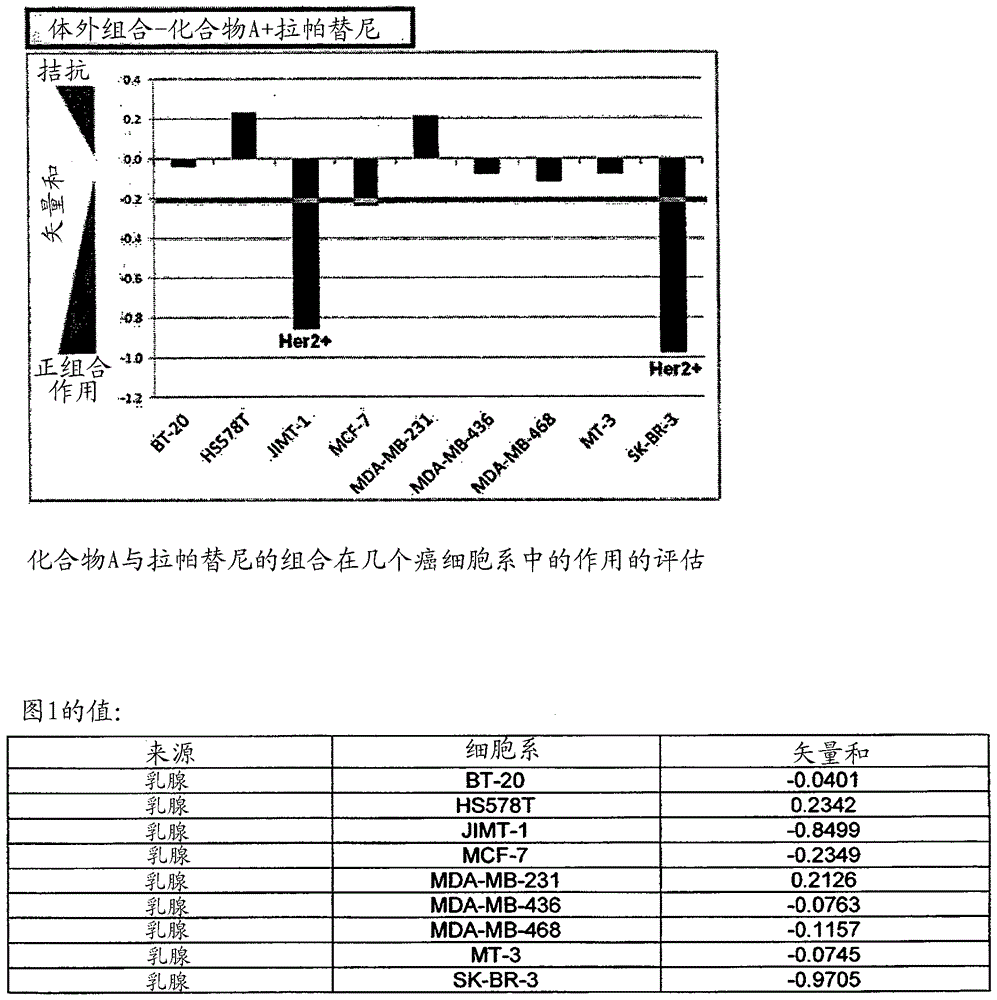
图1: 化合物A与拉帕替尼的组合在几个癌细胞系中的作用的评估。
图2: 化合物A与曲妥珠单抗的组合在乳腺癌的患者衍生移植瘤模型中的作用的评估。
发明详述
本发明涉及预防和/或治疗HER2过度表达的癌症(Her2+癌症)的方法,其包括将化合物A和/或其生理可接受盐和溶剂合物和一种或多种Her2抑制剂给药至对象。
化合物A和/或其生理可接受盐和溶剂合物和Her2抑制剂可以同时或相继给药。当同时给药时,化合物A和/或其生理可接受盐和溶剂合物和Her2抑制剂可以作为在一种药物组合物中的化合物混合物或作为分开的药物组合物给药。
在一个优选实施方案中,根据本发明的方法包括使用相继给药的化合物A和/或其生理可接受盐和溶剂合物和一种Her2抑制剂。在另一优选实施方案中,首先给药Her2抑制剂。
本发明特别涉及用于预防和/或治疗选自HER2+乳腺癌、胃癌或胃食管连接部腺癌的肿瘤的方法。但是,该治疗方法还涉及对曲妥珠单抗表现出响应的其它HER2+肿瘤类型,如膀胱癌、肺癌、卵巢癌、子宫内膜癌、食管癌、唾液腺癌等或基于它们的分子谱(molecular profile)响应曲妥珠单抗治疗的其它肿瘤。
在一个优选实施方案中,根据本发明的方法涉及癌症,且特别涉及上下文中描述的肿瘤的治疗。
此外,本发明涉及药物组合物,其包含活性药物成分(API)化合物A及其生理可接受盐和溶剂合物和一种Her2抑制剂的化合物混合物。如果Her2抑制剂是小化学分子,如拉帕替尼(而非生物分子,如抗体、抗体片段或抗体缀合物(conjugate)),该药物组合物中的化合物混合物还可包含这种小分子Her2抑制剂的生理可接受盐和溶剂合物。
合适的酸加成盐是所有生理或药物上可接受的酸的无机或有机盐,例如卤化物,特别是盐酸盐或氢溴酸盐,乳酸盐、硫酸盐、柠檬酸盐、酒石酸盐、马来酸盐、富马酸盐、草酸盐、乙酸盐、磷酸盐、甲磺酸盐、苯甲酸盐或对甲苯磺酸盐。
化合物A和小分子Her2抑制剂的溶剂合物被认为是指由于它们的相互吸引力而形成的惰性溶剂分子加合到化合物A上。溶剂合物是例如水合物,如一水合物或二水合物,或醇合物,即与醇,例如与甲醇或乙醇的加成化合物。
化合物A的优选盐形式是游离碱。还优选的是其盐酸盐、二盐酸盐、甲磺酸盐、琥珀酸盐或丙二酸盐。
表达“有效量”是指在组织、系统、动物或人体中产生例如研究人员或医师寻求或期望的生物或医学响应的药物或药物活性成分的量。
此外,表达“治疗有效量”是指与尚未接受这种量的相应对象相比具有下列结果的量:疾病、综合征、病症、不适、障碍的改进的治疗、治愈、预防或消除或副作用的预防,或减轻疾病、病症或障碍的进展。术语“治疗有效量”还包括有效提高正常生理机能的量。
根据本发明的药物组合物包含两种API的混合物,例如以1:1、1:2、1:3、1:4、1:5、1:10、1:100或1:1000的比率。
该药物组合物还包含至少一种固体、液体和/或半液体赋形剂或佐剂。因此,本发明还涉及包含根据本发明的所述API混合物和所述赋形剂和/或佐剂的药物组合物。
此外,本发明涉及所述药物组合物用于制备用于治疗癌症的药物的用途。
本发明还涉及套盒(set)(药盒(kit)),其由以下单独包装组成:
(a) 包含有效量的化合物A的药物组合物,
(b) 包含有效量的Her2抑制剂的药物组合物和任选的
(c) 包含有效量的第三癌症治疗剂的药物组合物。
该套盒包括合适的容器,如盒、独立瓶、袋或安瓿。该套盒可以例如包括单独的安瓿,各自含有溶解或冻干形式的包含有效量的化合物A和/或其可药用盐的药物组合物、包含有效量的Her2抑制剂和/或其可药用盐的药物组合物和任选的包含有效量的另一种癌症治疗剂的药物组合物。
可以根据本发明与化合物A和Her2抑制剂组合的癌症治疗剂可包括一种或多种,但优选一种下列试剂:
- 烷基化剂,如六甲蜜胺、苯达莫司汀、白消安、卡莫司汀、苯丁酸氮芥、氮芥、环磷酰胺、达卡巴嗪、异环磷酰胺、甲苯磺酸英丙舒凡、洛莫司汀、美法仑、二溴甘露醇、二溴卫矛醇、尼莫司汀、雷莫司汀、替莫唑胺、噻替派、苏消安、二氯甲基二乙胺(mechloretamine)、卡波醌、阿帕齐醌(apaziquone)、福莫司汀、葡磷酰胺、帕利伐米(palifosfamide)、哌泊溴烷、曲洛磷胺、尿嘧啶氮芥;
- 铂化合物,如卡铂、顺铂、依铂、米铂水合物、奥沙利铂、洛铂、奈达铂、吡铂、沙铂;
- DNA改变剂,如氨柔比星、比生群、地西他滨、米托蒽醌、甲基苄肼、曲贝替定、氯法拉滨、安吖啶、溴他里辛(brostallicin)、匹杉琼、laromustine;
- 拓扑异构酶抑制剂,如依托泊苷、伊立替康、雷佐生、索布佐生、替尼泊苷、拓扑替康、氨萘非特、贝洛替康、依利醋铵、伏利拉辛(voreloxin);
- 微管改进剂,如卡巴他赛、多西他赛、艾日布林、伊沙匹隆、紫杉醇、长春碱、长春新碱、长春瑞滨、长春地辛、长春氟宁、fosbretabulin、替司他赛(tesetaxel);
- 抗代谢物,如天冬酰胺酶、阿扎胞苷、左亚叶酸钙(caclium levofolinate)、卡培他滨、克拉屈滨、阿糖胞苷、依诺他滨、氟尿苷、氟达拉滨、氟尿嘧啶、吉西他滨、巯基嘌呤、氨甲喋呤、奈拉滨、培美曲塞、普拉曲沙、硫唑嘌呤、硫鸟嘌呤、卡莫氟、去氧氟尿苷、艾西拉滨、雷替曲塞、沙帕他滨(sapacitabine)、喃氟啶、三甲曲沙;
- 抗癌抗生素,如博来霉素、更生菌素、阿霉素、表阿霉素、伊达比星、左旋咪唑、米替福新、丝裂霉素C、罗米地辛、链脲菌素、戊柔比星、净司他丁、佐柔比星、柔红霉素(daunurobicin)、普卡霉素、阿柔比星、培洛霉素、吡柔比星;
- 激素/拮抗剂,如阿巴瑞克、阿比特龙、比卡鲁胺、布舍瑞林、卡普睾酮、氯烯雌醚、地加瑞克、地塞米松、雌二醇、氟考龙、氟甲睾酮、氟他米特、氟维司群、戈舍瑞林、组氨瑞林、亮丙瑞林、甲地孕酮、米托坦、那法瑞林、诺龙、尼鲁米特、奥曲肽、泼尼松龙、雷洛昔芬、他莫昔芬、促甲状腺素α、托瑞米芬、曲洛司坦、曲普瑞林、己烯雌酚、阿考比芬(acolbifene)、达那唑、德舍瑞林、环硫雄醇、orteronel、恩杂鲁胺;
- 芳香酶抑制剂,如氨鲁米特、阿那曲唑、依西美坦、法倔唑、来曲唑、睾内酯、福美坦;
- 小分子激酶抑制剂,如克唑替尼、达沙替尼、埃罗替尼、伊马替尼、拉帕替尼、尼洛替尼、帕唑帕尼、瑞格非尼、鲁索替尼、索拉非尼、舒尼替尼、凡德他尼、威罗菲尼、博舒替尼、吉非替尼、阿昔替尼、阿法替尼、阿利吉仑、达拉菲尼、达可替尼(dacomitinib)、dinaciclib、多韦替尼、恩扎妥林(enzastaurin)、尼达尼布(nintedanib)、乐伐替尼(lenvatinib)、利尼伐尼(linifanib)、linsitinib、马赛替尼、米哚妥林、莫替沙尼、来那替尼、orantinib、哌立福辛、帕纳替尼、拉多替尼、rigosertib、替吡法尼、tivantinib、tivozanib、曲美替尼、pimasertib、丙氨酸布立尼布、西地尼布、阿帕替尼、卡博替尼S-苹果酸盐、卡非佐米、依鲁替尼、埃克替尼;
- 光敏剂,如甲氧沙林、卟吩姆钠、他拉泊芬、替莫泊芬;
- 抗体,如阿仑单抗、贝西索单抗(besilesomab)、贝伦妥单抗维多汀(brentuximab vedotin)、西妥昔单抗、狄诺塞麦、易普利单抗(ipilimumab)、奥法木单抗、帕尼单抗、利妥昔单抗、托西莫单抗、曲妥珠单抗、贝伐单抗、卡妥索单抗、elotuzumab、依帕珠单抗、farletuzumab、mogamulizumab、necitumumab、尼妥珠单抗、阿托珠单抗(obinutuzumab)、ocaratuzumab、奥戈伏单抗(oregovomab)、雷莫芦单抗(ramucirumab)、利妥木单抗(rilotumumab)、可妥昔单抗(siltuximab)、托珠单抗(tocilizumab)、托鲁木单抗(zalutumumab)、扎木单抗(zanolimumab)、马妥珠单抗、达妥珠单抗(dalotuzumab)、onartuzumab、帕妥珠单抗、racotumomab、tabalumab;
- 细胞因子,如阿地白介素、干扰素α、干扰素α2a、干扰素α2b;他索纳明、替西白介素、奥普瑞白介素;
- 药物缀合物,如地尼白介素(denileukin diftitox)、替伊莫单抗(ibritumomab tiuxetan)、碘苄胍I123、泼尼莫司汀、曲妥珠单抗、雌莫司汀、吉妥珠单抗、奥佐米星、阿柏西普、cintredekin besudotox、依多曲肽(edotreotide)、奥英妥珠单抗(inotuzumab ozogamicin)、那莫单抗(naptumomab estafenatox)、奥莫珠单抗(oportuzumab monatox)、锝(99mTc) 阿西莫单抗、vintafolide;
- 疫苗,如前列腺癌疫苗(sipuleucel)、维特斯朋(vitespen)、emepepimut-S、oncoVAX、rindopepimut、troVax、stimuvax;
- 其它试剂,如阿利维A酸、贝沙罗汀、硼替佐米、依维莫司、伊班膦酸、咪喹莫特、来那度胺、香菇多糖、甲酪氨酸、米伐木肽、帕米膦酸、培门冬酶、喷司他丁、sipuleucel、西佐喃、他米巴罗汀、坦西莫司(temsirolimus)、沙利度胺、维A酸、维莫德吉(vismodegib)、唑来膦酸、沙利度胺、伏立诺他、塞来昔布、西仑吉肽、恩替诺特、依他硝唑、ganetespib、idronoxil、iniparib、ixazomib、氯尼达明、尼莫拉唑、帕比司他(panobinostat)、培瑞维A酸(peretinoin)、plitidepsin、泊马度胺、procodazol、ridaforolimus、他喹莫德(tasquinimod)、telotristat、胸腺法新、替拉扎明、托多司他(tosedostat)、trabedersen、乌苯美司、伐司朴达、今又生(gendicine)、溶链菌(picibanil)、reolysin、瑞他霉素盐酸盐、trebananib、维鲁利秦(virulizin)。
化合物A和Her2抑制剂的特别优选的组合搭档是Her3抑制剂,如MM-121(特异性阻断HRG1-(神经调节蛋白-1 I型多肽)结合到Her3上的完全人源化抗-Her3抗体)、MM-111(结合到两种不同的靶蛋白ErbB2和ErbB3上的双特异性抗体)或U3-1287(AMG888,第一完全人源化Her3单克隆抗体)或如WO 2011/144749中所述的Her3纳米抗体。
根据本发明的化合物和化合物混合物可适于通过任何所需的合适方法给药,例如通过经口(包括口腔或舌下)、直肠、经鼻、局部(包括口腔、舌下或经皮)、阴道或肠胃外(包括皮下、肌肉内、静脉内或皮内)方法给药。可以使用制药领域中已知的所有方法通过例如将活性成分与一种或多种赋形剂或一种或多种佐剂合并来制备这样的药物。
适合口服给药的化合物和化合物混合物可作为独立单位,例如胶囊或片剂;粉末或颗粒;在水性或非水性液体中的溶液或悬浮液;可食用泡沫或泡沫食品;或水包油液体乳剂或油包水液体乳剂给药。
因此,例如,在以片剂或胶囊形式口服给药的情况下,可以将该化合物或化合物混合物与口服、无毒和药物上可接受的惰性赋形剂,例如乙醇、甘油、水等合并。通过将该化合物研碎至合适的细尺寸并将其与以类似方式研碎的药物赋形剂,例如可食用的碳水化合物,例如淀粉或甘露醇混合制备粉末。还可能存在调味料、防腐剂、分散剂和染料。
通过如上所述制备粉末混合物并用其填充成形明胶壳生产胶囊。可以在填充操作之前将助流剂和润滑剂,例如固体形式的高度分散硅酸、滑石、硬脂酸镁、硬脂酸钙或聚乙二醇添加到该粉末混合物中。也可以添加崩解剂或增溶剂,例如琼脂、碳酸钙或碳酸钠以改进服用胶囊后该化合物或化合物混合物的可用率。
此外,如果需要或必要,也可以将合适的粘合剂、润滑剂和崩解剂以及染料并入该混合物中。合适的粘合剂包括淀粉、明胶、天然糖,例如葡萄糖或β-乳糖,由玉米制成的甜味剂、天然和合成胶(rubber),例如阿拉伯树胶、黄蓍胶或藻酸钠,羧甲基纤维素、聚乙二醇、蜡等。这些剂型中所用的润滑剂包括油酸钠、硬脂酸钠、硬脂酸镁、苯甲酸钠、乙酸钠、氯化钠等。崩解剂包括,但不限于,淀粉、甲基纤维素、琼脂、膨润土、黄原胶等。通过例如制备粉末混合物、粒化或干压该混合物、添加润滑剂和崩解剂并将整个混合物压成片剂来配制片剂。通过将以合适方式研碎的化合物与如上所述的稀释剂或基料和任选与粘合剂,例如羧甲基纤维素、藻酸盐、明胶或聚乙烯基吡咯烷酮,溶解阻滞剂,例如石蜡,吸收促进剂,例如季铵盐,和/或吸收剂,例如膨润土、高岭土或磷酸二钙混合,制备粉末混合物。可通过用粘合剂,例如糖浆、淀粉糊、acadia粘液或纤维素或聚合物材料的溶液润湿粉末混合物并将其压过筛子来粒化该粉末混合物。代替粒化,可以使该粉末混合物经过压片机,产生形状不均匀的团块,将其打碎以形成颗粒。可以通过添加硬脂酸、硬脂酸盐、滑石或矿物油来润滑颗粒以防止粘着到片剂铸模上。然后将润滑的混合物压成片剂。根据本发明的化合物和化合物混合物也可以与自由流动的惰性赋形剂合并,然后在不进行粒化或干压步骤的情况下直接压成片剂。可以存在由虫胶密封层、糖或聚合物材料层和蜡光泽层构成的透明或不透明保护层。可以将染料添加到这些包衣中以便能区分不同的剂量单位。
口服液体,例如溶液、糖浆和酏剂可以以剂量单位形式制备以使所给的量包含预定量的该化合物。可以通过将该化合物和化合物混合物溶解在含有合适调味料的水溶液中来制备糖浆,而酏剂使用无毒醇类载体制备。可以通过将该化合物分散在无毒载体中来配制悬浮液。也可以加入增溶剂和乳化剂,例如乙氧基化异硬脂醇和聚氧乙烯山梨糖醇醚,防腐剂、调味料添加剂,例如薄荷油,或天然甜味剂或糖精,或其它人工甜味剂等。
如果需要,用于口服给药的剂量单位制剂可包封在微胶囊中。也可以以使得延长或延迟释放的方式制备该制剂,例如通过将微粒材料包衣或包埋在聚合物、蜡等中。
根据本发明的化合物和化合物混合物及其盐和溶剂合物也可以以脂质体递送体系,例如小单层囊泡、大单层囊泡和多层囊泡的形式给药。脂质体可以由各种磷脂,例如胆甾醇、十八烷基胺或磷脂酰胆碱形成。
根据本发明的化合物和化合物混合物也可以使用单克隆抗体作为独立载体(该化合物分子偶联到其上)递送。该化合物和化合物混合物也可以偶联到作为靶向药物载体的可溶聚合物上。这样的聚合物可包括聚乙烯基吡咯烷酮、吡喃共聚物、聚羟丙基甲基丙烯酰氨基酚、聚羟乙基天冬酰氨基(aspartamido)酚或聚环氧乙烷聚赖氨酸,其被棕榈酰基取代。该化合物还可偶联到适合实现药物控释的一类可生物降解的聚合物,例如聚乳酸、聚ε-己内酯、聚羟基丁酸、聚原酸酯、聚缩醛、聚二羟基吡喃、聚氰基丙烯酸酯和水凝胶的交联或两亲嵌段共聚物上。
适合经皮给药的化合物和化合物混合物可作为独立的膏药给药以与受体的表皮长时间密切接触。因此,例如,可以通常如Pharmaceutical Research, 3(6), 318 (1986)中所述通过离子电渗从膏药递送活性成分。
适合局部给药的化合物和化合物混合物可配制为软膏、乳膏、悬浮液、洗剂、粉末、溶液、糊剂、凝胶、喷雾剂、气雾剂或油。
为了治疗眼睛或其它外部组织,例如口腔和皮肤,该制剂优选以局部软膏或乳膏的形式施加。在配制成软膏的情况下,该化合物或化合物混合物可以与石蜡或水混溶性膏基一起使用。或者,该化合物或化合物混合物可以用水包油膏基或油包水膏基配制成乳膏。
适合局部施加于眼睛的化合物和化合物混合物包括滴眼剂,其中将活性成分溶解或悬浮在合适的载体,特别是水性溶剂中。
适合在口腔中局部施加的化合物和化合物混合物包括锭剂、软锭剂和漱口剂。
适合直肠给药的化合物和化合物混合物可以以栓剂或灌肠剂的形式给药。
其中载体物质是固体的适合经鼻给药的化合物和化合物混合物包括粒度为例如20-500微米的粗粉末,其以鼻吸的方式给药,即通过经鼻腔通道从靠近鼻子放置的含有粉末的容器快速吸入。以液体作为载体物质的作为鼻喷剂或鼻滴剂给药的适合制剂包括在水或油中的活性成分溶液。
适合通过吸入给药的化合物和化合物混合物包含可由各种类型的含气雾剂的加压分配器、喷雾器或吹入器产生的细微粒状粉尘或雾剂。
适合阴道给药的化合物和化合物混合物可作为子宫托、棉塞、乳膏、凝胶、糊剂、泡沫或喷雾制剂给药。
适合肠胃外给药的化合物和化合物混合物包括包含抗氧化剂、缓冲剂、抑菌剂和溶质(借此使该制剂与待治疗的受体的血液等渗)的水性和非水性无菌注射液;和可包含悬浮介质和增稠剂的水性和非水性无菌悬浮液。该制剂可以在单剂或多剂容器,例如密封安瓿和管瓶中给药并以冷冻干燥(冻干)状态储存,以致只需在临使用前添加无菌载体液体,例如注射用水。可以由无菌粉末、颗粒和片剂制备根据配方制备的注射液和悬浮液。
无需说,除上文特别提到的成分外,根据本发明的药物还可包含本领域中关于药物制剂的特定类型常见的其它试剂;因此,例如,适合口服给药的化合物或化合物混合物可包含调味料。
本发明的化合物或化合物混合物的治疗有效量取决于许多因素,包括例如受体的年龄和体重、需要治疗的确切病症及其严重程度、制剂的性质和给药方法,并最终由治疗医生或兽医决定。但是,根据本发明的用于治疗疾病的API的有效量通常为0.1至100毫克/千克受体(哺乳动物)体重/天,且特别通常为1至10毫克/千克体重/天。因此,用于体重70千克的成年哺乳动物的每天实际量通常为70至700毫克,其中该量可作为每天单剂给药或更通常以每天一系列分剂量(例如2、3、4、5或6)给药,以使总日剂量相同。可作为根据本发明的化合物和化合物混合物本身的有效量的分数确定其盐或溶剂合物或生理功能衍生物的有效量。
根据本发明的药物配制品可用作人药和兽药中的药物。合适的赋形剂是适合肠内(例如口服)、肠胃外或局部给药并且不与该新型化合物反应的有机或无机物质,例如水、植物油、苄醇、聚乙二醇、明胶、碳水化合物如乳糖或淀粉、硬脂酸镁、滑石或凡士林。片剂、包衣片剂、胶囊、糖浆、汁液、滴剂或栓剂特别适合肠内给药,溶液,优选基于油的溶液或水性溶液,以及悬浮液、乳剂或埋植剂适合肠胃外给药,且软膏、乳膏或粉末适合局部给药。该化合物和化合物混合物也可冻干且所得冻干产物例如用于制备注射配制品。
所示配制品可以是无菌的和/或包含佐剂,如润滑剂、防腐剂、稳定剂和/或润湿剂、乳化剂、用于改变渗透压的盐、缓冲物质、染料、调味料和/或芳香物质。如果需要,它们也可包含一种或多种其它活性成分,例如一种或多种维生素。
实施例
下列实施例涉及药物配制品:
实施例A1: 注射管瓶
使用2N盐酸将100克根据本发明的化合物或化合物混合物和5克磷酸氢二钠在3升再蒸馏水中的溶液调节到pH 6.5,无菌过滤,转移到注射管瓶中,冻干并在无菌条件下密封。各注射管瓶含有5毫克活性成分。
实施例A2: 栓剂
将20克根据本发明的化合物或化合物混合物与100克大豆卵磷脂和1400克可可脂一起熔融,倾倒到模具中并使其冷却。各栓剂含有20毫克活性成分。
实施例A3: 溶液
由1克根据本发明的化合物或化合物混合物、9.38克NaH2PO4 x 2 H2O、28.48克NaH2PO4x 12 H2O和0.1克苯扎氯铵在940毫升再蒸馏水中制备溶液。将pH调节到6.8,并将该溶液补充到1升并通过辐照灭菌。该溶液可以以滴眼剂的形式使用。
实施例A4: 软膏
将500毫克根据本发明的化合物或化合物混合物与99.5克凡士林在无菌条件下混合。
实施例A5: 片剂
以传统方式压制1千克根据本发明的化合物或化合物混合物、4千克乳糖、1.2千克马铃薯淀粉、0.2千克滑石和0.1千克硬脂酸镁以产生片剂,以这样的方式使各片剂含有10毫克活性成分。
实施例A6: 包衣片剂
类似于实施例E压制片剂并随后以传统方式用蔗糖、马铃薯淀粉、滑石、黄蓍胶和染料的衣料包衣。
实施例A7: 胶囊
将2千克根据本发明的化合物或化合物混合物以传统方式引入硬明胶胶囊中,以这样的方式使各胶囊含有20毫克活性成分。
实施例A8: 安瓿
将1千克根据本发明的化合物或化合物混合物在60升再蒸馏水中的溶液转移到安瓿中,在无菌条件下冻干并在无菌条件下密封。各安瓿含有10毫克活性成分。
下列实施例涉及使用化合物A和Her2抑制剂的组合研究。
实施例B1: 在九个人类乳腺癌细胞系中的化合物A和拉帕替尼的组合
细胞培养和生长抑制测定的实验程序:
在供应商推荐的培养基中在与10% FCS (PAN, Germany)一起供应的100 U/ml青霉素G和100 μg/ml链霉素存在下使细胞系生长。
在96孔微滴定板中进行细胞生长和处理。将通过胰蛋白酶化从对数期培养物中收取的细胞在190微升培养基中以最佳接种密度培养(plate)。测定各细胞系的最佳接种密度以确保在实验期间的对数生长。如目视检查确定的,无抗癌剂时生长的所有细胞在该处理结束时都是近汇合的(sub-confluent)。在96孔硬质PCR板中进行在DMSO中的化合物稀释。然后将化合物在RPMI培养基中1:50稀释。190微升细胞在24小时预生长期后通过与10微升含化合物的培养基(以使最终DMSO浓度为0.1%)混合被处理。使细胞在37℃下生长72小时。此外,所有实验包括几个使用加工成在24小时恢复期后立即测量的细胞的板。这些板含有关于在处理前在时间零点存在的细胞数的信息并用于计算细胞毒性和/或生长抑制效应。在处理后,通过添加10% TCA沉淀细胞。在固定前,如所述抽出该培养基 [Pauwels等人, 2003]。在4℃下培养1小时后,用400微升去离子水将板洗涤两次。然后用100微升0.08% wt/v SRB将细胞染色。将板静置至少30分钟并用1%乙酸洗涤六次以除去未结合的染色 [Vichai和Kirtikara, 2006]。使板在室温下干燥并用100微升10 mM Tris碱溶解结合的SRB。在Victor 2酶标仪(plate reader)(Perkin Elmer, Germany)上在560纳米下进行光学密度的测量。
实验设计:
在体外组合研究之前,使用80个细胞系的组研究各试剂的活性。该浓度范围为选择特定细胞系的浓度范围提供指导。通过在96孔板中合并这些浓度的拉帕替尼和这些浓度的化合物A的矩阵(matrix),测试该组合。将这些试剂同时添加到细胞中。
所用化合物的浓度列在下表中:
。
在所有细胞系中使用6x6矩阵测试化合物A与拉帕替尼的成对组合。设计该筛选以测定潜在的协同组合。使用所有和/或一部分6x6矩阵设计该研究。
计算协同作用的方法可见于[Berenbaum, 1989]。计算下列参数
其中i = [1..n]是所用矩阵的值之一且理论值i如对Bliss Independence方法所述的计算[Berenbaum, 1989]
矢量(vector)和如下测定
在此术语中矢量和代表标量
低于-0.5的平均值表明强协同效应:(-0.5, -0.02] – 协同效应,(-0.02, 0.02) - 0效应(相加效应),(0.02, 0.5) - 潜在拮抗,和高于0.5 - 强拮抗。
文献
M.C. Berenbaum. What is synergy? Pharmacol Reviews, 41:93–141, 1989.
Bea Pauwels, Annelies E. C. Korst, Christel M. J. de Pooter, Greet G. O. Pattyn, Hilde A. J. Lambrechts, Marc F. D. Baay, Filip Lardon和Jan B. Vermorken. Comparison of the sulforhodamine b assay and the clonogenic assay for in vitro chemoradiation studies. Cancer chemotherapy and pharmacology, 51:221–226, 2003年3月.
Vanicha Vichai和Kanyawim Kirtikara. Sulforhodamine b colorimetric assay for cytotoxicity screening. Nature protocols, 1:1112–1116, 2006年8月.
实施例B2: 在乳腺癌的患者衍生移植瘤模型中的化合物A和曲妥珠单抗的组合
5-7周的雌性裸鼠(nude)(Harlan;nu/nu)皮下移植来自从宿主动物中收获的Her2+患者衍生乳腺模型"CTG-0033"(传代3,即该模型已从原始宿主连续传代/生长3次)的肿瘤片段。当宿主肿瘤达到1-1.5立方厘米时,收获肿瘤以再移植到用于疗效研究的动物中。当CTG-0033(传代4)肿瘤达到大约190立方毫米时,将动物根据肿瘤体积随机分成治疗组或对照组(n = 10)并在第0天开始给药。治疗是载体(盐水)、化合物A 30 mg/kg QD(每日)PO(口服)、Herceptin(30 mg/kg负荷剂量和15 mg/kg维持剂量) QW(每周)IV(静脉)和与Herceptin 15 mg/kg QW IV组合的化合物A 30 mg/kg QD PO。每周记录肿瘤体积两次。在第55天由于大肿瘤而终止载体治疗组。其它治疗在第75天停止并在小鼠不接受治疗的情况下使肿瘤重新生长超过2个月。在此研究中两个动物死亡。化合物A组中的一个动物在第9天死于疑似强饲错误,且化合物A + Herceptin组中的另一动物死于人为错误。
在第55天,用化合物A 30 mg/kg治疗(单药治疗组)导致50%肿瘤消退,而用化合物A + Herceptin治疗导致78%消退。在第55天,用Herceptin治疗导致37的%T/C。在第55天,用化合物A + Herceptin治疗与单试剂Herceptin和化合物A相比显著抑制肿瘤生长(P<.05;2 Way RM-ANOVA Bonferonni Post Hoc Test)。在载体组终止的同时,动物继续接受化合物A、Herceptin或化合物A + Herceptin直至第75天,并允许肿瘤重新生长。在第76天,用化合物A治疗导致-66%消退,而组合组完全消退(肿瘤体积小于40立方毫米)。在治疗停止后的71天再生长期后,化合物A治疗肿瘤中的6个重新生长,而组合治疗肿瘤无一重新生长。用化合物A与化合物A和Herceptin相比的治疗的肿瘤的重新生长的差异是统计上显著的(对数秩和检验p<.05)。组合组中的所有动物被视为治愈,因为肿瘤没有重新生长。
- 还没有人留言评论。精彩留言会获得点赞!