用于免疫调节的方法和组合物与流程
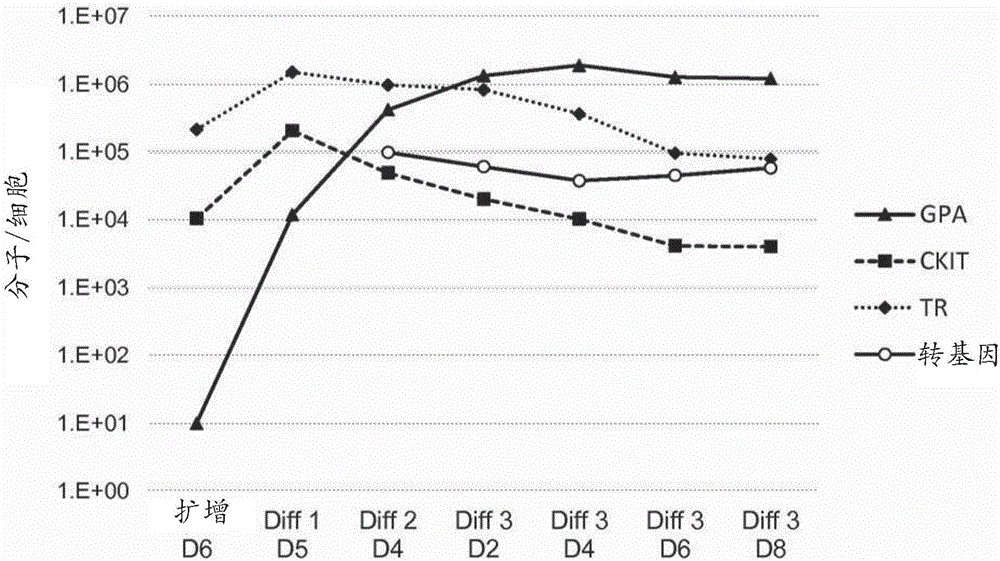
本发明的领域是用于治疗疾病和障碍的药物组合物。发明背景异常免疫激活是许多人类疾病和病症的标志。当身体免疫系统不当感测自体抗原作为异物并且攻击自身组织时,自身免疫性疾病发生。当身体免疫系统由常见食源性或环境抗原不当触发时,可能会出现炎性疾病和过敏症。用来治疗一系列人类疾病的多肽和蛋白经常被对其发生响应的免疫细胞破坏、中和、或以其他方式失去效力,就好像它们是外源抗原。目前对不适当的免疫激活的疾病的治疗涉及用以下各项进行免疫抑制:化学试剂像皮质激素,或炎症介质的抑制剂像抗组胺、抗体或细胞因子。这些一般性治疗与显著发病率有关,如对感染的易感性,因为它们广泛抑制免疫系统。针对一些严重的过敏症,通过暴露于随着时间缓慢增加剂量的侵入性蛋白进行临床测试来诱导对过敏原的“耐受性”。迄今为止,这些治疗缺少长期效力并且与严重的过敏反应的风险有关。存在对治疗这些疾病的新颖疗法的需求。发明概述在某些方面,本发明涉及表达抗原的分离的去核造血细胞。本文中将去核造血细胞称为“EHCs”(或以其单数形式:“EHC”)。在一些实施例中,去核造血细胞或EHC缺少核材料。例如,这些EHC可以是类红细胞或拟血栓细胞。在一些实施例中,缺乏核材料的EHC是血红细胞、红细胞、网织红细胞、或血小板。在一些实施例中,这些去核造血细胞或EHC是有核前体类红细胞或前体拟血栓细胞,这些细胞,例如,诱导失去其核材料或者功能上使其去核并且不能复制。在一些实施例中,表达外源抗原的EHC是循环细胞,如血红细胞。在一些实施例中,使用定义的因子从造血前体培养表达外源抗原的EHC。在一些实施例中,表达外源抗原的EHC是拟血栓细胞,如血小板。在一些实施例中,使用定义的因子从造血前体培养拟血栓细胞。在一些实施例中,该表达外源抗原的EHC是分离自患者的与抗原接触的原代细胞,用于自体或同种异体用途。本发明的某些方面涉及表达外源抗原的EHC,当向受试者给予(例如,以包括表达外源抗原的EHC的药物组合物的形式)时,这些EHC能够诱导免疫耐受。可以针对具体疾病、障碍或病症定制由EHC表达的外源抗原。表达外源抗原的EHC能以多个方式包括抗原,如,例如,感兴趣的抗原的表面展示、细胞内表达、细胞内荷载、或表面结合。表达外源抗原的EHC可以更有效管理异常的免疫激活的疾病和/或具有比现有治疗更少的副作用。例如,表达外源抗原的EHC可以选择性地调节免疫系统,同时使更广泛的免疫系统生理学基本上未受干扰。在一些实施例中,表达外源抗原的EHC可以诱导抗原特异性T和B淋巴细胞的破坏、钝化、和/或无反应性。可替代地或此外,表达外源抗原的EHC可以诱导抗原特异性调节性T淋巴细胞的增殖。本发明的某些方面涉及表达外源抗原的EHC,这些表达外源抗原的EHC包括被自身免疫性疾病中免疫细胞识别的外源抗原,这些自身免疫性疾病如,例如,多发性硬化症、1型糖尿病、类风湿性关节炎、和膜性肾炎。本发明的某些方面涉及表达外源抗原的EHC,这些表达外源抗原的EHC包括被炎性疾病中免疫细胞识别的外源抗原,这些炎性疾病如,例如,克罗恩病、溃疡性结肠炎、乳糜泻、或其他自发性的炎性肠病。本发明的某些方面涉及表达外源抗原的EHC,这些表达外源抗原的EHC包括被人白细胞抗原(HLA)错配介导的疾病中免疫细胞识别的外源抗原,这些人白细胞抗原(HLA)错配介导的疾病如,例如,移植物抗宿主疾病或器官移植排斥。本发明的某些方面涉及表达外源抗原的EHC,这些表达外源抗原的EHC包括被过敏性疾病中免疫细胞识别的外源抗原,这些过敏性疾病如,例如,哮喘、花生过敏、贝类过敏、花粉过敏、牛奶蛋白过敏、昆虫叮咬过敏、和乳胶过敏。本发明的某些方面涉及表达外源抗原的EHC,这些表达外源抗原的EHC包括是一种治疗性蛋白的外源抗原,该治疗性蛋白的疗效或效力被免疫细胞减少或损伤,该治疗性蛋白如,例如,血友病A中的凝固因子VIII、血友病B中的凝固因子IX、类风湿性关节炎及其他炎性疾病中的抗肿瘤坏死因子(TNFα)抗体、戈谢病中的葡糖脑苷脂酶、或急性成淋巴细胞白血病(ALL)中的天冬酰胺酶。本发明的某些方面涉及包括外源抗原的表达外源抗原的EHC,该外源抗原包括多肽的全长、截断、和嵌合融合,其a)介导补体调节,b)介导免疫复合物的结合和螯合,c)自身免疫抗体,或d)致病颗粒。在一些实施例中,外源抗原包括多肽的全长、截断、和嵌合融合,其在一个小分子底物转化成另一个小分子产物或一个多肽底物转化成第二多肽产物(包括,例如,多肽底物的裂解)中具有酶活性。在某些方面,本发明还提供了使用表达外源抗原的EHC和本文中提供的其药物组合物治疗疾病的方法。在一些方面,本文中披露了诱导免疫耐受的方法。该方法包括向患有或处于形成自身免疫性疾病、障碍或病症风险的人类受试者给予包括表达外源抗原的去核造血细胞的药物组合物,其中将该药物组合物以一定量进行给予,该量在受试者中有效地诱导对介导该自身免疫性疾病、障碍或病症的抗原的免疫耐受。在一些实施例中,该自身免疫性疾病选自下组,该组由以下各项组成:多发性硬化症、1型糖尿病、和表F中列出的那些。在一些实施例中,该方法进一步包括在治疗期内给予该药物组合物至少两次,这样治疗了自身免疫性疾病、障碍或病症,或者减少了其症状。在某些实施例中,该方法进一步包括在治疗期内给予该药物组合物至少两次,这样预防了自身免疫性疾病、障碍或病症。在其他实施例中,该方法进一步包括在治疗期内给予该药物组合物足够次数,这样使得在治疗期间实质上减少了抗原特异性免疫细胞的百分数。在一些实施例中,该免疫细胞是T细胞。在一些实施例中,该免疫细胞是B细胞。在一些实施例中,从取自受试者的生物样品中通过流式细胞术测量抗原特异性免疫细胞的浓度的减少。在一些实施例中,该生物样品是淋巴结活检、脾样品、或外周血。在一些实施例中,在部分或整个治疗期间,抗原特异性免疫细胞的浓度减少至少约1%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%、99.9%、99.99%、或大于99.99%。在其他实施例中,在约1、5、10、15、20、30、40、或50分钟,或约1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、或23小时,或1、2、3、4、5、或6天或约1、2、3、4、5、或6周的给予期内,抗原特异性免疫细胞的浓度减少至少约1%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%、99.9%、99.99%、或大于99.99%。在一些实施例中,在治疗期内给予该药物组合物足够次数,这样使得抗原特异性免疫细胞的浓度实质上减少持续至少约一周、两周、三周、四周、一个月、两个月、三个月、四个月、五个月、六个月、或大于六个月。在某些实施例中,在治疗期内给予该药物组合物足够次数,这样使得抗原特异性免疫细胞的浓度实质上减少持续至少与治疗期一样长的一段时间。在一些实施例中,在治疗期内给予该药物组合物足够次数,这样使得在治疗期间,实质上减少了循环中的抗原特异性的抗体的浓度。在一些实施例中,通过ELISA测量循环中抗原特异性抗体的浓度。在一些实施例中,在部分或整个治疗期间,抗原特异性循环抗体的浓度减少至少约1%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%、99.9%、99.99%、或大于99.99%。在一些实施例中,在约1、5、10、15、20、30、40、或50分钟,或约1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、或23小时,或1、2、3、4、5、或6天或约1、2、3、4、5、或6周的给予期内,抗原特异性的抗体的浓度减少至少约1%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%、99.9%、99.99%、或大于99.99%。在某些实施例中,在治疗期内给予该药物组合物足够次数,这样使得抗原特异性的循环抗体的浓度实质上减少持续至少约一周、两周、三周、四周、一个月、两个月、三个月、四个月、五个月、六个月、或大于六个月。在某些实施例中,在治疗期内给予该药物组合物足够次数,这样使得抗原特异性的循环抗体的浓度实质上减少持续至少与治疗期一样长的一段时间。在一些实施例中,在治疗期内给予该药物组合物足够次数,这样使得在治疗期间,实质上增加了抗原特异性的调节T细胞的百分数。在一些实施例中,从取自受试者的生物样品中通过流式细胞术测量抗原特异性免疫细胞的浓度的减少。在一些实施例中,该生物样品是淋巴结活检、脾样品、或外周血。在某些实施例中,在部分或整个治疗期间,抗原特异性调节T细胞的浓度增加至少约1%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%、99.9%、99.99%、或大于99.99%。在某些实施例中,在约1、5、10、15、20、30、40、或50分钟,或约1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、或23小时,或1、2、3、4、5、或6天或约1、2、3、4、5、或6周的给予期内,抗原特异性的调节T细胞的浓度增加至少约1%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%、99.9%、99.99%、或大于99.99%。在一些实施例中,在治疗期内给予该药物组合物足够次数,这样使得抗原特异性的调节T细胞的浓度实质上增加持续至少约一周、两周、三周、四周、一个月、两个月、三个月、四个月、五个月、六个月、或大于六个月。在一些实施例中,在治疗期内给予该药物组合物足够次数,这样使得抗原特异性的调节T细胞的浓度实质上增加持续至少与治疗期一样长的一段时间。在一些实施例中,在治疗期内给予该药物组合物足够次数,这样使得预防、减少或延迟自身免疫性疾病、障碍或病症的一种或多种症状。在一些实施例中,该治疗期不超过一年、六个月、三个月、两个月、一个月、两周、一周、三天、两天、一天。在某些实施例中,在治疗期内给予之间的时间间隔不超过该治疗期,其中表达外源抗原的去核造血细胞的数量减少至少于所给予的药物组合物中存在的表达外源抗原的去核造血细胞的数量的约5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、或95%。在一些实施例中,给予的频率足以有效减少抗原特异性免疫细胞的浓度到低于与自身免疫性疾病、障碍或病症的症状有关的水平。在某些实施例中,给予的频率足以有效减少抗原特异性循环抗体的浓度到低于与自身免疫性疾病、障碍或病症的症状有关的水平。在某些实施例中,给予的频率足以有效增加抗原特异性调节T细胞的浓度到高于与自身免疫性疾病、障碍或病症的症状有关的阈值水平。在一些实施例中,该去核造血细胞是类红细胞、拟血栓细胞、或其前体。在一些实施例中,该类红细胞是红细胞或网织红细胞。在一些实施例中,该拟血栓细胞是血小板。在一些实施例中,该去核造血细胞分离自供体。在一些实施例中,该去核造血细胞自体衍生自该受试者。在一些实施例中,该去核造血细胞是同种异体衍生的。在一些实施例中,该去核造血细胞是异基因衍生的。在一些实施例中,通过诱导排出其细胞核的基于培养的方法,该有核造血细胞衍生自有核前体细胞。在一些实施例中,该去核造血细胞产生自有核前体细胞,该有核前体细胞经化学上或物理上操纵以去除其细胞核。在一些实施例中,通过辐射或化学破坏有核前体细胞的细胞核来生产去核造血细胞。在一些实施例中,用细胞松弛素B进行化学破坏。在一些实施例中,用至少5Gy、7Gy、10Gy、15Gy、25Gy、30Gy、40Gy或至少50Gy进行辐射。在一些实施例中,该外源抗原是由外源核酸编码的多肽。在一些实施例中,该外源抗原与去核造血细胞的细胞膜有关。在一些实施例中,该外源抗原是该融合或嵌合多肽。在一些实施例中,该融合体或嵌合体包括S结构域、A结构域或U结构域中的至少一种,其中该S结构域是在去核造血细胞上的表面结构域,其中该A结构域是细胞膜中或其上的锚,其中该U结构域面向去核造血细胞的细胞内、非暴露面,并且其中该S结构域、A结构域、和/或U结构域具有不同多肽来源。在一些实施例中,该S结构域和/或A结构域包括至少5、6、7、8、9、10、15、20、25、30、40、50、60、70、80、90、100、150、200、250、或者至少500个氨基酸。在一些实施例中,该S结构域和/或A结构域包括至少500、750、或者至少1,000个氨基酸。在一些实施例中,该外源抗原选自下组,该组由以下各项组成:髓磷脂碱性蛋白、蛋白脂质蛋白、髓磷脂少突胶质细胞糖蛋白、胰β细胞抗原、胰岛素、以及表F、表6、和表8中所列出的那些。在一些实施例中,该去核造血细胞包括至少10个拷贝、100个拷贝、1,000个拷贝、10,000个拷贝、25,000个拷贝、50,000个拷贝、100,000个拷贝、500,000个拷贝、1,000,000个拷贝、或2,000,000个拷贝的外源抗原。在一些实施例中,该药物组合物进一步包括药学上有活性的试剂。在一些实施例中,该方法进一步包括以下步骤:给予药学上有活性的试剂,其中将该药学上有活性的试剂在药物组合物之前、之后、或与之同时给予。在一些实施例中,经静脉内给予该药物组合物。在一些实施例中,该药学上有活性的试剂选自:生物剂、小分子剂、或核酸剂。在一些实施例中,该药物组合物组合物进一步包含一种药学上可接受的载体。在某些实施例中,该方法进一步包括以下步骤:针对治选择疗患有或处于选自下组的自身免疫性疾病、障碍或病症的风险的受试者,该组由以下各项组成:血栓性血小板减少性紫癜、CAPS、APS、重症肌无力、古德帕斯彻氏综合征、膜性肾炎、1型糖尿病、类风湿性关节炎、多发性硬化症、克罗恩病或表F和表G中所列出的那些。在一些方面中,本文中所披露的是包括表达外源抗原的去核造血细胞的药物组合物,其中给予有效量的药物组合物能够诱导人类受试者中的免疫耐受,该人类受试者患有或处于形成自身免疫性疾病、障碍或病症的风险,该自身免疫性疾病、障碍或病症由以上权利要求中任一项所述的方法进行管理。在一些实施例中,该药物组合物组合物进一步包含一种药学上可接受的载体。在一些实施例中,该药物组合物包括表达外源抗原的造血细胞群。在一些实施例中,该药物组合物包括至少1x103个表达外源抗原的造血细胞。在药物组合物的某些实施例中,以约10nl、100nl、1μl、10μl、100μl、1ml、10ml、20ml、或50ml体积提供了表达外源抗原的造血细胞。在药物组合物的其他实施例中,以约1ml、10ml、20ml、50ml、100ml、250ml、或500ml体积提供了表达外源抗原的造血细胞。在药物组合物的一些实施例中,配制该组合物用于长期储存。在药物组合物的一些实施例中,该组合物是冷冻的。在一些实施例中,该药物组合物包括药学上有活性的试剂。在药物组合物的某些实施例中,该药学上有活性的试剂选自:生物剂、小分子剂、或核酸剂。在一些方面,本文中披露了一种剂型,该剂型包括本文中描述的配置为用于静脉内注射的液体悬浮液的组合物。在一些方面,本文中披露了一种医疗装置,该医疗装置包括容纳本文中所描述的药物组合物的容器,和一种向受试者静脉内注射该药物组合物的施药器。在一些方面,本文中披露了一种医疗试剂盒,该医疗试剂盒包括本文中所描述的药物组合物,和一种向受试者静脉内注射该药物组合物的医疗装置。在一些方面,本文中披露了通过本文中所描述的方法中任一种所给予的药物组合物的、表达外源抗原的造血细胞。在一些方面,本文中披露了表达本文中所披露的外源抗原的造血细胞群。在一些实施例中,将表达外源抗原的造血细胞群配制为一种液体。在一些实施例中,表达外源抗原的造血细胞群是冷冻的。在一些方面,本文中披露了由本文中所描述的造血细胞群所表达的分离抗原。在一些方面,本文中披露了编码本文中所描述的外源抗原的外源核酸。在一些方面,本文中披露了包括外源抗原的去核造血细胞,该外源抗原包括S结构域、A结构域或U结构域中至少一种,其中该S结构域是细胞外表面结构域,该A结构域是锚,并且该U结构域是细胞内定位的,并且其中当向受试者给予时,该去核造血细胞能够诱导免疫耐受。在本文中所披露的去核造血细胞的一些实施例中,该外源抗原是融合或嵌合多肽。在本文中所披露的去核造血细胞的一些实施例中,该S结构域、A结构域、和/或U结构域具有不同的多肽来源。在本文中所披露的去核造血细胞的某些实施例中,该S结构域和/或A结构域包括至少5、6、7、8、9、10、15、20、25、30、40、50、60、70、80、90、100、150、200、250、或者至少500个氨基酸。在本文中所披露的去核造血细胞的某些实施例中,该S结构域和/或A结构域包括至少500、750、或者至少1,000个氨基酸。在本文中所披露的去核造血细胞的一些实施例中,该外源抗原选自下组,该组由以下各项组成:髓磷脂碱性蛋白、蛋白脂质蛋白、髓磷脂少突胶质细胞糖蛋白、胰β细胞抗原、胰岛素、以及表F、表6、和表8中所列出的那些。在一些实施例中,该去核造血细胞包括至少10个拷贝、100个拷贝、1,000个拷贝、10,000个拷贝、25,000个拷贝、50,000个拷贝、100,000个拷贝、500,000个拷贝、1,000,000个拷贝、或2,000,000个拷贝的外源抗原。在某些实施例中,该去核造血细胞是类红细胞、拟血栓细胞、或其前体。在一些实施例中,该类红细胞是红细胞或网织红细胞。在一些实施例中,该拟血栓细胞是血小板。在一些实施例中,该去核造血细胞分离自供体。在一些实施例中,该去核造血细胞自体衍生自该受试者。在一些实施例中,该去核造血细胞是同种异体衍生的。在一些实施例中,该去核造血细胞是异基因衍生的。在某些实施例中,通过诱导排出其细胞核的基于培养的方法,该去核造血细胞衍生自有核前体细胞。在某些实施例中,该去核造血细胞产生自有核前体细胞,该有核前体细胞经化学上或物理上操纵以去除其细胞核。在某些实施例中,通过辐射或化学破坏有核前体细胞的细胞核来生产去核造血细胞。在一些实施例中,用细胞松弛素B进行化学破坏。在一些实施例中,用至少5Gy、7Gy、10Gy、15Gy、25Gy、30Gy、40Gy或至少50Gy进行辐射。在本文中所披露的去核造血细胞的一些实施例中,该外源抗原是由外源核酸编码的多肽。在本文中所披露的去核造血细胞的一些实施例中,该细胞衍生自人类来源。在本文中所披露的去核造血细胞的一些实施例中,该细胞衍生自选自下组的非人类来源,该组由以下各项组成:猪、黑猩猩、猕猴、非人类灵长动物、和非灵长类哺乳动物。在本文中所披露的去核造血细胞的一些实施例中,该多肽抗原是细胞内定位的。在本文中所披露的去核造血细胞的一些实施例中,该多肽抗原是在细胞表面上在细胞外定位的。在本文中所披露的去核造血细胞的一些实施例中,该多肽抗原融合到内源性细胞蛋白。在本文中所披露的去核造血细胞的一些实施例中,该多肽抗原融合到内源性跨膜蛋白质的胞内区。在本文中所披露的去核造血细胞的一些实施例中,该多肽抗原融合到内源性跨膜蛋白质的胞外区。在本文中所披露的去核造血细胞的一些实施例中,该多肽抗原融合到糖基磷脂酰肌醇(GPI)锚定蛋白。在一些方面,本文中披露了一批包括本文中所描述的去核造血细胞的组织培养。在一些方面,本文中披露了本文中所描述的去核造血细胞群。在一些方面,本文中披露了包括在此描述的细胞群的药物组合物。在一些方面,本文中披露了诱导免疫耐受的方法,该方法包括向对其有需要的受试者以足够在该受试者中诱导免疫耐受的量和/或频率给予本文中所描述的药物组合物。在一些方面,本文中披露了治疗免疫激活疾病的方法,该方法包括向对其有需要的受试者以足够治疗免疫激活疾病的量和/或频率给予本文中所描述的药物组合物。在一些实施例中,该疾病选自下组,该组由以下各项组成:自抗体介导的疾病、自身免疫性疾病、炎性疾病、过敏性疾病、HLA-不匹配介导的疾病、和可由免疫原性治疗性蛋白治疗的疾病。在一些方面,本文中披露了响应于治疗性蛋白治疗方案的、减少或缓解免疫激活的方法,该方法包括向对其有需要的受试者以足够实质上减少或缓解免疫激活的量和/或频率给予本文中所描述的药物组合物。在一些实施例中,该治疗性蛋白选自下组,该组由以下各项组成:表I、表J、和表7中所列出的那些。在一些方面,本发明所披露了包括编码内源性类红细胞蛋白的核酸序列的表达载体,该内源性类红细胞蛋白与选自下组的一种或多种外源性多肽抗原融合,该组由以下各项组成:表F、表G、表H、表I、表J、表6、表7、和表8中所列出的那些。在一些方面,本发明所披露了包括编码内源性类红细胞蛋白的核酸序列的信使RNA,该内源性类红细胞蛋白与选自下组的一种或多种外源性多肽抗原融合,该组由以下各项组成:表F、表G、表H、表I、表J、表6、表7、和表8中所列出的那些。在一些方面,本发明所披露了诱导免疫耐受的方法,该方法包括向患有或处于形成过敏原介导的疾病、障碍或病症风险的人类受试者给予包括表达外源抗原的去核造血细胞的药物组合物,其中将该药物组合物以一定量进行给予,该量在该受试者中有效地诱导对介导该疾病、障碍或病症的过敏原的免疫耐受。在某些实施例中,该外源抗原选自下组,该组由以下各项组成:Arah2、2S白蛋白、透明质酸酶、和表H中所列出的那些。在某些实施例中,该过敏原介导的疾病、障碍或病症选自下组,该组由以下各项组成:花生过敏、树坚果过敏、昆虫毒液过敏、和表H中所列出的那些。在一些方面,本发明所披露了诱导免疫耐受的方法,该方法包括向患有或处于形成人白细胞抗原(HLA)错配介导的疾病、障碍或病症风险的人类受试者给予包括表达外源抗原的去核造血细胞的药物组合物,其中将该药物组合物以一定量进行给予,该量在该受试者中有效地诱导对介导该疾病、障碍或病症的HLA的免疫耐受。在一些方面,本发明所披露了诱导免疫耐受的方法,该方法包括向患有或处于形成可以通过免疫原性治疗性分子治疗的疾病、障碍或病症风险的人类受试者给予包括表达外源抗原的去核造血细胞的药物组合物,其中将该药物组合物以一定量进行给予,该量在该受试者中有效地诱导对用于治疗该疾病、障碍或病症的免疫原性治疗性分子的免疫耐受。在一些实施例中,该治疗性分子选自下组,该组由以下各项组成:重组体(因子VIII)、贝赋(因子IX)、修美乐(抗-TNFα)、以及表I、表J、和表7中所列出的那些。附图简要说明图1是血红细胞的流式细胞术图的集合,该血红细胞与在脂质体中封装的荧光标记的IgG接触。显示出用无脂质体(左)、低剂量的脂质体(中)、和高剂量的脂质体(右)孵育的细胞。在底部直方图上,显示出了荧光细胞的百分比。图2是通过定量流式细胞术评估的细胞表面表达水平的图。该图显示出在类红细胞分化的过程中各种细胞表面受体-血型糖蛋白A(实心三角)、cKIT(虚线正方形)、转铁蛋白受体(点线菱形)-和外源表面转基因(空心环)。图3A-C、F、I-M、O-Z、和AA-AU是流式细胞术图和免疫印迹的集合,该集合证实在三种细胞类型(去核类红细胞、有核红细胞前体细胞、和红白血病细胞)中,作为融合体和作为完整蛋白质,在细胞质中,表面上的大量示例性抗原的表达。图3A-C、F、I-M、和O-S显示出去核培养的类红细胞上的表面和胞浆蛋白的外源性表达。图3A-通过共同翻译的GFP的表达评估的在胞质的C末端处具有HA表位标签的血型糖蛋白A的表达。图3B-通过抗HA染色评估的在该基因的前导序列和主体之间的N末端处具有HA表位标签的血型糖蛋白A的表达。图3C-通过抗HA染色评估的在N末端处具有HA表位标签的具有约70kDa的补体受体1-衍生的片段的表达。图3F-当N末端融合到血型糖蛋白A时,通过抗HA染色评估的抗体scFv的表达。图3I-通过抗HA染色评估的与具有71个氨基酸的凯尔(Kell)衍生的片段的C末端融合的抗体scFv的表达。图3J-通过抗HA染色评估的与具有79个氨基酸的凯尔衍生的片段的C末端融合的抗体scFv的表达。图3K-通过抗HA染色评估的在前导序列后细胞外N末端处具有HA表位标签的CD55的表达。图3L-通过抗HA染色评估的在前导序列后细胞外N末端处具有HA表位标签的CD59的表达。图3M-通过抗HA蛋白质印迹评估的与具有37个氨基酸的CD55衍生的片段的N末端融合的抗体scFv的表达。图3O-通过抗HA蛋白质印迹评估的与HA标签融合的腺苷脱氨酶的细胞质表达。预期大小约40kDa。图3P-通过抗HA蛋白质印迹评估的与HA标签融合的苯丙氨酸羟化酶的细胞质表达。预期大小约33kDa。图3Q-通过抗HA蛋白质印迹评估的与腺苷脱氨酶和HA标签融合的苯丙氨酸羟化酶的细胞质表达。图3R-通过抗HA蛋白质印迹评估的与血型糖蛋白A的细胞内C末端融合的腺苷脱氨酶的细胞质表达。预期大小约55kDa。图3S-通过抗HA蛋白质印迹评估的与血型糖蛋白A的细胞内C末端融合的苯丙氨酸羟化酶的细胞质表达。预期大小约50kDa。图3T-AO显示出有核、培养的类红细胞前体细胞上的表面和胞浆蛋白的外源性表达。图3T-通过共同翻译的GFP的表达评估的在胞质的C末端处具有HA表位标签的血型糖蛋白A的表达。图3U-通过抗HA染色评估的在该基因的前导序列和主体之间的N末端处具有HA表位标签的血型糖蛋白A的表达。图3V-通过抗-CR1染色评估的补体受体1的过量表达。图3W-通过抗HA染色评估的在N末端处具有HA表位标签的具有约70kDa的补体受体1-衍生的片段的表达。图3X-通过抗HA染色评估的在N末端处具有HA表位标签的具有约210kDa的补体受体1-衍生的片段的表达。图3Y-通过抗HA染色评估的在N末端处具有HA表位标签的、与血型糖蛋白A的N末端融合的、具有约230kDa的补体受体1-衍生的片段的表达。图3Z-当N末端融合到血型糖蛋白A时,通过抗HA染色评估的抗体scFv的表达。图3AA-通过抗HA染色评估的与凯尔细胞外C末端融合的抗体scFv的表达。预期大小约108kDa。图3AB-通过抗HA染色评估的与凯尔细胞外C末端融合的HA标签的表达。图3AC-通过抗HA染色评估的在C(细胞外)末端处具有HA标签的具有71个氨基酸的凯尔衍生的片段的表达。图3AD-通过抗HA染色评估的在C末端处具有HA标签的具有79个氨基酸的凯尔衍生的片段的表达。图3AE-通过抗HA染色评估的与具有71个氨基酸的凯尔衍生的片段的C末端融合的抗体scFv的表达。图3AF-通过抗HA染色评估的与具有79个氨基酸的凯尔衍生的片段的C末端融合的抗体scFv的表达。图3AG-通过抗HA染色评估的在前导序列后细胞外N末端处具有HA表位标签的CD55的表达。图3AH-通过抗HA染色评估的在前导序列后细胞外N末端处具有HA表位标签的CD59的表达。图3AI-通过抗HA染色评估的与具有37个氨基酸的CD55衍生的片段的N末端融合的抗体scFv的表达。图3AJ-通过抗HA染色评估的与CD59的N末端融合的抗体scFv的表达。图3AK-通过抗HA蛋白质印迹评估的与HA标签融合的腺苷脱氨酶的细胞质表达。预期大小约40kDa。图3AL-通过抗HA蛋白质印迹评估的与HA标签融合的苯丙氨酸羟化酶的细胞质表达。预期大小约33kDa。图3AM-通过针对来自共翻译的GFP的荧光的流式细胞术评估的与腺苷脱氨酶和HA标签融合的苯丙氨酸羟化酶的细胞质表达。图3AN-通过抗HA蛋白质印迹评估的与血型糖蛋白A的细胞内C末端融合的腺苷脱氨酶的细胞质表达。预期大小约55kDa。图3AO-通过抗HA蛋白质印迹评估的与血型糖蛋白A的细胞内C末端融合的苯丙氨酸羟化酶的细胞质表达。预期大小约50kDa。图3AP-AU显示出K562红白血病细胞上的表面和胞浆蛋白的外源性表达。图3AP-通过抗-CR1染色评估的补体受体1的过量表达。图3AQ-当N末端融合到血型糖蛋白A时,通过抗HA染色评估的抗体scFv的表达。图3AR-通过抗HA染色评估的与具有37个氨基酸的CD55衍生的片段的N末端融合的抗体scFv的表达。图3AS-通过抗HA染色评估的与CD59的N末端融合的抗体scFv的表达。图3AT-通过抗HA蛋白质印迹评估的与HA标签融合的腺苷脱氨酶的细胞质表达。预期大小约40kDa。图3AU-通过抗HA蛋白质印迹评估的与HA标签融合的苯丙氨酸羟化酶的细胞质表达。预期大小约33kDa。图4是测量原代血小板中荧光的流式细胞术直方图的集合,这些原代血小板已经用编码荧光蛋白(GFP)的mRNA进行了转染。(A)未转染的血小板。(B)用3ugGFPmRNA转染的血小板。(4C)用6.8ugGFPmRNA转染的血小板。图5显示出培养物中转基因类红细胞的蛋白质表达和酶活性。(A)是从有核前体细胞(“DiffID5”)到去核类红细胞(“DiffIIID8”),在分化过程中,用抗HA抗体检测的外源性表达的腺苷脱氨酶的蛋白质印迹。(B)是由表达完整腺苷脱氨酶的293T细胞从腺苷生产的肌苷的条形图。(C)是从有核前体细胞(“DiffID5”)到去核类红细胞(“DiffIIID8”),在分化过程中,在各时间点处,用抗HA抗体检测的外源性表达的苯丙氨酸羟化酶的蛋白质印迹。(D)通过所培养的表达苯丙氨酸羟化酶的去核类红细胞的裂解物从苯丙氨酸产生的酪氨酸的条形图。图6显示出通过所培养的过量表达补体受体1(CR1)的类红细胞,免疫复合物捕获并且转移到巨噬细胞。(A)是流式细胞术图,其显示出通过所培养的过量表达CR1的类红细胞,荧光免疫复合物(白色直方图)和补体缺陷型免疫复合物(阴影直方图)的捕获。(B)是流式细胞术数据的条形图,该数据评估了通过巨噬细胞(左组)或用所培养的过量表达CR1的类红细胞孵育的巨噬细胞(右组),荧光免疫复合物(阴影条)、补体缺陷型免疫复合物(灰色条)或无免疫复合物(黑色条)的吸收。图7显示出在培养的类红细胞的表面上结合乙型肝炎表面抗原(scFv)的抗体scFv的活性。(A)是流式细胞术直方图,该直方图显示出450nM抗原(白色直方图)或无抗原(灰色直方图)的结合。(B)是针对一系列抗原浓度通过流式细胞术评估的结合信号的滴度。(C-D)是来自小鼠的血细胞的流式细胞术图,该小鼠已经注射了荧光抗原和培养的(C)不表达或(D)表达scFv的类红细胞。该y轴数值为抗原荧光。该x轴数值为所培养的细胞的荧光。图8显示出通过体内表达外源抗原的EHC介导的循环抗体的特异性清除。(A)是一组流式细胞术图,其显示出循环Dylight650标记的小鼠抗HA抗体与CFSE标记的、培养的、分离自受体小鼠的人类红细胞的未结合(顶部)和结合(底部),该受体小鼠在其表面上不表达(顶部)或表达(底部)HA表位标签。该x轴数值为CFSE荧光。该y轴数值为抗HA抗体Dylight650荧光。(B)是来自HA表位标签底物ELISA的数据,该数据比较了随着时间收集自小鼠的血浆中的抗HA抗体水平,该小鼠注射了抗HA抗体(空心圆、实线),抗HA抗体之后是不表达HA表位标签的所培养的人类红细胞(短划线),或抗HA抗体之后是表达HA表位标签的所培养的人类红细胞(点线)。(C)是一组流式细胞术图,其显示出Dylight650标记的小鼠抗生物素抗体与CFSE标记的原代人红细胞的未结合(顶部)和结合(底部),这些红细胞与在它们表面上的生物素未结合(顶部)或结合(底部)。该x轴数值为CFSE荧光。该y轴数值为抗生物素抗体Dylight650荧光。(D)是来自生物素底物ELISA的数据,该数据将收集自小鼠的血浆中随着时间变化的抗生物素抗体水平进行比较,该小鼠注射了抗生物素抗体(空心圆、实线)、抗生物素抗体之后是所培养的未与生物素结合的人类红细胞(短划线)、或抗生物素抗体之后是所培养的与生物素结合的人类红细胞(点线)。图9显示出小鼠中所培养的人类红细胞的清除率。(A)是针对人血型糖蛋白A(y轴)和CFSE(x轴)染色的所抽的血的代表性流式细胞术点图,其中培养的人细胞是双阳性的。(B)是在NSG小鼠注射以下各项后双阳性细胞仍存在的百分比随着时间的清除率的图:人血红细胞(实心圆)、培养的去核类红细胞(虚线菱形)、表达细胞内外源蛋白的、培养的去核类红细胞(点线正方形)和表达表面外源蛋白的、培养的去核类红细胞(空心三角形)。图10是在将培养的人类红细胞注射到小鼠中后不良事件的评估。(A-B)显示出在注射以下各项后20分钟(黑色)、6小时(灰色)、和48小时(白色)从小鼠收集的血浆中通过ELISA评估的(A)纤维蛋白肽A和(B)纤维蛋白肽B的水平:(1)人血红细胞、(2)培养的人类红细胞、(3)表达外源胞浆蛋白的、培养的人类红细胞、(4)表达外源表面转基因的、培养的人类红细胞、或(5)重组蛋白。(C-D)显示出注射以下各项的小鼠的脾的组织学染色区域的显微镜图像:(C)培养的人类红细胞和(D)重组蛋白。图11跟踪循环中培养的类红细胞上的外源蛋白的表达。(A)是从小鼠采血的流式细胞术数据,该小鼠注射了表达外源表面蛋白的、培养的人类红细胞,显示出随着时间变化HA阳性的培养的人类红细胞的百分比。(B)是从两只小鼠采血的蛋白质印迹,其中一只小鼠注射了表达外源胞浆蛋白的、培养的人类红细胞,并且其中另一只小鼠注射了在任何细胞不存在下、纯化的、重组产生的外源蛋白,显示出血液中随着时间变化含HA蛋白的水平。图12是培养的人类红细胞的扩增和分化的评估。(A)是包含转基因的体外分化类红细胞(短划线和点线)和不包含转基因的细胞(实线)的不同培养物的扩增速率的图。(B)是不包含(左)或包含(右)转基因的培养的人类红细胞的不同培养物的细胞表面标志GPA和CKIT的流式细胞术图。(C)是不包含(左)或包含(右)转基因的培养的人类红细胞的流式细胞术图,其中将这些细胞用DNA着色剂DRAQ5(y轴)和抗血型糖蛋白A(x轴)进行染色,这鉴定了(1)去核细胞、(2)有核细胞、和(3)细胞核的不同群。图13A是三种方式的原理图,其中抗原可以定位在表达外源抗原的EHC中。B是三种方式的原理图,其中定位在表达外源抗原的EHC中或其上的抗原可以作用于循环中的靶标。C是使用SpyTag-SpyCatcher机理将内源多肽锚与抗原自催化融合的原理图。发明详细描述在某些方面,本发明提供了分离的细胞,将这些分离的细胞工程化为或修饰为包含感兴趣的外源抗原。在某些方面,本发明的分离的EHC包括一种或多种抗原,这些抗原包括多肽,或由其组成。在一些实施例中,该抗原是一种全长蛋白。在一些实施例中,该抗原由全长蛋白内包含的一种或多种多肽构成,该多肽具有大于约7个氨基酸的任何长度。包括该抗原的多肽可以包括一种或多种免疫性表位,这些免疫性表位可以是构象表位或者可以是线性表位。该抗原可以由来自一种或多种不同蛋白的一种或多种多肽构成。在某些方面,本发明的EHC包括一种或多种抗原,这些抗原包括碳水化合物,或由其组成。在某些方面,本发明的EHC包括一种或多种抗原,这些抗原包括脂质,或由其组成。在某些方面,本发明的EHC包括一种或多种抗原,这些抗原包括一种或多种多肽、脂质、和/或碳水化合物、及其任何组合,或由其组成。这些细胞可以是循环细胞,如EHC。这些EHC可以使用所定义的因子从造血前体来培养,这些所定义的因子如:干细胞因子、细胞因子如IL-3和IL-6、胰岛素、转铁蛋白、促红细胞生成素、氢化可的松、和雌激素。本发明的方面涉及培养EHC来包括感兴趣外源抗原的方法。这些感兴趣的外源抗原可以通过多种方法来引入,像例如,细胞内表达、细胞表面表达、与内源性蛋白质的融合、通过化学或酶促方式与细胞表面蛋白结合、或物理荷载到胞内空间。本发明包括抗原的细胞可以用作治疗剂。本发明的方面涉及包括抗原的细胞在通过诱导外周耐受的免疫激活的疾病的治疗中的用途。在一些方面,诱导外周耐受意指抗原特异性免疫细胞的缺失或失活,像例如,CD8T淋巴细胞(CD8T细胞)、CD4T淋巴细胞(CD4T细胞)、CD4T调节性淋巴细胞(Treg)、或B淋巴细胞(B细胞)。免疫激活疾病包括自身免疫性疾病,像例如,多发性硬化症、1型糖尿病、类风湿性关节炎、和膜性肾炎。免疫激活疾病还包括炎性疾病,像例如,克罗恩病、溃疡性结肠炎、乳糜泻、或其他自发性炎性肠病。免疫激活疾病还包括过敏性疾病,像例如,哮喘、花生过敏、贝类过敏、花粉过敏、牛奶蛋白过敏、昆虫叮咬过敏、和乳胶过敏。免疫激活疾病还包括响应于治疗性蛋白的免疫激活(其减轻了该治疗性蛋白的功效),该治疗性蛋白被给予来治疗主要病症,像例如,血友病A中的凝血因子VIII、血友病B中的凝血因子IX、类风湿性关节炎及其他炎性疾病中的抗肿瘤坏死因子α(TNFa)抗体、戈谢病中的葡糖脑苷脂酶、或急性成淋巴细胞白血病(ALL)中的天冬酰胺酶。免疫耐受的生物学身体已经形成了用于预防异常的免疫激活和自身免疫性疾病的复杂机制,统称为免疫耐受。中枢耐受是指在中枢淋巴器官(例如,胸腺和骨髓)中在形成期间的自身反应性T细胞和B细胞的抗原特异性缺失。外周耐受是指在中枢淋巴器官外,成熟T和B淋巴细胞的缺失或失活。外周耐受包括通过调节T细胞(Treg)抑制自身反应性淋巴细胞或者在抗原特异性效应淋巴细胞中通过在共刺激“危险”信号不存在下暴露于持续低剂量抗原诱导无反应性或无应答。Treg激活和淋巴细胞无反应性都可以通过分泌抑制因子(像例如TGF-β、IL-10、和IL-4)来诱导。响应于抗原的免疫激活通常需要二次“危险”信号,如toll样受体配体,其通常衍生自微生物病原体或病毒病原体(马青格(Matzinger),免疫学年度回顾(AnnuRevImmuno)1994)。此类危险信号包括双链RNA、单链DNA、脂多糖、细菌脂蛋白、鞭毛蛋白、酵母聚糖、及其他。接受抗原和危险信号的抗原递呈细胞展示出在其表面上的共刺激分子,像CD80和CD86,除抗原肽之外。识别抗原肽和共刺激分子的T细胞被激活。仅接受抗原肽信号的那些变得无变应性。已经研发了在危险信号不存在下利用抗原呈递来诱导免疫耐受的治疗策略,用于许多食物过敏的实验性治疗。这些研究采取对增加剂量的过敏原的迁延照射的形式,意图诱导耐受。自2007年,已经有13次研究以此形式研究了各种常见的食物过敏原像花生、奶、和蛋。50%-100%的患者变得敏感,即,能够在无过敏反应的情况下在食品激发中存活。然而,长期耐受不太成功,并且只有25%-50%的患者在停止治疗一个月后能够耐受抗原。参见,例如,伯克斯(Burks)等人,新英格兰医学杂志(NewEnglandJournalofMedicine)2012。据认为,过敏症是IgE介导的,并且激活肥大细胞和嗜碱细胞。低剂量的过敏原的口服给予,如连续供给,通过抗原的CD11c+树突细胞呈递和TGF-β、IL-10、和Il-4的分泌诱导Treg。高剂量口服给予通过浆细胞样树突状细胞诱导抗原特异性T细胞缺失和无反应性。在人类研究中,过敏原的口服给予导致IgE、肥大细胞、和嗜碱细胞的减少,IgG4、TGF-β、IL-10、和在治疗开始时Treg暂时上升的增加。参见,例如,赫尔佐克(Herzog),先进的药物控释评测(AdvDrugDelivRev)2013。虽然不希望受任何具体作用机制的限制,但据信可以通过来自凋亡细胞的自身抗原来诱导外周免疫耐受(格里菲思(Griffith)和弗格森(Ferguson),免疫(Immunity)2011;格林(Green)等人,自然免疫学综述(NatRevImmunol)2009)。虽然从分子角度没有完全理解精确机制,但是自身蛋白(如HSP90及其他损伤相关分子模式)促进由树突细胞吸收。树突细胞受体样CD205识别了这些信号,交叉呈递的抗原,并且诱导致耐受性细胞因子并且抑制共刺激蛋白质表达(博尼法斯(Bonifaz),实验医学杂志(JExpMed)2002)。正在调查利用凋亡细胞的致耐受性潜力诱导外周免疫耐受的治疗策略。这些策略典型地涉及感兴趣的抗原对细胞表面的化学偶联。在研究中,在小鼠、大鼠、和豚鼠中,各种蛋白抗原化学附着于脾细胞和白细胞的表面。参见,例如,米勒(Miller)等人,实验医学杂志(JExpMed)1979;布雷利-马伦(Braley-Mullen)等人,细胞免疫学(CellImmunol)1980;罗(Luo)等人,PNAS2008;斯马(Smarr)等人,免疫学杂志(JImmunol)2011。在近期人类临床研究I期中,将与多发性硬化症相关的肽抗原的混合物化学偶联到自体外周血单核细胞上,并且重新注入患者中(卢特罗蒂(Lutterotti)和马丁(Martin),科学转译医学(SciTransMed)2013)。这些细胞耐受性良好,并且有抗原特异性T细胞响应减少的证据。EHC是死细胞的主要来源。每天,在凋亡样程序性细胞死亡(称为红细胞衰亡)后,清理了大量红细胞(人类中每天大于1%,约1x1011个细胞)。尽管红细胞清除的确切触发物尚未明朗,但是衰亡的(eryptotic)红细胞特征在于磷脂酰丝氨酸不对称、膜异质性、和膜联蛋白V结合、类似于凋亡的有核细胞。EHC在体内也是持续存在的。在成人中,红细胞循环持续长达120天。如此,包括感兴趣的抗原的EHC可以能够使抗原持续暴露于宿主。如以上所描述的,尽管精确的分子机制没有完全理解,但是认为持续暴露于抗原可以通过在共刺激信号不存在下通过抗原呈递诱导外周耐受,导致调节T细胞的扩增、效应T和B细胞的缺失和无反应性、以及抗炎性的和促致耐受性细胞因子的分泌。还已经通过实验探讨通过利用红细胞诱导外周耐受。在前期工作中,已经显示出当非共价附接到红细胞上(康托斯(Kontos)等人,PNAS2013)或经渗透荷载到红细胞中(克里梅尔(Cremel)和高德福林(Godfrin),国际药物杂志(IntJPharm)2013)时,该模型抗原卵白蛋白诱导抗原特异性CD8T细胞缺失和抗原特异性Treg诱导。包括感兴趣的外源抗原的本发明的培养的EHC可以具有超过抗原的明显优势,该抗原非共价附接到红细胞上,例如,通过多肽结合域。一个优势可以是,包括感兴趣的外源抗原的EHC的生物分布比具有靶向结构域的抗原的多肽组合物的生物分布更明确。包括感兴趣的外源抗原的EHC将局限于脉管系统和红细胞典型残留的位置,例如,脾。包括感兴趣的外源抗原的EHC将不被肾滤出或进入外周组织,这是当给予多肽抗原组合物时可能出现的问题。如果培养包括感兴趣的外源抗原的细胞,每EHC外源抗原剂量可以,比如果直接将多肽抗原注射到血流中并且在血流中在整个约10万亿红细胞中分布,显著更高。在一些情况下,可以优选的是具有EHC胞内隔室中所限制的感兴趣的外源抗原。例如,如果该抗原是免疫原性的,那么细胞内定位可以是有利的,因为它可以修饰来自免疫系统的免疫原性抗原,并且因此预防或减少免疫激活。这种构型在多肽抗原组合物的情况下是不可能的。所培养的表达感兴趣外源抗原的EHC可以具有超过经渗透荷载到EHC中的抗原的明显优势。与其中大孔破坏了细胞膜和细胞骨架的整体的渗透荷载过程的产物相反,所培养的包括感兴趣外源抗原的EHC将具有基本上不变的细胞膜和细胞骨架。EHC的形态学和生物物理特性是生物分布、循环、和与脉管系统和免疫细胞的相互作用的关键决定因素(例如,普里斯(Pries)等人,心血管研究(Cardiovasc.Res.)1996),并且因此维持细胞的完整性对保留功效性可以是关键的。物理附接(例如,通过与内源胞浆蛋白的直接融合或与内源跨膜蛋白的融合)到培养的EHC上的外源抗原将不会从细胞中泄露出来并且暴露于免疫系统,直到EHC被消耗。如果使用渗透荷载过程(其中可能损坏该细胞膜)将该细胞与该抗原接触,那么可能出现泄漏的问题。可以直接将所培养的表达感兴趣抗原的EHC给予到需要抗原的受试者中。在制造产物期间,不需要抗原的分离和纯化。这与渗透荷载产物相反(其中该抗原必须分别合成并且纯化,并且然后与该细胞组合),并且这可以在制造产物中提供显著的成本和时间的优势。通过在培养物中扩增,可以将所培养的表达感兴趣抗原的EHC按比例放大。可以生产大型工业规模的批次,以产生既定抗原的EHC的基本上均匀的药物组合物,该既定抗原可以用来普遍治疗许多受试者。与之相反,渗透荷载一般受限于一个供体对一个受试者的规模。细胞采集本发明的表达外源抗原的去核造血细胞可以通过本文中所描述的任何方法来生产。在一些实施例中,这些步骤包括将衍生自造血干细胞的任选分离的培养细胞与抗原接触。造血干细胞产生哺乳动物血液中所发现的所有血细胞类型,这些血细胞类型包括髓细胞(单核细胞和巨噬细胞、中性细胞、嗜碱细胞、嗜酸细胞、红细胞、巨核细胞/血小板、树突细胞)和淋巴系(T细胞、B细胞、NK细胞)。造血干细胞可以分离自成人骨中的骨髓,这些成人骨包括,例如,股骨、髋、肋骨、或胸骨。细胞可以直接从髋获得,例如,通过使用针头和注射器用抽吸从骨髓去除细胞。可替代地,在用细胞因子(像例如,粒细胞集落刺激因子(G-CSF))预处理后,造血干细胞可以分离自正常外周血。G-CSF动员来自骨髓隔室的细胞释放到外周循环中。造血干细胞的其他来源包括脐带血和胎盘。可以离体培养、扩增和分化分离的造血干细胞,以提供各种源材料来产生表达外源抗原的EHC。例如,分离自骨髓、细胞因子刺激的外周血或脐带血的造血干细胞可以离体扩增并且分化成成熟红细胞((Giarratana)等人,自然生物技术(NatureBiotech.)23:69-74(2005);美国专利申请2007/0218552)。如此,使用,例如,磁微珠粒选择和迷你MACS柱(德国美天旎生物技术公司(MiltenyiBiotech)),将CD34+细胞从骨髓或外周血或脐带血分离。在一个实施例中,随后在补充以下各项的改性无血清介质中培养这些细胞:1%牛血清白蛋白(BSA)、120μg/ml铁饱和人转铁蛋白、900ng/ml硫酸亚铁、90ng/ml硝酸铁和10μg/ml胰岛素,并且在37℃下在空气中的5%二氧化碳中保持。细胞培养物的扩增和分化可以在多个步骤中发生。例如,在分离后的初始生长步骤中,这些细胞可以在本文中所描述的介质中,在多种生长因子存在下进行扩增,这些生长因子包括,例如,氢化可的松、干细胞因子、IL-3、和促红细胞生成素。在第二阶段,这些细胞任选地可以进行共培养,例如,在促红细胞生成素存在下在粘附性基质层上。在第三阶段,这些细胞可以在外源因子不存在下在培养介质中在粘附性基质层上进行培养。例如,该粘附性基质层可以是鼠MS-5基质细胞。可替代地,该粘附性基质层可以是衍生自成人骨髓的间充质基质细胞。例如,该粘附性基质层可以维持在补充以10%胎牛血清的RPMI中。在一些实施例中,在非EHC的情况下,红细胞前体细胞及衍生自其的细胞群不是共培养的,例如,在粘附性基质层的情况下,即,它们在非EHC不存在下进行培养。在一些实施例中,包括抗原的EHC在非EHC不存在下进行培养,并且分化,这样使得大于10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%或大于98%的EHC是去核的并且无需如下富集步骤即获得去核细胞群,如有核细胞的重力分离、磁性或荧光分选、辐射、中毒,诸如此类来选择去核细胞。在一些情况下,可能希望扩增并且使CD34+造血干细胞体外部分分化,并且允许体内发生最终分化为成熟红细胞(参见,例如,尼尔德兹-纽伦(Neildez-Nguyen)等人,自然生物技术(NatureBiotech.)20:467-472(2002))。在包含各种因子的介质中,在粘附性基质层不存在下,可以体外扩增分离的CD34+造血干细胞,这些因子包括,例如,Flt3配体、干细胞因子、促血小板生成素、促红细胞生成素、和胰岛素生长因子。产生的红细胞前体细胞可以表征为CD36和GPA的表面表达,并且可以输注到受试者中,其中允许进行最终分化为成熟红细胞。在一些实施例中,该EHC群包括多种去核EHC,这些去核EHC包括在去核过程中保留的抗原多肽。得到的分离的去核EHC(其包括抗原多肽)表现出基本上与对应的分离的、非修饰的、未培养的EHC相同的渗透膜脆性。在一些实施例中,在分化和/或细胞循环阶段的基本相同阶段中,该EHC群包括多个红细胞前体细胞,其中该前体细胞包括编码抗原的重组核酸。包括编码抗原的重组核酸的红细胞前体细胞的大部分能够分化成成熟红细胞,这些成熟红细胞在没有保留重组核酸的情况下保留了抗原。在一些实施例中,这些原代细胞可以通过静脉穿刺、毛细管穿刺、或动脉穿刺来进行收集。然后可以从所收集的全血,使用如下技术之一或组合来分离红细胞、血小板或其他细胞,这些技术包括血浆耗竭、密度梯度、羟乙基淀粉,PrepaCyte-CB和离心。同种异体/和自体来源在一些实施例中,产生表达外源抗原的EHC包括将任选分离的培养细胞与抗原接触,这些培养细胞对受试者来说是自体的和/或同种异体的。例如,与受试者同种异体的红细胞包括一种或多种血型特异性的红细胞或一种或多种万能供血者的红细胞。在一些实施例中,表达外源抗原的EHC可以通过红细胞的融合来生产,例如,在与受试者自体的红细胞和一种或多种同种异体红细胞、脂质体、和/或人工囊泡之间。在某些实施例中,表达外源抗原的EHC的自体输注包括从受试者分离红细胞、网织红细胞或造血干细胞,通过本文中描述的方法通过将该细胞与抗原接触产生适合的表达外源抗原的EHC,并且给予(例如,通过输注)表达外源抗原的EHC到相同受试者中。在某些实施例中,表达外源抗原的EHC的异体输注包括从供体分离红细胞、网织红细胞或造血干细胞,通过本文中描述的方法通过将该细胞与抗原接触产生适合的表达外源抗原的EHC,并且给予(例如,通过输注)表达外源抗原的EHC到不同于供体的受试者中。在同种异体细胞用于输血时,需要小心使用兼容的ABO血型来预防急性血管内溶血性输血反应,该血管内溶血性输血反应表征为补体激活和不兼容红细胞的溶解。基于血型抗原A和B的存在或不存在定义ABO血型,在红细胞表面上与糖蛋白和糖脂有关的寡糖链末端处发现单糖碳水化合物结构(在刘(Liu)等人,自然生物技术(Nat.Biotech.)25:454-464(2007)中综述)。O型红细胞缺乏这两种抗原单糖结构。具有A型红细胞的受试者具有对B型红细胞的天然发生抗体,而具有B型红细胞的受试者具有对A型红细胞的抗体。AB血型受试者没有二者抗体中的人任一种,并且O血型个体二者都有。具有抗A和/或抗B抗体的受试者不能接受包含对应抗原的血液的输注。因为O型红细胞既不包含A抗原也不包含B抗原,它们能够安全输注到具有任何ABO血型的受体中,例如,A、B、AB、或O型受体。O型红细胞被认为是通用的,并且可以用于所有输注。相比之下,可以将A型红细胞给到A和AB型受体,可以将B型红细胞给到B和AB型受体,并且仅可以将AB型红细胞给到AB型受体。在实施例中,其中表达外源抗原的EHC是通过将红细胞或其前体与抗原接触来产生的,将源红细胞或其前体针对于受体的兼容性进行匹配。在一些情况下,可能有益的是将包括非O型红细胞的表达外源抗原的EHC转化为通用血型。酶促去除A型和B型红细胞表面上的免疫显性单糖可以用来产生表达O型样外源抗原的EHC群(参见,例如,刘(Liu)等人,自然生物技术(Nat.Biotech.)25:454-464(2007))。可以使用衍生自绿咖啡豆的α半乳糖苷酶转化表达B型外源抗原的EHC。可替代地或此外,如果存在于表达外源抗原的EHC上的话,衍生自脑膜炎黄杆菌(E.Meningosepticum)细菌的α-N-乙酰半乳糖胺酶和α-半乳糖苷酶酶活性可以用来分别去除免疫显性A和B抗原(刘(Liu)等人,自然生物技术(Nat.Biotech.)25:454-464(2007))。在一个实例中,在26℃下,将如本文中描述的堆积的红血细胞在α-N-乙酰半乳糖胺酶或α-半乳糖苷酶(约300μg/ml堆积的血红细胞)存在下在200mM甘氨酸(pH6.8)和3mMNaCl中孵育60min。在处理后,在盐水中通过3-4次冲洗洗涤这些血红细胞,伴随离心并且根据标准血库技术进行ABO分类。在具体实施例中,本文中所描述的表达外源抗原的EHC可以按以下方式来产生。首先,分离红细胞前体细胞。这些细胞可替代地可以是患者自体的或基本上来自万能供血者血液。例如,这些细胞可以是ABOO型、Rh因子Rhr/r、Duffy-/-、和大凯尔抗原K1阴性。在从红细胞前体细胞到EHC的分化过程中,引入编码抗原的重组核酸。该编码抗原的重组核酸可以是在红细胞特异性启动子(如,GATA-1启动子)控制下(参见,例如,瑞皮克(Repik)等人,临床与实验免疫学(ClinExpImmunol)2005,140:230)。可按本领域中任何已知方式引入编码该抗原的重组核酸,例如,以质粒DNA、病毒、或mRNA。核酸引入可以通过各种标准方法来实现,例如,转染、转导、或电穿孔。血小板衍生与成熟在具体实施例中,本文中所描述的表达外源抗原的EHC可以通过将血小板与抗原接触产生。每天,成人生产2×1011个血红细胞、和约同样数量一半的白细胞和血小板。在人类中,几乎所有的血细胞生产在红骨髓中发生,这表示由造血干细胞、中间祖细胞和定型为各谱系的成熟细胞构成的分级发育系统。尽管在其整个初始发育阶段中所有主要血细胞类型的形态学是类似的,但是巨核细胞(定型为血小板生产的细胞)是通过超越分化生长为大部分其他骨髓和血细胞直径10倍的尺寸、并且包含多达128倍的正常染色体补体的母细胞水平的明显结构和功能背离来标记的,这些细胞产生血小板。在一系列正常细胞分裂后,发育的巨核细胞前体进入了一种独特的细胞周期,该细胞周期特征为简短的(约1小时)G1期、典型的(7小时)S期、非常短暂(约45分钟)G2期、随后是核内有丝分裂期(中止的M期)。一旦细胞形成了一个高度多倍体核,它还会形成胞质分裂所必需的分隔膜。该事件伴随有糖蛋白GPIIbIIIa(血小板纤维蛋白原受体;帕帕亚诺普洛(Papayannopoulou)等人,实验血液学(Exp.Hematol.),24:660-9,1996)和GPIb(von-Willibrand因子受体;考杉斯基(Kaushansky)等人,自然(Nature),369:568-571,1994)的表达,该蛋白是包含ADP、血清素、-血小板球蛋白、及其他成熟血小板功能关键物质的颗粒。最后,高度多倍体巨核细胞经受细胞质分割,允许数千血小板释放(蔡(Choi)等人,血液(Blood),85:402-413,1995;克莱默(Cramer)等人,血液,89:2336-2346,1997)。像所有血细胞前体,巨核细胞衍生自多能性骨髓干细胞,这些细胞保留了广泛自我更新的能力,或者分化成血液的所有成分。血小板生产部分是通过信号机制来调节的,该信号机制通过血小板生成素(TPO)及其细胞受体TPOR/MPUc-MPL之间的相互作用来诱导的。血小板生成素(TPO)是一种参与巨核细胞刺激和血小板生产的造血生长因子。TPO在肝和肾中表达,并且,响应于血小板需求,其表达还可以在骨髓微环境中进行上调(加藤(Kato)等人,干细胞(StemCells),16:322-328,1998;麦卡蒂(McCarty)等人,血液(Blood),86:3668-3675,1995)。由于TPO表达大多是组成型的,TPO水平被认为是通过由血小板螯合来调节的(菲尔德(Fielder)等人,血液,87:2154,1996)。已经克隆并表征了编码TPO的基因(库特(Kuter)等人,美国国家科学院院刊(Proc.Natl.Acad.Sci.USA),91:11104-11108,1994;巴特利(Bartley)等人,细胞(Cell),77:1117-1124,1994;考杉斯基(Kaushansky)等人,自然(Nature),369:568-571,1994;温德灵(Wendling)等人,自然,369:571-574,1994,和德索瓦热(deSauvage)等人,自然,369:533-538,1994)。人TPO(hTPO)cDNA编码353个氨基酸长的多肽。从哺乳动物细胞所分泌的全长hTPO在信号肽裂解后由332个氨基酸组成。尽管这种蛋白预测的分子量是38kDa,但是从来自重组细胞的血清或培养液中的材料的测量所报道的分子量从18至85kD变化(糖基化,和翻译后蛋白酶解加工)。TPO的细胞表面受体(TPOR/MPL/c-MPL)是原癌基因c-mpl、v-mpl同系物、显示出诱导泛骨髓障碍的骨髓增生性白血病病毒的包膜蛋白(MPLV)的产物(温德灵,病毒学(Virol.),149:242-246,1986)。人类c-mpl基因编码具有71kD预测分子量的635aa的蛋白质(维根(Vigon)等人,美国国家科学院院刊(Proc.Natl.Acad.Sci.USA),89:5640-44,1992;米格诺特(Mignotte)等人,基因组学(Genomics),20:5-12,1994)。使得表达TPO或其受体(TPOR/MPL/c-MPL)失效的小鼠表现出严重的血小板减少表现型(格尼(Gurney)等人,科学(Science),265:1445,1994;考杉斯基(Kaushansky)等人,临床研究杂志(J.Clin.Invest.),96:1683,1995;德索瓦热(deSauvage)等人,实验医学杂志(J.Exp.Med.),183:651,1996)。多种细胞因子(例如,干细胞因子[SCF]、IL-1、IL-3、IL-6、IL-11、白血病抑制因子[LIF]、G-CSF、GM-CSF、M-CSF、红细胞生成素(EPO)、kit配体、和干扰素)已经显示出具有血小板生成活性。血小板激活产生的血小板是小圆片形细胞片段,当他们遇到血管损伤位点时,其经历快速转型。它们变得更圆整并且挤出伪足,其纤维蛋白原受体激活,导致聚集,并且它们释放了其颗粒内含物,并且最终它们形成了负责初级止血的塞子(西斯(Siess),W.,生理学评论(Physiol.Rev.)69:58-178,1989)。血小板激活还涉及不稳定型心绞痛、心肌梗塞和中风的发病机理(帕卡姆(Packham),M.A.,加拿大生理学药学期刊(CanJ.PhysiolPharmacol.)72:278-284)。激活血小板中涉及到若干生理性物质,如胶原蛋白,其在内皮下表面暴露于凝血酶(由凝血级联产生),和血栓烷A2(TXA2)和ADP(释放自激活的血小板)。胶原蛋白结合至包括整联蛋白α2β1的若干血小板膜蛋白,导致通过TXA2和ADP的释放的血小板激活(沙蒂尔(Shattil),S.J.等人,细胞生物学新观点(Curr.Opin.CellBiol.)6:695-704,1994)。相比之下,凝血酶、TXA2、和ADP直接激活G蛋白偶联受体,并且诱导血小板聚集和颗粒释放(胡拉尼(Hourani),S.M,和丘萨克(Cusack),N.J.,药理学评论(Pharmacol.Rev.)43:243-298,1991)。据信,血小板激活中涉及的主要事件是激活磷酸脂酶C的β-亚型(PLC)的结果,导致产生肌醇1,4,5三磷酸酯和二酰基甘油。血小板主要包含两种亚型,PLC-β2和PLC-β3。介导血小板粘附和聚集的血小板受体位于两种主要血小板表面糖蛋白复合物上。这些复合物是糖蛋白Ib-IX复合物(其通过结合到vonWillebrand因子(vWF)上来促进血小板粘附),和糖蛋白IIb-IIIa复合物(其通过结合到纤维蛋白原来将血小板链接到聚集体中)。患有巨血小板综合征(先天性出血性障碍)的患者由于糖蛋白Ib-IX复合物的缺乏显示出缺陷型血小板粘附,该糖蛋白Ib-IX复合物结合vWF、轻度血小板减少、和大淋巴细胞血小板。糖蛋白V(GPV)是大型的(约等于12,000分子/血小板)、高度糖基化血小板膜蛋白(Mr82,000)。将血小板暴露于凝血酶释放了称为GPVfl的69kDa可溶性片段。GPV可以非共价地与GPIb-IX复合物相互作用,该复合物是一种通过GPIb(由二硫键连接到GPIbβ(24kDa蛋白)的GPIbα(145kDa蛋白)组成)与GPIX(22kDa蛋白)非共价结合形成的复合物。在GPIb-IX复合物上的vonWillebrand因子和凝血酶的结合位点已经定位于GPIbα上。由于现在已知凝血酶通过切割凝血酶受体(一种G蛋白偶联受体)来激活血小板(乌(Vu)等人,细胞64:1057-1068(1990)),因此未知因为凝血酶结合至GPIbα,凝血酶是否偶然切割GPV,或者此切割是否具有生理作用。GPIBα、GPIBβ、和GPIX包含一个或多个同源的24个氨基酸的富亮氨酸结构域。这些结构域还发现于富亮氨酸糖蛋白(LRG)的大家族中。GPV是巨核细胞细胞谱系的标志物。针对GPV具有特异性的单克隆抗体(SW16)不能结合到红细胞、白细胞内皮细胞、或细胞系如HEL或MEG-01上,这些已知表达血小板巨核细胞标志物。成熟GPV由543个氨基酸组成,其包含单跨膜结构域、短胞质域(16个残基)和具有8个潜在的N-糖基化位点的大细胞外结构域。分析细胞外结构域揭示了与GPIbα具有同源性的具有24个氨基酸的15个串联富Leu的重复序列的存在,并且鉴定了凝血酶的靠近C末端的切割位点,该切割位点与纤维蛋白原的Aα链具有同源性。培养条件用于产生本文中所描述的表达外源抗原的EHC的来源包括循环细胞如EHC。合适的细胞源可以分离自如在此描述的受试者,衍生自患者的造血或红祖细胞、衍生自永生的EHC系、或衍生自诱导性多能干细胞,任选地进行培养和分化。使用细胞培养技术用于生产红细胞的方法在本领域中是所熟知的,例如,贾拉塔纳(Giarratana)等人,血液(Blood)2011,118:5071,黄(Huang)等人,分子治疗(MolTher)2013,电子版先于印刷版9月3日,或栗田(Kurita)等人,公共科学图书馆综合(PLOSOne)2013,8:e59890。根据产生的细胞所表征的生长因子、起始细胞系、培养期、和形态特征来变化方案。还已经针对血液生产建立了培养系统,该培养系统可以替代供体输注(费巴赫(Fibach)等人,1989血液73:100)。近来,CD34+细胞分化至网织红细胞阶段,随后成功输注到人类受试者(贾拉塔纳(Giarratana)等人,血液(Blood)2011,118:5071)。本文中提供了EHC和衍生自EHC的表达外源抗原的EHC的方法。EHC可以从造血祖细胞进行培养,这些造血祖细胞包括CD34+造血祖细胞(贾拉塔纳(Giarratana)等人,血液(Blood)2011,118:5071)、诱导型多能干细胞(栗田(Kurita)等人,公共科学图书馆综合2013,8:e59890)、和胚胎干细胞(广濑(Hirose)等人,2013干细胞报告(StemCellReports)1:499)。适用于扩增和分化祖细胞的生长因子和分化因子的混合物在本领域中是已知的。适合的扩增和分化因子的实例包括但不限于:干细胞因子(SCF),白细胞介素(IL)如IL-1、IL-2、IL-3、IL-4、IL-5、IL-6、IL-7、IL-8、IL-9、IL-11、IL-12、CSF、G-CSF、血小板生成素(TPO)、GM-CSF、红细胞生成素(EPO)、Flt3、Flt2、PIXY321、和白血病抑制因子(LIF)。EHC可以从造血祖细胞(如CD34+细胞)通过将祖细胞与在多步骤培养方法中所定义的因子接触来进行培养。例如,EHC可以从三步法中的造血祖细胞来培养。该第一步骤可以包括将培养中的细胞与1-1000ng/mL的干细胞因子(SCF)、1-100U/mL的红细胞生成素(EPO)、以及0.1-100ng/mL的白细胞介素-3(IL-3)接触。第一步骤任选地包括将培养中的细胞与结合并且激活核激素受体的配体接触,像例如,糖皮质素受体、雌激素受体、孕酮受体、雄激素受体、或孕酮X受体。这些受体的配体包括,例如,皮质类固醇,像例如,10nM-100μM的地塞米松或10nM-100μM的氢化可的松;雌激素,像例如,10nM-100μM的β-雌二醇;孕激素,像例如,10nM-100μM的黄体酮、10nM-100μM的羟孕酮、10nM-100μM的5a-二氢孕酮、10nM-100μM的11-脱氧皮质酮,或合成孕酮,像例如,10nM-100μM的氯地孕酮;雄激素,像例如,10nM-100μM的睾丸素、10nM-100μM的双氢睾酮或10nM-100μM的雄烯二酮;或者孕酮X受体配体,像例如,10nM-100μM的利福平、10nM-100μM的贯叶金丝桃素,10nM-100μM的圣约翰草(金丝桃素),或维生素E样分子,像例如,10nM-100μM的生育酚。该第一步骤还可以任选地包括将培养中的细胞与胰岛素样分子接触,像例如,1-50μg/mL的胰岛素、1-50μg/mL的胰岛素样生长因子1(IGF-1)、1-50μg/mL的胰岛素样生长因子2(IGF-2)、或1-50μg/mL的力生长因子。该第一步骤进一步可以任选地包括将培养中的细胞与0.1-5mg/mL的转铁蛋白接触。该第一步骤可以任选地包括将培养中的细胞与一种或多种白细胞介素(IL)或生长因子接触,像例如,IL-1、IL-2、IL-4、IL-5、IL-6、IL-7、IL-8、IL-9、IL-11、IL-12、粒细胞集落刺激因子(G-CSF)、巨噬细胞集落刺激因子(M-CSF)、粒细胞巨噬细胞集落刺激因子(GM-CSF)、血小板生成素、成纤维细胞生长因子(FGF)、血小板源性生长因子(PDGF)、转化生长因子β(TGF-B)、肿瘤坏死因子α(TNF-A)、巨核细胞生长和发育因子(MGDF)、白血病抑制因子(LIF)、和Flt3配体。每种白细胞介素或生长因子可以典型地以0.1-100ng/mL浓度来提供。该第一步骤还可以任选地包括将培养中的细胞与血清蛋白或非蛋白分子接触,像例如,胎牛血清(1%-20%)、人血浆(1%-20%)、人血浆蛋白粉(1%-20%)、人血清(1%-20%)、白蛋白(0.1-100mg/mL)、或肝素(0.1-10U/mL)。该第二步骤可以包括将培养中的细胞与1-1000ng/mL的干细胞因子(SCF)和1-100U/mL的红细胞生成素(EPO)接触。该第二步骤还可以任选地包括将培养中的细胞与胰岛素样分子接触,像例如,1-50μg/mL的胰岛素、1-50μg/mL的胰岛素样生长因子1(IGF-1)、1-50μg/mL的胰岛素样生长因子2(IGF-2)、或1-50μg/mL的力生长因子。该第二步骤可以进一步任选地包括将培养中的细胞与0.1-5mg/mL的转铁蛋白接触。该第二步骤还可以任选地包括将培养中的细胞与血清蛋白或非蛋白分子接触,像例如,胎牛血清(1%-20%)、人血浆(1%-20%)、人血浆蛋白粉(1%-20%)、人血清(1%-20%)、白蛋白(0.1-100mg/mL)、或肝素(0.1-10U/mL)。该第三步骤可以包括将培养中的细胞与1-100U/mL的红细胞生成素(EPO)接触。该第三步骤可以任选地包括将培养中的细胞与1-1000ng/mL的干细胞因子(SCF)接触。该第三步骤可以进一步任选地包括将培养中的细胞与胰岛素样分子接触,像例如,1-50μg/mL的胰岛素、1-50μg/mL的胰岛素样生长因子1(IGF-1)、1-50μg/mL的胰岛素样生长因子2(IGF-2)、或1-50μg/mL的力生长因子。该第三步骤还可以任选地包括将培养中的细胞与0.1-5mg/mL的转铁蛋白接触。该第三步骤还可以任选地包括将培养中的细胞与血清蛋白或非蛋白分子接触,像例如,胎牛血清(1%-20%)、人血浆(1%-20%)、人血浆蛋白粉(1%-20%)、人血清(1%-20%)、白蛋白(0.1-100mg/mL)、或肝素(0.1-10U/mL)。该培养方法可以任选地包括通过本领域中已知的方法将细胞与一种分子接触,例如,DNA分子、RNA分子、mRNA、siRNA、microRNA、lncRNA、shRNA、荷尔蒙、或小分子,这激活或敲低一个或多个基因。靶基因可以包括,例如,编码转录因子、生长因子、或生长因子受体的基因,包括但不限于,例如:GATA1、GATA2、CMyc、hTERT、p53、EPO、SCF、胰岛素、EPO-R、SCF-R、转铁蛋白-R、胰岛素-R。在一个实施例中,在总计22天的三个独立的分化阶段中,将CD34+细胞置于包含不同量的IMDM、FBS、谷氨酰胺、BSA、全转铁蛋白(holotransferrin)、胰岛素、地塞米松、β-雌二醇、IL-3、SCF、和红细胞生成素的培养物中。在一个实施例中,在总计14天的三个独立的分化阶段中,将CD34+细胞置于包含不同量的IMDM、FBS、谷氨酰胺、BSA、全转铁蛋白(holotransferrin)、胰岛素、地塞米松、β-雌二醇、IL-3、SCF、和血小板生成素的培养物中。在一个实施例中,在总计15天的三个独立的分化阶段中,将CD34+细胞置于包含不同量的IMDM、FBS、谷氨酰胺、BSA、全转铁蛋白(holotransferrin)、胰岛素、地塞米松、β-雌二醇、IL-3、SCF、和GCSF的培养物中。在某些实施例中,包括感兴趣外源抗原的细胞可以包括或从多种循环细胞衍生,这些循环细胞包括但不限于:表A中列出的那些。在一个优选的实施例中,本发明的循环细胞是EHC,像例如,有核血红细胞、血红细胞前体或去核血红细胞。例如,这些EHC是脐带血干细胞、CD34+细胞、造血干细胞(HSC)、脾集落形成(CFU-S)细胞、髓共同祖细胞(CMP)、胚细胞集落形成细胞、爆发集落形成单位红细胞(BFU-E)、巨核细胞红祖细胞(MEP)、红细胞菌落形成单位(CFU-E))、网织红细胞、红细胞、诱导性多能干细胞(iPSC)、间充质干细胞(MSC)、多染性成红细胞、晚正成红细胞(orthochtomraticnormoblast)、或表A1中所列出的那些,或其组合。在一些实施例中,这些EHC是永生的或是永生化细胞,例如,通过逆转录病毒转导CD34+定向造血干细胞产生的,来表达Oct4、Sox2、Klf4、cMyc,和抑制TP53的永生成红细胞(例如,(Huang)等人,分子治疗(MolTher)2013,电子版先于印刷版9月3日)。本文中提供了红细胞组合物,其中多种红细胞表达感兴趣的外源抗原或其片段。这些细胞可以从衍生自患者的造血或红祖细胞、衍生自永生的EHC系、或衍生自诱导性多能干细胞进行培养。在细胞培养中用于生产红细胞的方法在本领域中是已知的,例如,贾拉塔纳(Giarratana)等人,血液(Blood)2011,118:5071,黄(Huang)等人,分子治疗(MolTher)2013,或栗田(Kurita)等人,公共科学图书馆综合(PLOSOne)2013,8:e59890。可以通过转染单个或多个拷贝的基因,用病毒转导,或在DNA或RNA的存在下电穿孔来引入外源抗原。在哺乳动物细胞中表达外源蛋白的方法在本领域中是熟知的。例如,在造血细胞中表达外源因子IX是通过CD34+祖细胞的病毒转导来诱导的,参见常(Chang)等人,自然生物技术(NatBiotechnol)2006,24:1017。本文中所描述的红细胞组合物可以按以下方式来产生。首先,分离红细胞前体细胞。这些细胞可替代地可以是患者自体的或基本上来自万能供血者血液。例如,这些细胞可以是ABOO型、Rh因子Rhr/r、杜菲-/-、和大凯尔抗原K1阴性。在从红细胞前体细胞到EHC的分化过程中,引入编码外源抗原的核酸。该编码外源抗原的核酸可以是在红细胞特异性启动子(如,GATA-1启动子)控制下(参见,例如,瑞皮克(Repik)等人,临床与实验免疫学(ClinExpImmunol)2005,140:230)。可按本领域中任何已知方式引入编码外源抗原的核酸,例如,以质粒DNA、病毒、或mRNA。核酸引入可以通过各种标准方法来实现,例如,转染、转导、或电穿孔。祖细胞的修饰。可以将核酸(如用于产生感兴趣抗原的DNA表达载体或mRNA)引入祖细胞,这些核酸可以分离自初始来源或者可以通过如本文中所提供的常规重组技术如上进行扩展来获得。在一些情况下,可以设计该表达载体,这样使得它们可以通过同源或非同源重组、通过本领域中已知的方法掺入细胞的基因组中。在一些情况下,使用编码可以选择性靶向并且切除该基因组的多肽的核酸,例如CRISPR/Cas9、转录激活因子样效应核酸酶(TALEN)、或锌指核酸酶,来引导表达载体的核酸有效负载插入具体基因组位点,例如CR1基因座(1q32.2)、血红蛋白基因座(11p15.4)、或另一种有关红细胞的蛋白,其包括但不限于表C中所列出的那些。在一些情况下,该核酸是RNA分子、或编码RNA分子的DNA分子,其沉默或抑制靶基因的表达。例如,该分子可以是小干扰RNA(siRNA)、反义RNA分子、或短发夹RNA(shRNA)分子。用于将表达载体转移到祖细胞中的方法包括但不限于:病毒介导的基因转移、脂质体介导的转移、转化、基因枪、转染和转导,例如使用病毒介导的基因转移如基于DNA病毒(如腺病毒、腺相关病毒和疱疹病毒)的载体、连同基于逆转录病毒的载体。基因转移模式的例子包括裸DNA、CaPO4沉淀、DEAE右旋糖酐、电穿孔、原生质融合、lipofecton、和细胞显微注射。本文中所描述的任何基因修饰的祖细胞可以在允许分化成成熟去核血红细胞的适合的条件下进行培养,例如,本文中所描述的体外培养方法。产生的去核血红细胞展示出并且表达与成熟红细胞有关的蛋白,例如,血红蛋白、血型糖蛋白A,其可以通过标准方法(例如,蛋白质印迹法或FACS分析)来验证并且量化。用于外源抗原表达的策略本文中提供了通过表达外源抗原的EHC所表现的抗原。在一些实施例中,抗原能够与靶标相互作用,例如,来与靶标缔合或结合至靶标。抗原可以包括多肽,或者可以基本上由多肽组成。在一些实施例中,该抗原包括多肽、碳水化合物、核酸、脂质、小分子、或其组合。在一些实施例中,抗原不与靶标相互作用,而是作为有效负载,通过表达外源抗原的EHC递送到受试者体内的细胞、组织或其他位点。抗原多肽、嵌合体和融合体在一些实施例中,抗原包括多肽。受体多肽大小范围可以是从6个氨基酸至3000个氨基酸,并且可以超过6、10、15、20、25、30、35、40、50、60、70、80、90、100、150、200、300、400或可以超过500个氨基酸。受体多肽大小范围可以是从约20个氨基酸至约500个氨基酸,从约30个氨基酸至约500个氨基酸或者从约40个氨基酸至约500个氨基酸。在一些实施例中,该抗原多肽包括嵌合或融合蛋白,该蛋白可以包括两个或更多个不同的蛋白质结构域。这些嵌合抗原是异源或外源的,这个意义上讲各种结构域衍生自不同来源,并且如此,在自然界中没有一起发现并且能够,例如,通过重组核酸来编码。抗原多肽可以通过该多种方法来生产,这些方法中许多在本领域中是所熟知的并且也在本文中进行了描述。例如,抗原多肽可以通过提取(例如,从分离的细胞)、通过表达编码抗原多肽的重组核酸、或者通过化学合成来获得。抗原多肽可以通过,例如,重组技术来生产,并且编码该多肽的表达载体被引入宿主细胞(例如,通过转化或转染)用于表达所编码的抗原多肽。在不改变活性的情况下,存在通常可以对氨基酸序列进行的多种保守改变。这些改变称为保守性置换或突变;即,属于一组具有具体大小、电荷或其他特征的氨基酸的氨基酸可以取代为另一种氨基酸。氨基酸序列的取代可以选自该氨基酸所属类别的其他成员。例如,非极性(疏水性)氨基酸包括丙氨酸、亮氨酸、异亮氨酸、缬氨酸、脯氨酸、苯丙氨酸、色氨酸、蛋氨酸、和酪氨酸。极性中性氨基酸包括甘氨酸、丝氨酸、苏氨酸、半胱氨酸、酪氨酸、天冬酰胺和谷氨酰胺。荷正电(碱性)氨基酸包括精氨酸、赖氨酸和组氨酸。荷负电(酸性)氨基酸包括天冬氨酸和谷氨酸。不期望此类改变实质性地影响如通过聚丙烯酰胺凝胶电泳或等电点所确定的表观分子量。保守取代还包括将序列的光学异构体取代为其他光学异构体,确切地说针对序列的一个或多个残基用D型氨基酸取代L型氨基酸。此外,序列中的所有氨基酸可经历D到L的异构体取代。示例性保守取代包括但不限于:Lys取代Arg,并且反之亦然,来维持正电荷;Glu取代Asp,并且反之亦然,来维持负电荷;Ser取代Thr,从而维持游离-OH;并且Gln取代Asn,来维持游离NH2。此外,在一些情况下,在没有多肽或核酸片段的功能损失的情况下,可以进行多肽序列或对应的核酸序列的点突变、缺失、和插入。取代可以包括,例如,1、2、3、或更多残基。具体氨基酸序列或编码多肽的重组核酸的任何教导,或其名称的教导包括这些多肽序列或对应的核酸序列和在数据库中针对蛋白质或基因所保藏的任何序列的任何保守取代、点突变、缺失、和插入,这些可以在没有损失多肽或核酸片段的功能的情况下进行。在一些实施例中,该抗原多肽与表达外源抗原的EHC的膜缔合。在其他实施例中,该抗原多肽不与表达外源抗原的EHC的膜缔合。在一个实施例中,在表达外源抗原的EHC中脂质与抗原的质量比小于1:1000,约1:1000、约1:500、约1:250、约1:100、约1:50、约1:25、约1:10、约1:9、约1:8、约1:7、约1:6、约1:5、约1:4、约1:3、约1:2、约1:1、约2:1、约3:1、约4:1、约5:1、约6:1、约7:1、约8:1、约9:1、约10:1、约25:1、约50:1、约100:1、约250:1、约500:1、约1000:1、约10,000:1、约100,000:1、约1,000,000:1、约10,000,000:1、约100,000,000:1、约1,000,000,000:1或大于约1,000,000,000:1。在一个实施例中,在表达外源抗原的EHC中非外源抗原多肽与抗原的质量比小于1:1000,约1:1000、约1:500、约1:250、约1:100、约1:50、约1:25、约1:10、约1:9、约1:8、约1:7、约1:6、约1:5、约1:4、约1:3、约1:2、约1:1、约2:1、约3:1、约4:1、约5:1、约6:1、约7:1、约8:1、约9:1、约10:1、约25:1、约50:1、约100:1、约250:1、约500:1、约1000:1、约10,000:1、约100,000:1、约1,000,000:1、约10,000,000:1、约100,000,000:1、约1,000,000,000:1或大于约1,000,000,000:1。在某些实施例中,该多肽抗原位于表面上,并且暴露于表达外源抗原的EHC周围的环境中。在一些实施中,该多肽抗原位于表达外源抗原的EHC内并且面向其未暴露侧。在某些实施例中,该多肽抗原包括以下结构域中的至少一种:S结构域(表面)、A结构域(锚)、和/或U结构域(未暴露的),其中该S结构域是暴露于表达外源抗原的EHC周围环境的表面结构域,其中该A结构域是锚、并且其中该U结构域位于表达外源抗原的EHC内和/或面向表达外源抗原的EHC的未暴露侧。任选地,该抗原多肽包括i)一个或多个另外的S结构域,称为S’结构域,或ii)一个或多个另外的U结构域,称为U’结构域。在一些实施例中,该S结构域和该A结构域形成部分相同多肽链。在一些实施例中,该A结构域和该U结构域形成部分相同多肽链。在一些实施例中,将该S、A、U结构域中的任何一种或多种从外部添加到表达外源抗原的EHC中。在一些实施例中,在表达外源抗原的EHC内,产生该S、A、U结构域中的任何一种或多种。在一些实施例中,该S、A、U结构域中的任何一种或多种是多肽。在一些实施例中,该S、A、U结构域中的任何一种或多种不是多肽。在图13A、13B、和13C中显示出了在表达外源抗原的EHC内或上的抗原的示例性构象的图解。A结构域在某些实施例中,该A结构域是膜多肽。该A结构域可以是,例如,整合膜多肽或膜相关多肽。该A结构域可以选自以下类别之一,这些类别包括但不限于,例如:α-螺旋双面体,α-螺旋凸多面体,β-桶状跨膜、所有α单面体/外周、所有β单面体/外周、α/β单面体/外周、α+β单面体/外周、α螺旋肽、β发夹肽、β螺旋肽、1型跨膜蛋白(N末端细胞外)、2型跨膜蛋白(N末端细胞内)、3型跨膜蛋白、4A型跨膜蛋白、4B型跨膜蛋白、脂质锚定蛋白、糖基磷脂酰肌醇(GPI)锚定蛋白、异戊二烯基链锚定蛋白、或非正规结构肽。在某些实施例中,该A结构域是内源的,例如,与EHC、血小板、或造血细胞是内源的。在一些实施例中,该A结构域与哺乳动物细胞是内源的。在某些实施例中,该A结构域是外源的,例如,与EHC、血小板、或造血细胞是外源的。在一些实施例中,该A结构域与哺乳动物细胞是外源的。该A结构域可以选自以下分子或其片段,其包括但不限于:CD1、CD2、CD3、CD4、CD5、CD6、CD7、CD8、CD9、CD10、CD11a、CD11b、CD11c、CD12w、CD13、CD14、CD15、CD16、CDw17、CD18、CD19、CD20、CD21、CD22、CD23、CD24、CD25、CD26、CD27、CD28、CD29、CD30、CD31、CD32、CD33、CD34、CD35、CD36、CD37、CD38、CD39、CD40、CD41、CD42、CD43、CD44、CD45、CD46、CD47、CD48、CD49a、CD49b、CD49c、CD49d、CD49e、CD49f、CD53、CD54、CD55、CD56、CD57、CD58、CD59、CD61、CD62E、CD62L、CD62P、CD63、CD68、CD69、CD71、CD72、CD73、CD74、CD80、CD81、CD82、CD83、CD86、CD87、CD88、CD89、CD90、CD91、CD95、CD96、CD100、CD103、CD105、CD106、CD107、CD107a、CD107b、CD109、CD117、CD120、CD122、CD123、CD127、CD132、CD133、CD134、CD135、CD138、CD141、CD142、CD143、CD144、CD147、CD151、CD152、CD154、CD155、CD156、CD158、CD163、CD165、CD166、CD168、CD184、CDw186、CD195、CD197、CDw199、CD209、CD202a、CD220、CD221、CD235a、CD271、CD279、CD303、CD304、CD309、CD326、Ras-相关蛋白1A、信号素(semaporin)7A前体、钙和整合素结合蛋白1、55kDa红细胞膜蛋白、脂阀结构蛋白-1、脂阀结构蛋白-2、红细胞膜相关蛋白、真核翻译起始因子2C2、细胞色素b5还原酶、细胞分裂控制蛋白质42同系物、KIAA1363蛋白、带3、膜联蛋白VII、水通道蛋白、Ecto-ADP-核糖基转移酶4、凯尔、LFA-3、溶质载体家族2成员1、LGALS3蛋白、尿素转运体、Rh血CE组抗原多肽、Rh-相关糖蛋白、成束蛋白、ABO血型、水通道蛋白3、Aubergers、带3、基础免疫球蛋白、C41、CD44、CisAB、科尔顿抗原、补体成分4、CR1、DAF、Diego、Duffy、Hh/孟买抗原、ii抗原、印度血型、凯尔、Kidd、Lewis抗原、路德抗原、MNS抗原系统、成本组、Er组、束蛋白、Stomatin、原肌球蛋白、葡萄糖转运体、内收蛋白、Rab亲和蛋白、C1四氢叶酸合酶、维尔血型、Lan抗原、At抗原、Jr抗原、AnWj抗原、Sd抗原、Batty、Bilkes、Box、Christiansen、HJK、HOFM、JFV、JONEs、Jensen、Katagiri、Livesay、Milne、Oldeide、Peters、Rasmussen、Reid、REIT、SARA、恒河猴血液D组、醛缩酶、原肌球调节蛋白、精氨酸酶、肌酸激酶、B-Cam蛋白、Rap1A、Bennett-Goodspeed、P抗原系统、RhXg血型抗原系统、XK蛋白、Yt/Cartwright抗原系统、CD58、Rh、Scianna、Radin、DARC(Duffy)、CR1Knops-McCoy、DAFCromer、Gerbich(GYPC)、CD47、血型糖蛋白A、带3(AE3)、GYPBSs、C4A、C4BChido、补体的RodgersC4成分、HLABgHLAI类、RHAGRh-相关氨转运体、糖蛋白、科尔顿(Co)水通道蛋白、ACHECartwright(Yt)乙酰胆碱酯酶、谷胱甘肽转移酶、血型糖蛋白C、水通道蛋白、成红细胞相关膜蛋白、CD44、小突触泡蛋白2、核糖核酸酶、十二指肠细胞色素B、ABO糖基转移酶、CD59、CD44印度(In)、AnWj粘附受体、MER2、DOKDombrockADP-核糖转移酶、SEMA7AJMH假定的粘附受体、UMODSdaTamm-Horsfall蛋白(尿调素)、Diego(Di)、Wright(Wr)阴离子通道蛋白(带3、AE1)、Kidd(Jk)尿素转运体、FUT3Lewis(Le)α(1,3)岩藻糖转移酶、OKOka内皮素、假定的粘附分子、LW粘附受体、FUT2分泌腺(Se)α(1,2)岩藻糖转移酶、FUT1Hhα(1,2)岩藻糖转移酶、LULutheran(Lu)粘附受体、P1糖基转移酶、XKKx假定的神经递质转运体、XGXg以前称为PBDX、MIC2、血红蛋白、锚蛋白、血影蛋白、KEL凯尔(形成K,k,Kp,Js)金属蛋白酶、Torkildsen抗原、辅酶Q10、Rab35、RalA結合蛋白、透明带结合蛋白、LynB蛋白、KIaa1741蛋白、DC38、钙转运ATP酶、GPIX、GPIba、GPIbb、GPV、GPIb-IX-V、GPVI、GPIa/IIa、GPIIb/IIIa、GPV/IIa。S结构域在一些实施例中,该S结构域是蛋白或多肽。在其他实施例中,该S结构域是核酸。在一些实施例中,该S结构域是一种化学品。在某些实施例中,该S结构域是小分子。在一些实施例中,该S结构域是一种多肽,该多肽选自或衍生自以下类型中的一种或多种,这些以下类型包括但不限于:柔性连接物、表位标签、酶、蛋白酶、核酸酶、抗原、抗体样分子、抗体配体、生长因子、细胞因子、趋化因子、生长因子受体、细胞因子受体、趋化因子受体、酶识别序列、转肽酶识别序列、蛋白酶识别序列、可切割结构域、内含肽、DNA結合蛋白、和RNA結合蛋白、补体调节分子、补体级联分子、凝血级联分子、螯合剂、补体调节结构域、SCR结构域、CCP结构域、免疫球蛋白或免疫球蛋白样结构域、犰狳重复序列、亮氨酸拉链、死亡效应结构域、钙粘蛋白重复序列、EF手、磷酸酪氨酸结合结构域、普列克底物蛋白同源结构域、SCR同源2结构域、锌指结构域、环肽、细胞穿透肽。在一些实施例中,该S结构域是非多肽分子,例如,核酸、碳水化合物、或小分子。在一些实施例中,该S结构域是选自以下类别中的一种或多种的核酸,这些以下类别包括但不限于:DNA适体、RNA适体、siRNA、shRNA、单链RNA探针、单链DNA探针、mRNA、化学修饰的寡核苷酸。在一些实施例中,该S结构域是选自以下类别中的一种或多种的小分子,这些以下类别包括但不限于:螯合剂、DOTA、放射性核素、同位素、显像剂、荧光分子、化学发光分子、气体。U结构域在一些实施例中,该U结构域是蛋白或多肽。在其他实施例中,该U结构域是核酸。在一些实施例中,该U结构域是一种化学品。在某些实施例中,该U结构域是小分子。在一些实施例中,该U结构域是一种多肽,该多肽选自或衍生自以下类型中的一种或多种,这些以下类型包括但不限于:柔性连接物、表位标签、酶、蛋白酶、核酸酶、抗原、抗体样分子、抗体配体、生长因子、细胞因子、趋化因子、生长因子受体、细胞因子受体、趋化因子受体、酶识别序列、转肽酶识别序列、蛋白酶识别序列、可切割结构域、内含肽、DNA結合蛋白、和RNA結合蛋白、补体调节分子、补体级联分子、凝血级联分子、螯合剂、补体调节结构域、SCR结构域、CCP结构域、免疫球蛋白或免疫球蛋白样结构域、犰狳重复序列、亮氨酸拉链、死亡效应结构域、钙粘蛋白重复序列、EF手、磷酸酪氨酸结合结构域、普列克底物蛋白同源结构域、SCR同源2结构域、锌指结构域、环肽、细胞穿透肽、激酶结构域、磷酸酶结构域、细胞骨架蛋白、与细胞骨架蛋白相互作用的蛋白、G蛋白偶联受体、酪氨酸激酶、ITIM结构域、ITAM结构域。在一些实施例中,该U结构域是非多肽分子,例如,核酸、碳水化合物、或小分子。在一些实施例中,该U结构域是选自以下类别中的一种或多种的核酸,这些以下类别包括但不限于:DNA适体、RNA适体、siRNA、shRNA、单链RNA探针、单链DNA探针、mRNA、化学修饰的寡核苷酸。在一些实施例中,该U结构域是选自以下类别中的一种或多种的小分子,这些以下类别包括但不限于:螯合剂、DOTA、放射性核素、同位素、显像剂、荧光分子、化学发光分子、气体。抗原多肽的实例抗原多肽的实例包括:该抗原多肽包括在N末端处具有HA表位标签的血型糖蛋白A;该多肽抗原包括血型糖蛋白A前导序列、HA表位标签、和血型糖蛋白A的主体序列;该多肽抗原包括补体受体1(CR1);该多肽抗原包括CR1的前导序列、HA表位标签、CR1的主体序列;该多肽抗原包括CR1的前导序列、HA表位标签、CR1的LHR-A和LHR-B的六个SCR结构域、CR1的膜近端的两个SCR结构域、CR1的跨膜区、和CR1的胞内区;该多肽抗原包括CR1的前导序列、HA表位标签、CR1的LHR-A和LHR-B和LHR-C的九个SCR结构域、CR1的膜近端的两个SCR结构域、CR1的跨膜区、和CR1的胞内区;该多肽抗原包括CR1的前导序列、CR1的LHR-A、CR1的LHR-B、CR1的LHR-C、CR1的膜近端的两个SCR结构域、CR1的跨膜区、和CR1的胞内区;该多肽抗原包括CR1的前导序列、CR1的LHR-A、CR1的LHR-B、CR1的LHR-C、CR1的膜近端的两个SCR结构域、血型糖蛋白A的跨膜区和胞内区;该多肽抗原包括血型糖蛋白A的前导序列、针对乙型肝炎表面抗原(scFv)的抗体scFv、(Gly3Ser)2柔性连接物、HA表位标签、和血型糖蛋白A的主体;该多肽抗原包括凯尔、(Gly3Ser)2柔性连接物、HA表位标签、和scFv;该多肽抗原包括凯尔和HA表位标签;该多肽抗原包括凯尔的71个氨基酸的N末端片段和HA表位标签;该多肽抗原包括凯尔的71个氨基酸的N末端片段、(Gly3Ser)2柔性连接物、和HA表位标签;该多肽抗原包括凯尔的79个氨基酸的N末端片段和HA表位标签;该多肽抗原包括凯尔的79个氨基酸的N末端片段、(Gly3Ser)2柔性连接物、和HA表位标签;该多肽抗原包括凯尔的71个氨基酸的N末端片段、(Gly3Ser)2柔性连接物、scFv、和HA表位标签;该多肽抗原包括凯尔的79个氨基酸的N末端片段、(Gly3Ser)2柔性连接物、scFv、和HA表位标签;该多肽抗原包括CD55的前导序列、scFv、HA表位标签、和CD55的末端37个氨基酸;该多肽抗原包括CD55的前导序列、HA表位标签、和CD55的主体;在一个实施例中,该多肽抗原包括CD59的前导序列、scFv、HA表位标签、和CD59的主体;该多肽抗原包括CD59的前导序列、和HA表位标签、和CD59的主体;该多肽抗原包括腺苷脱氨酶和HA表位标签;该多肽抗原包括苯丙氨酸羟化酶和HA表位标签;该多肽抗原包括腺苷脱氨酶、(Gly3Ser)2柔性连接物、苯丙氨酸羟化酶、和HA表位标签;该多肽抗原包括血型糖蛋白A、在胞质C末端处的腺苷脱氨酶、和HA表位标签;该多肽抗原包括血型糖蛋白A、在胞质C末端处的苯丙氨酸羟化酶、和HA表位标签。在某些实施例中,该抗原能够与巨噬细胞相互作用。该抗原多肽可以包括以下中的一种或多种:补体受体(瑞乌(Rieu)等人,细胞生物学杂志(J.CellBiol.)127:2081-2091(1994))、清道夫受体(布拉瑟尔(Brasseur)等人,光化学和光生物学(Photochem.Photobiol.)69:345-352(1999))、转铁蛋白受体(德雷尔(Dreier)等人,生物共轭化学(Bioconjug.Chem.)9:482-489(1998);汉布林(Hamblin)等人,光化学与光生物学杂志(J.Photochem.Photobiol.)26:4556(1994));Fc受体(罗让萨库(Rojanasakul)等人,药学研究(Pharm.Res.)11:1731-1733(1994));和甘露糖受体(弗兰克尔(Frankel)等人,碳水化合物研究(Carbohydr.Res.)300:251-258(1997);查克拉巴蒂(Chakrabarty)等人,原生动物学杂志(J.Protozool.)37:358-364(1990))。其他能够与巨噬细胞相互作用的抗原包括:低密度脂蛋白(曼克孜(Mankertz)等人,生物化学和生物物理研究通讯(Biochem.Biophys.Res.Commun.)240:112-115(1997);冯·拜尔(vonBaeyer)等人,国际临床药理学、治疗和毒理学(Int.J.Clin.Pharmacol.Ther.Toxicol.)31:382-386(1993))、极低密度脂蛋白(塔巴斯(Tabas)等人,细胞生物学杂志(J.CellBiol.)115:1547-1560(1991))、甘露糖残基及其他碳水化合物部分(皮泰(Pittet)等人,核医学与生物学(Nucl.Med.Biol.)22:355-365(1995))、聚阳离子分子,如聚左旋赖氨酸(汉布林(Hamblin)等人,光化学与光生物学学报(J.Photochem.Photobiol.26:45-56(1994)),脂质体(巴克-沃登博格(Bakker-Woudenberg)等人,药物靶向杂志(J.DrugTarget.)2:363-371(1994);贝蒂盖里(Betageri)等人,药学与药理学杂志(J.Pharm.Pharmacol.)45:48-53(1993))和2-巨球蛋白(朱(Chu)等人,免疫学杂志(J.Immunol.)152:1538-1545(1994))。本文中提供了包含EHC的组合物,这些EHC包括具有功能活性的抗原,该抗原i)不存在于同谱系的天然EHC中,或者ii)与包括该抗原的EHC相比,以降低的水平或降低的活性水平存在于同谱系的天然EHC中。这类功能活性包括补体抑制、免疫复合物清除、人工抗原呈递、凝血级联的调整、氧传递、药物递送、细胞毒素吸附、吞噬作用的避免、循环时间的延长。在一些实施例中,由于包含CR-1抗原,EHC比同谱系的天然EHC具有更高水平的补体受体多肽,如CR1。在替代性实施例中,这些包括抗原的EHC比同谱系的天然EHC具有更高水平的补体受体激动剂多肽或补体相关多肽,这些多肽包括但不限于表6和表8中所列出的多肽。该补体受体抗原多肽包括人类补体受体-1(CR1)多肽、变体、或其功能片段。该CR1抗原多肽可以衍生自CR1的一个或不只一个天然等位基因,例如,A等位基因(又称为F等位基因或CR1*1等位基因)、B等位基因(又称为S等位基因或CR1*2等位基因)、C等位基因(又称为F’等位基因或CR1*3等位基因)、或D等位基因(又称为CR1*4等位基因)。表3中提供了这些原生形式的序列和数据库登录号。在一些实施例中,该CR1抗原多肽包含CR1多肽的结构域。例如,该CR1多肽可以包括一个或多个短共有重复(SCR)结构域,又称为补体对照蛋白(CCP)模块或寿司(Sushi)结构域,例如,Genbank登录号AAV65577.1。在一个实施例中,该CR1抗原多肽包括一个或多个短共有重复序列(SCR),例如,1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44或大于44个SCR。在另一个实施例中,该CR1抗原多肽包括CR1的一个或多个长同源重复(LHR)单元,例如,LHR-A、LHR-B、LHR-C、或LHR-D,例如,1、2、3、4、5、6或大于6个LHR结构域。在另一个实施例中,该CR1抗原多肽可以包括一个或不只一个融合至另一个细胞膜蛋白的CR1的细胞外结构域,这些细胞膜蛋白例如:血型糖蛋白A、血型糖蛋白B、血型糖蛋白C、血型糖蛋白D、凯尔、带3、水通道蛋白1、glut1、kidd抗原蛋白、Rh抗原,其包括但不限于表1和表6中所列出的细胞表面部分。在一些实施例中,EHC包含编码补体受体抗原多肽的重组核酸,或者可选地或组合地,补体受体激动剂抗原多肽或补体相关抗原多肽,该多肽包括但不限于:表8中列出的多肽、和针对这些多肽的激动剂。在一些实施例中,这些EHC进一步包括外源衰变加速因子(CD59,GenBank:CAG46523.1)多肽、或外源膜辅因子(CD46,GenBank:BAA12224.1)多肽、或其变体或功能片段、或其组合。Cr1活性包括与包含C3b的免疫复合物的结合和这些免疫复合物从循环到肝脏以及网状内皮系统的脾巨噬细胞的穿梭。当遇到网状内皮系统的细胞时,免疫复合物被吞噬细胞吞噬,但血红细胞得以幸免以继续其循环。有时,免疫复合物的去除导致来自血红细胞表面的CR1的蛋白酶剪切。为了测量结合活性,人们可以进行在EHC和免疫复合物之间的体外结合测定。为了测量EHC的节约,人们可以用吞噬细胞和荷载免疫复合物的EHC进行体外吞噬作用测定。为了测量循环免疫复合物到肝脏的体内清除,人们可以使用放射性标记的免疫复合物进行清除和生物分布测定。提供了包含含有抗原的EHC的组合物,该含有抗原的EHC包括天然多肽,EHC水平比同谱系不包括该抗原多肽的造血细胞的水平更高。例如,EHC群包含抗原,如补体受体1水平比缺乏CR1抗原多肽的同谱系的对应造血细胞多出至少约1.1,例如,1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9、2、3、4、5、6、7、8、9、10、15、20、25、30、35、40、45、50、60、70、80、90、100、150、200、250、300、350、400、450、500、600、700、800、900、1000、2000、3000、4000、5000、6000、7000、8000、9000、10000、或超过10000倍。网织红细胞和红细胞的CR1水平典型地在50-2000个分子/细胞之间(洛奇-特里非李尔夫(Lach-Trifilieff),免疫学杂志(JImmunol)1999,162:7549)。提供了包含EHC群的组合物,并且CR1水平为至少约2500、5000、6000、7000、8000、9000、10000、15000、20000、25000、30000、40000、50000、100000、200000、300000、400000、500000、600000、700000、800000、900000、1000000、或大于1000000个分子/细胞。可以测量在野生型和表达外源抗原的EHC中的CR1水平,并且通过,例如,流式细胞术用针对CR1具有特异性的抗体进行量化。在一些实施例中,本文提供了包括抗原的EHC、包括抗原的EHC群、和包括抗原的EHC的组合物。在一些实施例中,该抗原与循环病原体,如病毒或细菌,相互作用。在一些实施例中,该EHC表达编码针对循环病原体具有特异性的抗体、scFv、或纳米抗体的重组基因。可以将抗体、scFv、或纳米抗体表达为融合蛋白。在其他实施例中,将对循环病原体具有亲和力的抗体、scFv、或纳米抗体抗原或其他抗原荷载到EHC中或其上。可以将对循环病原体具有亲和力的抗体、scFv、或纳米抗体抗原或其他抗原在细胞内或细胞外定位。在一些实施例中,该抗原针对病毒或细菌性抗原,如表面、被膜或衣壳抗原是具有特异性的。在某些实施例中,本文提供了包括抗原的EHC、包括抗原的EHC群、和包括抗原的EHC的组合物。在一些实施例中,该抗原与毒素,优选地外源毒素相互作用,如衍生自病原体或以其他方式衍生自环境的毒素。在一些实施例中,该EHC表达编码包括氨基酸序列的抗原的重组基因,该氨基酸序列衍生自脂多糖结合蛋白(LBP)、杀菌增透性蛋白(BPI)、淀粉样蛋白P组分、或阳离子蛋白。可以将毒素结合抗原表达为融合蛋白。在其他实施例中,可以将毒素结合抗原荷载到EHC中或其上。可以将毒素结合抗原在细胞内或细胞外定位。在一些实施例中,该毒素结合抗原针对细菌毒素如肉毒菌或炭疽是具有特异性的。此外,表达外源抗原的EHC可以表达能够提高其螯合目标的能力的抗原。潜在的螯合增强抗原包括多肽转运体,其包括但不限于表1中所列出的那些。在一个实施例中,该抗原包括一种多肽,该多肽包括衍生自趋化因子的Duffy抗原受体(DARC)的氨基酸序列。在一个实施例中,该EHC表达编码衍生自趋化因子的Duffy抗原受体(DARC)的氨基酸序列的重组基因。该DARC抗原可以表达为全长蛋白或其片段。可以将DARC表达为融合蛋白。在其他实施例中,将DARC蛋白荷载到EHC中或其上。在一些实施例中,将所荷载的DARC进行另外的功能化或以其他方式进行修饰。该DARC抗原分子可以在细胞内或细胞外定位。DARC被鉴定为有效的多配体趋化因子受体。DARC属于视紫红质样七螺旋跨膜蛋白家族。除红细胞以外,DARC在后毛细血管小静脉内皮细胞中表达,该后毛细血管小静脉内皮细胞是大部分组织中白细胞迁移的主要位点。DARC提供了CC和CXC趋化因子的高度特异性结合位点。DARC被认为针对ELR基序CXC趋化因子具有更高亲和性。CXC趋化因子是中性粒细胞化学引诱物并且可能是促血管生成的。DARC和CXCL8之间的相互作用已经证实解离常数(Kd)为5nmol/L并且受体结合位点估计为1000-9000/红细胞(哈德利(Hadley),血液(Blood),1997)。不像其他七种跨膜趋化因子受体,DARC缺乏位于第二胞质环中的高度保守的G蛋白偶联基序(梅尼(Meny),免疫血液学(Immunohematology),2010)。DARC不是G蛋白偶联的并且没有已知的替代信号机制。DARC的生物作用尚未完全了解。DARC被认为是a)多特异性的;b)不能启动细胞内信号,并且c)与红细胞表面结合的趋化因子据信是不能接近其正常靶炎症细胞(尼欧特(Neote),生物化学杂志(JBiolChem),1993)。红细胞可以在通过DARC的存在调节炎性过程中发挥一定作用。炎症信号分子,如细胞因子,当以高浓度存在时,可以引发局部和全身组织损伤。细胞因子的突释涉及细菌性败血症、类风湿性关节炎、和其他炎性疾病的发病机理。外源表达的天然细胞因子受体或合成抗体样受体模拟物的经修饰的EHC可以螯合炎性细胞因子。一种示例性趋化因子受体是DARC。本文提供了包括抗原的EHC,该抗原是细胞因子受体或趋化因子受体,包括但不限于DARC。例如,表达DARC抗原的EHC(从而增加天然红细胞上存在的量)可以用来调节循环中和/或身体外周组织内趋化因子水平。包括DARC抗原的EHC可以被标记用于销毁或者可以缓慢释放炎症介质回到循环中,但是以低和分散的浓度。包括抗原的EHC可以作为信号转导肽的储库,该抗原包括趋化因子或细胞因子受体。在一个实施例中,该抗原包括一种多肽,该多肽包括衍生自抗体的氨基酸序列。在一个实施例中,该EHC表达编码衍生自抗体的氨基酸序列的重组基因。该抗体抗原可以表达为全长蛋白或其片段。可以将该抗体表达为融合蛋白。在其他实施例中,将该抗体蛋白荷载到EHC中或其上。在一些实施例中,将所荷载的抗体进行另外的功能化或以其他方式进行修饰。该抗体抗原可以在细胞内或细胞外定位。在一个实施例中,该抗原包括针对所希望靶标具有特异性的抗体氨基酸序列。在一些实施例中,该抗体是scFv。在其他实施例中,该抗体是纳米抗体。在某些实施例中,该EHC包括一种抗原,该抗原包括针对靶标具有特异性的抗体或其片段并且位于细胞表面。例如,针对肉毒杆菌毒素结合具有特异性的抗体的可变片段(Fv)在EHC表面上进行表达。肉毒杆菌毒素结合抗体在本领域中是已知的(艾默思道夫(Amersdorfer),信息与免疫(InfandImmunity),1997),抗体Fv部分的表达也是如此(赫德马克(Hoedemaeker),生物化学杂志(JournofBioChemistry),1997)。当结合时,该毒素由EHC通过Fv区保留下来,经循环系统螯合并且穿梭到肝脏用于从身体清除。在一个实施例中,该抗原包括一种多肽,该多肽包括衍生自scFv抗体的氨基酸序列。在一个实施例中,该EHC表达编码衍生自scFv抗体的氨基酸序列的重组基因。该scFv抗体抗原可以表达为全长蛋白或其片段。可以将该scFv抗体表达为融合蛋白。在其他实施例中,将该scFv蛋白荷载到EHC中或其上。通过EHC表达的适合的scFv抗原多肽包括但不限于表6中所列出的那些。scFv抗体主要从杂交瘤、来自免疫小鼠的脾细胞、和来自人类的B淋巴细胞进行构建。通过V(H)和V(L)结构域的可变结构域的非共价异二聚体形成抗体的可变区,然后可以将其用于重组scFv抗体的构建中。scFv的生产在本领域中是已知的,并且需要首先将mRNA从杂交瘤中分离(或者还可以从脾、淋巴细胞、和骨髓)随后反转录到cDNA中用作用于抗体基因扩增的模板(PCR)。用这种方法,可以产生具有多变化抗体衍生性scFv集合的大文库(一个可比得上从中建立scFv的原始抗体的集合)。可以使该scFv抗原对任何靶分子具有特异性,这些靶分子包括但不限于表4中的那些。在一个实例中,针对炭疽毒素具有特异性的scFv抗原可以在EHC上进行表达。当向对其有需要的受试者给予时,有效量的包括针对炭疽毒素具有特异性的抗原分子的EHC群可以用于捕获并且螯合该炭疽毒素。该EHC迁移至进行清除的肝脏。在某些实施例中,红细胞包括抗原,该抗原包括在细胞表面上表达的源自骆驼的纳米抗体。纳米抗体通常是12-15kDa。它们比抗体和scFv小得多。因此纳米抗体可以更容易转染,并且该纳米抗体抗原将更容易进行表达、翻译和/或转运到EHC中的细胞表面。在某些实施例中,将纳米抗体抗原用于最小化由特异性抗原引起的免疫效应。纳米抗体,由于其小尺寸,将提供降低的免疫原性潜力。在某些实施例中,因为抗原纳米抗体限制了EHC的质膜的机械和形态行为的变化,因此使用抗原纳米抗体。这可以允许EHC显示正常的循环血红细胞行为。在某些实施例中,因为与标准抗体相比,抗原纳米抗体具有增加的识别隐藏或罕见表位的能力,因此使用抗原纳米抗体。例如,它们可以结合至靶标的小酶腔处,并且调节靶的分子行为。在某些实施例中,EHC包括对靶向人补体系统中分子表位具有特异性的抗原纳米抗体。可以将这类EHC给予对其有需要的受试者,以选择性消耗补体系统的一种或多种过度活性因子。例如,可以通过包括对靶向C5的表位具有特异性的抗原纳米抗体的EHC来靶向C5,并且当向受试者给予时通过EHC从系统中清除。该方法适用于提供治疗效果,例如,针对补体障碍,如阵发性睡眠性血红蛋白尿症。在某些实施例中,EHC包括对靶向分子表位具有特异性的抗原纳米抗体,这些分子包括但不限于表4中列出的那些。在一些实施例中,该抗原包括多肽,这些多肽包括衍生自以下各项之一的氨基酸序列:蛋白酶、核酸酶、淀粉酶、裂解酶(蔗糖酶)或水解酶(DNA酶、脂肪酶)。在一个实施例中,该EHC表达编码衍生自以下各项之一的氨基酸序列的重组基因:蛋白酶、核酸酶、淀粉酶、裂解酶(蔗糖酶)或水解酶(DNA酶、脂肪酶)。抗原蛋白酶、核酸酶、淀粉酶、裂解酶和水解酶可以表达为全长蛋白或其片段。抗原蛋白酶、核酸酶、淀粉酶、裂解酶和水解酶可以表达为融合蛋白。在其他实施例中,将抗原蛋白酶、核酸酶、淀粉酶、裂解酶或水解酶荷载到EHC中或其上。在一些实施例中,将该所荷载的抗原蛋白酶、核酸酶、淀粉酶、裂解酶或水解酶进行另外地功能化或者以其他方式进行修饰。该抗原蛋白酶、核酸酶、淀粉酶、裂解酶或水解酶抗原分子可以在细胞内或细胞外定位。在某些实施例中,EHC可以包括以下各项的抗原:蛋白酶、核酸酶、淀粉酶、裂解酶或水解酶。包括蛋白酶、核酸酶、淀粉酶、裂解酶或水解酶抗原的EHC能够独立于循环清除降解EHC上的靶标,例如,通过肝脏中的巨噬细胞。在某些实施例中,可以将包括包含蛋白酶、核酸酶、淀粉酶、裂解酶或水解酶的抗原的EHC给予对其有需要的受试者,通过选择性降解癌细胞生长所必需的代谢产物来治疗癌症。例如,使用天冬酰胺酶来降低局部天冬酰胺水平,以治疗急性成淋巴细胞白血病和急性髓细胞性白血病。适合的抗原可以,例如,包括两种主要类型酶(裂解酶和水解酶)中的一种或两种,这些酶能够降解靶分子。在某些实施例中,提供了包括抗原的EHC,该EHC包含分子,该分子包括但不限于表6中所列出的那些。在某些实施例中,红细胞包括包含裂解酶的抗原。在一个实施例中,该裂解酶是缬氨酸脱羧酶。缬氨酸脱羧酶抗原可以在EHC的胞内空间进行表达。可以将包括缬氨酸脱羧酶抗原的EHC给予对其有需要的受试者,以调节血液中的缬氨酸水平。包括缬氨酸脱羧酶抗原的EHC适用于治疗缬氨酸血症,缬氨酸血症是一种增加血液中氨基酸缬氨酸的水平的遗传性疾病。受影响的个体通常会发展呕吐、生长不良、智力残疾、和疲劳。缬氨酸血症是由缬氨酸转氨酶缺陷引起的并且具有常染色体隐性遗传模式。在某些实施例中,红细胞包括包含水解酶的抗原。在一个实施例中,该水解酶是脱氧核糖核酸酶I(DNA酶I)。DNA酶I抗原可以在EHC的表面上进行表达。可以将包括DNA酶I抗原的EHC给予对其有需要的受试者,来优先切割在邻近嘧啶核苷酸的磷酸二酯键处的循环DNA,产生在位置3’处上具有游离羟基基团的5’-磷酸-末端的聚核苷酸。平均来说,产生四核苷酸。包括DNA酶I抗原的EHC适用于治疗由循环中高水平的免疫原性DNA恶化的病症,如系统性红斑狼疮(SLE)。在某些实施例中,该抗原能够响应于外部刺激,例如,当结合配体或接触刺激时,其中响应需要,例如,移动,重折叠,改变构象,形成二聚体,形成同源二聚体,形成异二聚体,形成多聚体,转导信号,以可检测形式(例如,荧光)发射能量,与另一抗原功能性相互作用或与非外源抗原多肽功能性相互作用。靶标本文提供了表达外源抗原的EHC,这些EHC包括能够与靶标相互作用的外源抗原多肽。本文进一步提供了表达外源抗原的EHC,这些EHC包括能够与靶标相互作用的非多肽外源抗原。可以将表达外源抗原的EHC给予对其有需要的受试者,以调节存在于受试者循环系统中的靶标的量或浓度。可以选择适合的外源抗原来与特异性靶标相互作用。适合靶标包括与具体疾病、障碍、或病症相关联的实体。然而,还可以独立于具体疾病、障碍、或病症选择靶标。在一些实施例中,该靶标是抗体或抗体样分子,例如,自身免疫或自抗体、或外源抗体、或治疗性抗体,包括但不限于,例如:针对β-2-糖蛋白1的抗体、针对I/i抗原的抗体、针对胶原a3(IV)的NC1结构域的抗体、针对血小板糖蛋白的抗体、针对磷脂酶A2受体的抗体、针对红细胞血型糖蛋白A、B、或C的抗体、或针对红细胞Rh抗原的抗体。在一些实施例中,该靶标是补体级联的分子,例如,C1、C1r、C1s、C1q、C2、C2a、C2b、C3、C3a、C3b、C4、C4b、C4a、C3bBb、C3bBb3b、C4b2b、C4b2b3b、C5、C5a、C5b、C6、C7、C8、C9、聚-C9、膜攻击复合物因子B、因子D、备解素、C3、C3a、C3b、iC3b、C3c、C3dg、C3dk、C3e、Bb、因子I、C1q、C1r、C1s、C4、C4a、C4b、C2、C4bp、甘露糖结合凝集素(MBL)、MBL-相关的丝氨酸蛋白酶1(MASP1)、MBL-相关的丝氨酸蛋白酶2(MASP2)、C5、C5a、C6、C7、C8、C9、CR1、CR2、CR3、CR4、C3aR、C3eR、Decay-加速因子(DAF)、膜辅因子蛋白(MCP)、CD59、C3β链受体、C1抑制剂、C4结合蛋白、因子I、因子H。在一些实施例中,该靶标是免疫复合物,例如,IgG免疫复合物、IgA免疫复合物、IgM免疫复合物。在一些实施例中,该靶标是淀粉样蛋白斑块,例如,由一下各项构成的斑块:β淀粉样蛋白、IAPP(淀粉不溶素)、α-突触核蛋白、PrPSc、亨廷顿蛋白、降血钙素、心房利纳因子、载脂蛋白AI、血清淀粉样蛋白A、medin、催乳激素、甲状腺素运载蛋白、溶菌酶、β2微球蛋白、凝胶溶素、角膜上皮蛋白、胱抑素、免疫球蛋白轻链AL、S-IBM。在一些实施例中,该靶标是细菌,例如,肠球菌属、链球菌属、或分枝杆菌、立克次氏体、支原体、脑膜炎奈瑟球菌、淋病双球菌、军团菌属、霍乱弧菌、链球菌属、金黄色葡萄球菌、表皮葡萄球菌、铜绿假单胞菌、白喉棒杆菌、梭菌属、肠毒性大肠杆菌、和炭疽杆菌。已经报告菌血症处于一定水平的其他病原体包括以下各项:立克次氏体、亨氏巴尔通氏体、五日热巴尔通体、伯内特科克斯立克次体体、衣原体、麻风分枝杆菌、沙门氏菌属;志贺氏菌属;小肠结肠炎耶尔森氏菌;假结核耶尔森菌;嗜肺军团菌;结核分支杆菌;单核细胞增多性李斯特氏菌;支原体属;荧光假单胞菌;霍乱弧菌;流感嗜血杆菌;炭疽杆菌;梅毒螺旋体;钩端螺旋体属;包柔氏螺旋体;白喉杆菌;弗朗西斯氏菌属;羊布鲁氏杆菌;空肠弯曲杆菌;肠杆菌属;奇异变形杆菌;变形杆菌属;和肺炎克雷白氏杆菌。在一些实施例中,该靶标是一种病毒,包括但不限于当结合至细胞表面受体时其感染涉及将遗传物质注射到宿主细胞中的那些,其感染受细胞表面受体介导的病毒。这些病毒的非限制性实例选自:副黏液病毒科(例如,肺炎病毒、麻疹病毒、偏肺病毒、呼吸道病毒或腮腺炎病毒)、腺病毒科(例如,腺病毒)、沙粒病毒科(例如,沙粒病毒如淋巴细胞性脉络丛脑膜炎病毒)、动脉炎病毒科(例如,呼吸和生殖综合征病毒或马动脉炎病毒)、布尼亚病毒科(例如,白蛉病毒或汉坦病毒)、杯状病毒科(例如,诺沃克病毒)、冠状病毒科(例如,日冕形病毒或念状病毒)、丝状病毒科(例如,埃博拉病毒)、黄病毒科(例如,肝炎病毒或黄病毒)、疱疹病毒科(例如,单纯疱疹病毒、水痘病毒、巨细胞病毒、玫瑰疹病毒属、或淋巴隐性病毒)、正粘病毒科(例如,流感病毒或索哥托病毒)、细小病毒科(例如,细小病毒)、小RNA病毒科(例如,肠道病毒或肝病毒)、痘病毒科(例如,正痘病毒、禽痘病毒、或野兔痘病毒)、逆转录病毒科(例如,慢病毒或泡沫病毒)、呼肠孤病毒科(例如,轮状病毒)、弹状病毒科(例如,狂犬病病毒、粒外弹状病毒、或水泡病毒)、和披膜病毒科(例如,甲病毒或风疹病毒)。这些病毒的具体实例包括人呼吸道冠状病毒、流感病毒A-C、肝炎病毒A至G、和单纯疱疹病毒1-9。在一些实施例中,该靶标是一种寄生虫,包括但不限于,例如:肠道或血源性寄生虫、原生动物、锥体虫;能够引起疟疾的haemoprotozoa和寄生虫;包括带绦虫(taeniidcestodes)的肠道和系统性绦虫;肠道球虫(entericcoccidians);肠道鞭毛原生动物;丝虫线虫;胃肠道和系统性线虫和钩虫。在一些实施例中,该靶标是一种真菌,包括但不限于,例如,白色念珠菌、光滑假丝酵母、曲霉属、光滑球拟酵母、热带假丝酵母、克柔念珠菌和近平滑念珠菌。在一些实施例中,该靶标是细菌毒素,包括但不限于,例如:AB毒素、α毒素、炭疽毒素、细菌素、肉毒杆菌毒素、胆固醇依赖性细胞溶素、肉毒杆菌C3毒素、艰难梭菌毒素A、艰难梭菌毒素B、梭菌肠毒素、产气荚膜梭菌α毒素、产气荚膜梭菌β毒素、索状因子、Cry1Ac、念珠藻素、δ-内毒素、白喉毒素、B型肠毒素、红细胞毒素、表皮剥脱素、溶血素E、热不稳定肠毒素、热稳定肠毒素、溶血素、杀白细胞素、脂多糖、李斯特菌素O、小菌素、Panton-Valentine杀白细胞素、毒力岛、酚可溶性调节蛋白、肺炎球菌溶血素、成孔毒素、假单胞菌外毒素、RTX毒素、米酒乳杆菌素、金黄色葡萄球菌α毒素、金黄色葡萄球菌β毒素、金黄色葡萄球菌δ毒素、链球菌溶血素、SymplocamideA、野火菌毒素、破伤风菌溶血素、破伤风痉挛毒素、硫醇激活的细胞溶素、托拉氏毒素、毒性休克综合征毒素、毒黄素、海藻糖二霉菌酸酯、vero细胞毒素、和弧菌素。在一些实施例中,该靶标是一种朊病毒蛋白,包括但不限于,例如:PRP、PRPc、PRPsc、PRPres。在一些实施例中,该靶标是一种细胞因子或趋化因子或生长因子,包括但不限于,例如:酰化刺激蛋白、脂肪因子、白蛋白干扰素(albinterferon)、CCL1、CCL11、CCL12、CCL13、CCL14、CCL15、CCL16、CCL17、CCL18、CCL19、CCL2、CCL20、CCL21、CCL22、CCL23、CCL24、CCL25、CCL26、CCL27、CCL28、CCL3、CCL5、CCL6、CCL7、CCL8、CCL9、集落刺激因子、CX3CL1、CX3CR1、CXCL1、CXCL10、CXCL11、CXCL13、CXCL14、CXCL15、CXCL16、CXCL17、CXCL2、CXCL3、CXCL5、CXCL6、CXCL7、CXCL9、促红细胞生成素、Gc-MAF、粒细胞集落刺激因子、粒细胞巨噬细胞集落刺激因子、肝细胞生长因子、IL10家族、IL17家族、IL1A、IL1B、干扰素、干扰素β1a、干扰素β1b、干扰素γ、干扰素I型、干扰素II型、干扰素III型、白介素、白细胞介素1家族、白细胞介素1受体拮抗剂、白细胞介素10、白细胞介素12、白细胞介素12亚基β、白细胞介素13、白细胞介素16、白细胞介素2、白细胞介素23、白细胞介素23亚基α、白细胞介素34、白细胞介素35、白细胞介素6、白细胞介素7、白细胞介素8、白细胞介素-36、白血病抑制因子、白细胞促进因子、淋巴因子、淋巴毒素、淋巴毒素α、淋巴毒素β、巨噬细胞集落刺激因子、巨噬细胞炎症蛋白、巨噬细胞活化因子、单核因子、肌细胞、肌粘连蛋白、烟酰胺磷酸核糖基转移酶、制瘤素M、奥普瑞白介素、血小板因子4、促炎细胞因子、promegapoietin、RANKL、基质细胞衍生因子1、talimogenelaherparepvec、肿瘤坏死因子α、肿瘤坏死因子、XCL1、XCL2、XCR1、血管生成素、碱性成纤维细胞生长因子、β细胞素、骨形态生成蛋白、脑衍生神经营养因子、CCN细胞内信号传导蛋白、CTGF、达贝泊汀α、内皮糖蛋白、表皮生长因子、依泊汀α、依泊汀β、促红细胞生成素、FGF15、FGF15/19、成纤维细胞生长因子、成纤维细胞生长因子23、非格司亭、GLIA成熟因子、粒细胞集落刺激因子、粒细胞巨噬细胞集落刺激因子、生长分化因子-9、heberprot-P、造血生长因子、肝素结合EGF样生长因子、肝细胞生长因子、胰岛素样生长因子、胰岛素样生长因子1、胰岛素样生长因子2、角质形成细胞生长因子、肌肉生长抑制素、神经生长因子、神经营养因子-3、神经营养因子-4、癌调蛋白、osteopromotive、帕利夫明、PDGFB、胎盘生长因子、血小板α颗粒、血小板衍生生长因子、血小板衍生生长因子受体、增殖指数、血小板生成素、转化生长因子、血管内皮生长因子。在一些实施例中,该靶标是一种小分子,例如,化学品、氨基酸、原子、元素、有机酸,<2000Da、<1000Da、<500Da,包括但不限于,例如:铁、铜、钙、钾、乙醇、甲醇、甘氨酸、丙氨酸、缬氨酸、亮氨酸、异亮氨酸、丝氨酸、半胱氨酸、硒代半胱氨酸、苏氨酸、甲硫氨酸、脯氨酸、苯丙氨酸、酪氨酸、色氨酸、组氨酸、赖氨酸、精氨酸、天冬氨酸、谷氨酸、天冬酰胺、谷氨酰胺。在一些实施例中,该靶标是脂质、脂质复合物、蛋白脂质复合物、或胆固醇,包括但不限于例如:LDL、VLDL、HDL、HDL2B、甘油三酸酯、LP(a)、胆固醇。在一些实施例中,该靶标是哺乳动物细胞,包括但不限于,例如:人细胞、循环细胞、免疫细胞、嗜中性粒细胞、嗜酸性粒细胞、嗜碱性粒细胞、淋巴细胞、单核细胞、B细胞、T细胞、CD4+T细胞、CD8+T细胞、γ-δT细胞、调节性T细胞、天然杀伤细胞、天然杀伤T细胞、巨噬细胞、枯否细胞、树突细胞、癌细胞、癌干细胞、循环肿瘤细胞、来自以下癌症之一的癌细胞,这些癌症包括但不限于,急性淋巴细胞白血病(ALL)、急性髓细胞白血病(AML)、肛门癌、胆管癌、膀胱癌、骨癌、肠癌、脑肿瘤、乳腺癌、未知原发癌症、扩散至骨的癌症、扩散至脑的癌症、扩散至肝的癌症、扩散至肺的癌症、良性肿瘤、宫颈癌、绒毛膜癌、慢性淋巴细胞白血病(CLL)、慢性粒细胞白血病(CML)、结肠癌、结肠直肠癌、子宫内膜癌、眼癌、胆囊癌、胃癌、妊娠滋养细胞瘤(GTT)、毛细胞白血病、头颈癌、霍奇金淋巴瘤、肾癌、喉癌、白血病、肝癌、肺癌、淋巴瘤,黑素瘤皮肤癌、间皮瘤、男性癌症、葡萄胎妊娠、口腔和口咽癌、骨髓瘤、鼻和鼻窦癌、鼻咽癌、非霍奇金淋巴瘤(NHL)、食管癌、卵巢癌、胰腺癌、阴茎癌、前列腺癌、罕见癌症、直肠癌、唾液腺癌、继发性癌症、皮肤癌(非黑色素瘤)、软组织肉瘤、胃癌、睾丸癌、甲状腺癌、未知原发癌、子宫癌、阴道癌、和外阴癌。抗原表达、共轭、荷载在某些实施例中,该多肽抗原是在表达外源抗原的EHC内进行表达。多肽抗原可以展示在表达外源抗原的EHC的表面上,或者可以驻留在表达外源抗原的EHC内。在某些实施例中,该多肽抗原共轭至表达外源抗原的EHC上。该多肽抗原通常共轭至表达外源抗原的EHC的表面上。共轭可以通过本领域中已知的并且本文中所描述的方法经化学或酶促实现。还可以将非多肽抗原共轭至表达外源抗原的EHC上。在一些实施例中,该抗原没有共轭至表达外源抗原的EHC上。在某些实施例中,该多肽抗原被荷载至表达外源抗原的EHC中。还可以将非多肽抗原荷载至表达外源抗原的EHC内。在一些实施例中,该抗原没有被荷载至表达外源抗原的EHC中或其上。在一些实施例中,该表达外源抗原的EHC包括一种抗原,该抗原任选地从重组核酸进行表达,共轭至EHC,荷载到EHC中或上,及其任何组合。任选地,该表达外源抗原包括治疗剂或其他有效负载。在一些实施例中,该表达外源抗原的EHC是通过将适合的分离细胞(例如,EHC、网织红细胞、EHC前体、血小板、或血小板前体)与编码抗原多肽的重组核酸接触而产生。在一些实施例中,抗原多肽通过DNA进行编码,该DNA与有核红细胞前体细胞或有核血小板前体细胞接触。在一些实施例中,抗原多肽通过RNA进行编码,该RNA与血小板、有核EHC、有核血小板前体细胞、或网织红细胞接触。在一些实施例中,该抗原是多肽,该多肽与原代血小板、有核EHC、有核血小板、前体细胞、网织红细胞、或红细胞接触。抗原多肽可以从转基因来表达,该转基因通过电穿孔、化学或聚合转染、病毒转导、机械膜破坏、或其他方法引入EHC中;抗原多肽从mRNA来表达,该mRNA通过电穿孔、化学或聚合转染、病毒转导、机械膜破坏、或其他方法引入细胞中;抗原多肽通过引入外界因子(例如,转录激活因子、转录阻遏蛋白、或分泌途径增强子)从天然基因座过表达;和/或抗原多肽从生产细胞或其他外部系统合成、提取、或产生并且合并到EHC中。在一些实施例中,该抗原是一种全长蛋白。在一些实施例中,该抗原由全长蛋白内包含的一种或多种多肽构成,该多肽具有大于约7个氨基酸的任何长度。例如,这些多肽可以是7、8、9、10、11、12、13、14、15、16、17、18、19、20、或超过20个氨基酸,例如,30、40、50、60、70、80、90、100、或超过100个氨基酸。包括该抗原的多肽可以包括一种或多种免疫性表位,这些免疫性表位可以是构象表位或者可以是线性表位。该抗原可以由来自一种或多种不同蛋白的一种或多种多肽构成。可以通过融合到内源性细胞蛋白在循环细胞中表达感兴趣的抗原,这些内源性细胞蛋白包括但不限于表B和表C中所列出的那些。融合至内源性蛋白质可能是必须的,因为认为在分化和去核的天然过程中,该EHC流出并丢弃红细胞生成所需的但成熟红细胞功能(像例如,c-Kit(SCF受体)和转铁蛋白)所不需要的许多内源性蛋白。参见,例如,克司娃桑(Keerthivasan)等人,国际干细胞(StemCellsInternational)2011;米利亚乔(Migliaccio),血液病学(Haematologica)2010。保留的蛋白质包括某些膜蛋白,像例如,血型糖蛋白A,带3,和水通道蛋白;某些胞浆蛋白,像例如,血红蛋白α、血红蛋白β、和腺苷脱氨酶;和细胞骨架蛋白。可以通过多种方法在EHC的胞内空间内表达感兴趣的抗原,这些方法包括直接表达转基因,融合至内源性细胞内蛋白(像例如,血红蛋白),融合至内源性细胞表面蛋白(像例如,带3、血型糖蛋白A、凯尔)的细胞内结构域,或融合至红细胞骨架的结构组分。可以通过多种方法在EHC的细胞外表面上表达感兴趣的抗原,这些方法包括如果它包含跨膜结构域或其他膜附着结构域那么直接表达转基因,融合至内源性红细胞膜蛋白或至所述蛋白的跨膜结构域(像例如,带3、血型糖蛋白A、或凯尔);或融合至内源性红细胞GPI连接的细胞表面蛋白(像例如,乙酰胆碱酯酶、CD55、CD58或CD59)的GPI-连接物受体肽(参见,例如,库伊曼(Kooyman)等人,学科(Science)1995)。通过各种化学和酶手段可以将感兴趣抗原共轭至培养的EHC的表面,这些手段包括但不限于表D、表D1、和表E中所列出的那些。这些方法包括与双功能的交联剂(像例如,NHS酯-马来酰亚胺异双功能交联剂)的化学偶联,来将伯胺基团与还原性硫醇基基团连接。这些方法还包括酶促策略,像例如,由分选酶介导的转肽酶反应,来将包含受体序列LPXTG或LPXTA的一个多肽与包含N末端序列GGG的多肽连接,参见,例如,(Swee)等人,PNAS2013。这些方法还包括组合方法,像例如,分别在抗原和细胞上的点击化学处理(叠氮化物和炔烃)的分选酶介导的共轭,随后进行环化加成反应以将抗原化学结合至细胞,参见,例如,内维斯(Neves)等人,生物共轭化学(BioconjugateChemistry),2013。若需要,可以将催化键形成多肽结构域在EHC上或其中进行细胞内或细胞外表达。存在许多催化键形成多肽,包括转肽酶、分选酶、和异肽酶,包括衍生自Spy0128(从酿脓链球菌分离的蛋白)的那些。已证明拆分自催化Spy0128的异肽键形成亚基(CnaB2结构域)产生两种不同的多肽,这两种不同的多肽保留了对彼此具有特异性的催化活性。该系统中的多肽称为SpyTag和SpyCatcher。当混合时,SpyTag和SpyCatcher经受了在SpyTag上的Asp117和在SpyCatcher上的Lys31之间的异肽键形成(阿克里(Zakeri)和豪沃思(Howarth),JACS2010,132:4526)。该反应与细胞环境兼容并且针对蛋白/肽共轭是具有高度特异性的(阿克里(Zakeri),B.;菲尔德(Fierer),J.O.;克里克(Celik),E.;希托克(Chittock),E.C.;施瓦兹-利内克(Schwarz-Linek),U.;莫伊(Moy),V.T.;豪沃思(Howarth),M.,美国科学院院刊(Proc.Natl.Acad.Sci.U.S.A.)2012,109,E690-E697)。SpyTag和SpyCatcher已经显示出在弹性蛋白样蛋白中的直接翻译后拓扑修饰。例如,在N末端处的SpyTag和在C末端处的SpyCatcher的放置指导圆形弹性蛋白样蛋白的形成(张(Zhang)等人,美国化学学会杂志(JournaloftheAmericanChemicalSociety),2013)。可以将组分SpyTag和SpyCatcher进行交换,这样使得其中分子A融合至SpyTag并且分子B融合至SpyCatcher的系统功能上相当于其中分子A融合至SpyCatcher并且分子B融合至SpyTag的系统。出于此文献的目的,当使用SpyTag和SpyCatcher时,应当理解的是互补分子可以在其位置被取代。催化键形成多肽,如SpyTag/SpyCatcher系统,可以用来将感兴趣的外源抗原附着于EHC的表面。该SpyTag多肽序列可以在EHC的细胞外表面上表达。该SpyTag多肽可以,例如,融合至1型或3型跨膜蛋白,例如,血型糖蛋白A,融合至2型跨膜蛋白的C末端,例如,凯尔,插入细胞外末端处的框架或者插入多途径跨膜蛋白的细胞外环中,例如,带3,融合至GPI受体多肽,例如,CD55或CD59,融合至脂质链锚定多肽,或者融合至外周膜蛋白。可以在EHC内表达编码SpyTag融合体的核酸序列。可以将感兴趣的外源抗原融合至SpyCatcher。可以表达编码SpyCatcher融合体的核酸序列,并且将其从表达SpyTag融合体的相同EHC中分泌出来。可替代地,可以将编码SpyCatcher融合体的核酸序列进行外源生产,例如在细菌、真菌、昆虫、哺乳动物、或无细胞产生系统中。当SpyTag和SpyCatcher多肽反应时,将形成将感兴趣的外源抗原附接到EHC表面的共价键。在一个实施例中,该SpyTag多肽可以在EHC中的Gata1启动子的控制下表达为与血型糖蛋白A的N末端的融合体。融合至SpyCatcher多肽序列的感兴趣的外源抗原,例如,表F、表G、表H、表I和表J中所列出的外源抗原可以在相同EHC中的Gata1启动子控制下进行表达。当表达这两种融合多肽时,将在SpyTag和SpyCatcher多肽之间形成异肽键,在EHC表面和感兴趣的外源抗原之间形成共价键。在另一个实施例中,该SpyTag多肽可以在EHC中的Gata1启动子的控制下表达为与血型糖蛋白A的N末端的融合体。融合至SpyCatcher多肽序列的感兴趣的外源抗原,例如,表F、表G、表H、表I和表J中所列出的外源抗原可以在适合的哺乳动物细胞表达系统,例如,HEK293细胞中进行表达。当在EHC上表达SpyTag融合多肽时,可以使SpyCatcher融合多肽与细胞相接触。在适合的反应条件下,将在SpyTag和SpyCatcher多肽之间形成异肽键,在EHC表面和感兴趣的外源抗原之间形成共价键。催化键形成多肽,如SpyTag/SpyCatcher系统,可以用来将感兴趣的外源抗原锚定于EHC的胞内空间。可以通过多种方法在EHC的胞内空间内表达SpyTag多肽序列,这些方法包括直接表达转基因,融合至内源性细胞内蛋白(像例如,血红蛋白),融合至内源性细胞表面蛋白(像例如,带3、血型糖蛋白A、凯尔)的细胞内结构域,或融合至红细胞骨架的结构组分。该SpyTag序列不限于多肽末端并且可以整合在内源多肽的内部序列内,这样使得多肽翻译和定位不受干扰。可以将感兴趣的外源抗原融合至SpyCatcher。可以在表达SpyTag融合体的相同EHC内表达编码SpyCatcher融合体的核酸序列。当SpyTag和SpyCatcher多肽反应时,将形成共价键,其用于将感兴趣的抗原锚定在EHC的胞内空间中。在一个实施例中,EHC可以在细胞内表达与血红蛋白β融合的SpyTag。可以将该EHC用基因序列从遗传学上进行修饰,该基因序列包括血红蛋白启动子、β珠蛋白基因和SpyTag序列,这样使得当翻译时,将细胞内β珠蛋白融合至C末端处的SpyTag上。此外,该EHC表达编码SpyCatcher的Gata1启动子引导的基因,驱动苯丙氨酸羟化酶(PAH)表达,这样使得当翻译时,将细胞内PAH融合至在其N末端的SpyCatcher上。当表达两种蛋白时,在胞内空间中,该SpyTag结合的β珠蛋白通过异肽键连接到SpyCatcher结合的PAH,允许PAH被锚定至β珠蛋白并且在成熟过程中保留。在另一个实施例中,该SpyTag多肽可以表达为在EHC内与感兴趣外源抗原的融合体。该SpyCatcher多肽可以表达为在相同EHC内与血型糖蛋白A的C末端(细胞内)的融合体。当表达两种融合多肽时,在SpyTag和SpyCatcher多肽之间形成异肽键,在膜锚定的内源性红细胞多肽和感兴趣的外源抗原之间形成共价键。在另一个实施例中,通过多种方法过可以将感兴趣的外源抗原物理荷载到培养的EHC中(与表达相反),包括渗透荷载或低渗-高渗循环,其中外源抗原通引入到EHC膜中的孔(参见,例如,克莱梅尔(Cremel)和高德福林(Godfrin),国际药剂学杂志(IntJPharm)2013)并且与细胞穿透肽(如,衍生自细菌毒素的一种)的融合来扩散,参见,例如,(Kwon)等人,控制释放杂志(JContrRel)2009。感兴趣的外源抗原可以从转基因来表达,该转基因通过电穿孔、化学或聚合转染、病毒转导、机械膜破坏、或其他方法引入EHC中;外源抗原从mRNA来表达,该mRNA通过电穿孔、化学或聚合转染、病毒转导、机械膜破坏、或其他方法引入细胞中;外源抗原多肽通过引入外界因子(例如,转录激活因子、转录阻遏蛋白、或分泌途径增强子)从天然基因座过表达;外源抗原从生产细胞或其他外部系统合成、提取、或产生并且合并到EHC中。可以任选地通过对细胞施加受控损伤持续预定量的时间,用材料(有效负载)荷载本发明的EHC,这些材料如:肽、蛋白、DNA、RNA、siRNA、及其他大分子,以引起细胞膜中的干扰,这样使得这些材料可以被递送至细胞(例如,细胞质内)。在优选地实施例中,该EHC是网织红细胞。例如,可以通过受控的细胞损伤用编码外源抗原的mRNA荷载网织红细胞。根据需要,mRNA可以是裸露的或修饰的。改进mRNA稳定性和/或减少免疫原性的mRNA修饰包括,例如,ARCA:抗反转帽类似物(m27.3’-OGP3G)、GP3G(非甲基化帽类似物)、m7GP3G(一甲基化帽类似物)、m32.2.7GP3G(三甲基化帽类似物)、m5CTP(5’-甲基-胞苷三磷酸)、m6ATP(N6-甲基-腺苷-5’-三磷酸)、s2UTP(2-硫代-尿苷三磷酸)、和ψ(假尿苷三磷酸)。在另一个优选的实施例中,该EHC是红细胞。例如,可以通过受控的细胞损伤用外源抗原荷载红细胞。该细胞损伤可以是由,例如,由机械应变或剪切力诱导的压力引起,使细胞经受变形,收缩,快速拉伸,快速压缩或高剪切速率的脉冲。受控的细胞损伤导致材料(有效负载)从周围细胞介质吸收到细胞的细胞质中。基于细胞变形(例如,当细胞通过收缩部时的机械变形),使用受控的细胞损伤导致通过扩散而不是内吞作用吸收材料(有效负载)。在受控损伤时的细胞吸收后,材料(有效负载)存在于细胞质中而不是核内体中,从而使得材料对细胞来说是容易获得的。受控的细胞损伤,例如,通过受控变形,保持细胞活力(例如,大于50%、70%、或大于90%)。在某些实施例中,受控的细胞损伤,例如,通过受控变形,保持细胞分化和活性的状态。如果需要,使用组合治疗,例如通过在例如电穿孔或另一种细胞膜通透性增加方法之后或之前的变形的受控损伤。任选地,可以使用表面活性剂。机械变形方法特别适用于不能很好地耐受其他膜渗透性增加方法的细胞,例如,在进行这些方法后显示降低的存活力或不同的分化状态。机械变形方法也适用于不能很好地耐受其他膜渗透性增加方法的材料(有效负载)。可替代地或此外,有效负载可能不能使用可替代方法充分引入到细胞中,因为例如,有效载荷的电荷、疏水性或大小。例如,在PCT申请号WO2013059343,细胞内递送(INTRACELLULARDELIVERY)中描述了变形引起的受控损伤的一种示例性方法和适于此类方法的装置,将该申请通过引用结合在此。在一个具体的实施例中,提供已经通过受控变形受到受控细胞损伤的网织红细胞群。细胞可以,例如,通过穿过直径小于个体网织红细胞的微通道而被压缩并且变形,从而引起细胞膜中的干扰,这样使得膜变得多孔。通过施加压力使细胞移动(例如推动)穿过通道或导管。压缩和变形发生在包括有效负载(例如,外源多肽或寡核苷酸(例如,DNA、RNA,如mRNA))的递送介质中。例如,该递送介质可包括列于表F、表G、表H、表I和表J中所列出的外源抗原或其编码mRNA。当变形时,网织红细胞吸收并保留外源材料。在通过收缩、拉伸和/或高剪切速率的脉冲对细胞进行受控损伤后,任选地将细胞在包含材料(有效负载)的递送介质中孵育。细胞可以在递送介质中保持几分钟以恢复,例如,以闭合穿过收缩所造成的损伤。这可以在室温下进行。如本文中使用的受控的细胞损伤包括:i)病毒介导的转染(例如,单纯性疱疹病毒、腺病毒、腺相关病毒、痘苗病毒、或辛德毕斯病毒),ii)化学介导的转染,例如,阳离子型聚合物、磷酸钙、阳离子脂质、聚合物、和纳米粒子,如环糊精、脂质体、正离子脂质体、DEAE-右旋糖酐、聚乙亚胺、树枝状聚合物、二甲基、磷酸钙、lipofectin、DOTAP、lipofectamine、CTAB/DOPE、DOTMA;和iii)物理介导的转染,包括直接注射、生物射弹颗粒递送、电穿孔、激光辐射、声孔效应、磁性纳米颗粒和受控变形(例如,细胞挤压),如通过显微针、纳米针、毫微微注射器、原子力显微镜检术(AFM)尖端、基因枪(例如,金奈米粒子)、AmaxaNucleofector、光转染(多光子激光)、刺穿染、和磁转染、及本领域中已知的其他适合方法举例说明的。使用任何适合的方法来获得本文中所描述的EHC,这些EHC包括一个或多个所希望的DNA、RNA(例如,mRNA)、或包括抗原的多肽。可以在本发明的EHC上检测感兴趣的外源抗原。使用标准分子生物学方法,例如,蛋白质印迹法或FACS分析,可以验证和鉴定外源抗原的存在。细胞内环境中存在的外源抗原可以在细胞溶解或使用荧光检测时进行量化。制造在一些实施例中,使用前体造血细胞,例如,CD34+细胞、红细胞、血小板、巨核细胞、或嗜中性粒细胞作为来源来生产EHC。在一些实施例中,该前体造血细胞通过GMP兼容方法分离自人类供体。在一些实施例中,这些起始细胞来自自体供体。在一些实施例中,这些起始细胞来自同种异体供体。可以针对血细胞抗原多态性将供体进行归类,和/或针对血细胞抗原将供体进行基因归类。该供体可以是通用血液供体。在一些实施例中,该供体具有孟买表型,即不表达H抗原。在一些实施例中,该供体具有ABO血型O并且是Rh-阴性。在一些实施例中,使用CD34+造血祖细胞、动员外周CD34+细胞、或骨髓衍生的CD34+细胞作为起始材料来源来生产EHC。在一些实施例中,这些起始细胞衍生自脐带血、是诱导性多能干细胞或者是胚胎干细胞。可以培养表达外源抗原的EHC。可以将培养的EHC从台式规模到生物反应器规模按比例放大。例如,培养EHC直到它们达到饱和密度,例如,1x105、1x106、1x107、或大于1x107EHC/ml。任选地,当达到饱和密度时,可以将这些EHC转移到较大体积的新鲜介质中。可以将表达外源抗原的EHC在生物反应器,像例如,波型生物反应器、搅拌釜生物反应器中培养。生物反应器的各种构型在本领域中是已知的,并且可以根据需要选择合适的构型。无需过度实验,本领域技术人员可以很容易确定适用于培养和/或扩增表达外源抗原的EHC群的构型。该生物反应器可以是充了氧的。该生物反应器可以任选地包含一个或多个叶轮,再循环流,介质入口流和对照组分,以调节介质和营养素的流入,或者来调节介质、营养素、和废物的流出。在一些实施例中,该生物反应器可以包含包括外源抗原的人EHC群,在培养过程中这些EHC流出其细胞内DNA。例如,生物反应器可以包含培养后的人类EHC群、去核EHC、和红细胞。在一个具体实施例中,这些人类EHC和去核EHC包括外源抗原,并且该外源抗原被去核EHC保留,而编码外源抗原的重组核酸没有被去核细胞保留。在某些实施例中,去核EHC(其包括外源抗原)表现出基本上与对应的分离的、非修饰的、未培养的EHC相同的渗透膜脆性。在一个实施例中。在14天或更多天内,从生物反应器中的EHC或EHC前体产生的表达外源抗原的EHC群经历总计大于20,000倍的扩增。在一些实施例中,将该外源抗原引入培养的或新鲜分离的EHC前体中,并且在引入编码外源抗原的重组核酸后,从生物反应器中的EHC前体产生的表达外源抗原的EHC在生物反应器中从前体细胞扩增了大于20,000倍。在一些实施例中,该生物反应器是波型生物反应器或叶轮驱动的搅拌器。该生物反应器可以借助喷雾器来充气。在一个实施例中,该生物反应器是一次性的。在一个实施例中,该生物反应器是CIP(清洁到位)。可以在如本文中描述的生物反应器中获得的表达外源抗原的EHC的最终数量可以大于109、1010、1011、1012、1013或大于1013个EHC。在培养期间,可以通过血细胞计数法计数或通过在600nm下的光密度读数测量细胞密度来监测表达外源抗原的EHC的密度。任选地,针对pH水平、充氧、搅拌速率、和/或再循环率监测培养方法。过程和属性可以通过体外测定评估表达外源抗原的EHC的一致性。例如,通过计数群体中的EHC的数量评估表达外源抗原的EHC的一致性,例如,通过显微镜检术、通过流式细胞术、或通过血细胞计数法。可替代地或此外,通过分析EHC的蛋白含量评估表达外源抗原的EHC的一致性,例如,通过流式细胞术、蛋白印迹、免疫沉淀反应、荧光光谱、化学发光、质谱、或吸收光谱。在一个实施例中,所测定的蛋白含量是非表面蛋白,例如,膜内在蛋白质、血红蛋白、成年血红蛋白、胎儿血红蛋白、胚胎血红蛋白、细胞骨架蛋白。在一个实施例中,所测定的蛋白含量是表面蛋白,例如,分化标志物、受体、辅助受体、转运体、糖蛋白。在一个实施例中,该表面蛋白选自该列表,该列表包括但不限于血型糖蛋白A、CKIT、转铁蛋白受体、带3、凯尔、CD45、CD46、CD47、CD55、CD59、CR1。在一些实施例中,通过分析EHC的外源抗原含量评估表达外源抗原的EHC的一致性,例如,通过流式细胞术、蛋白印迹、免疫沉淀反应、荧光光谱、化学发光、质谱、或吸收光谱。例如,可以通过EHC的mRNA含量评估表达外源抗原的EHC的一致性,例如,通过RT-PCR、流式细胞术、或RNA印迹。可以通过核材料含量评估表达外源抗原的EHC的一致性,例如,通过流式细胞术、显微镜检术、或DNA印迹,使用,例如,核染色或核酸探针。可替代地或此外,通过EHC的脂质含量评估表达外源抗原的EHC的一致性,例如,通过流式细胞术、液相色谱法、或通过质谱。在一些实施例中,通过EHC的代谢活性评估表达外源抗原的EHC的一致性,例如,通过质谱、化学发光、荧光光谱、吸收光谱。通过ATP消耗率来评估代谢活性和/或测量表达外源抗原的EHC中2,3-二磷酸甘油酸(2,3-DPG)水平来评估代谢活性。该代谢活性可以被评估为以下各项之一的代谢速率,这些以下各项包括但不限于:乙酰水杨酸、N-乙酰半胱氨酸、4-氨基酚、硫唑嘌呤、布诺洛尔、卡托普利、氯丙嗪、氨苯砜、道诺霉素、脱氢表雄酮、双脱氧腺苷、多巴胺、肾上腺素、艾司洛尔、雌二醇、雌激素酮、依托泊苷、氟哌啶醇、海洛因、胰岛素、异丙肾上腺素、硝酸异山梨酯、LY217896、6-巯嘌呤、醚醇哨唑、硝酸甘油、去甲肾上腺素、对氨基苯甲酸。在一些实施例中,通过由EHC分配底物评估表达外源抗原的EHC的一致性,例如,通过质谱、化学发光、荧光光谱、或吸收光谱。该底物可以是以下各项之一,这些以下各项包括但不限于:乙酰唑胺、熊果苷、布美他尼、肌酸酐、Darstine、去乙基多佐胺(Desethyldorzolamide)、地高辛洋地黄毒苷、地高辛-16′-葡糖苷酸、肾上腺素、庆大霉素、马尿酸、二甲双胍、去甲肾上腺素、对氨基马尿酸、罂粟碱、青霉素G、酚红、血清素、磺基水杨酸、他克莫司、四环素、妥卡雷琐、和万古霉素。在一个实施例中,从前体细胞分化表达外源抗原的EHC群。在此实施例中,通过体外测定评估表达外源抗原的EHC群的分化状态。该体外测定包括本文中描述用于测定EHC一致性的那些,其包括但不限于:扩增速率、数量、蛋白质含量或表达水平、mRNA含量或表达水平、脂质含量、底物的分配、催化活性、或代谢活性。在一些实施例中,培养表达外源抗原的EHC,并且在培养过程中在多个时间点处评估EHC的分化状态。可以使用网织红细胞作为起始材料的来源生产表达外源抗原的EHC。可以使用显微镜检术评估分离的网织红细胞的纯度,因为网织红细胞表征为核糖体RNA的网状(网格样)网络,该核糖体RNA在显微镜下用某些着色剂如新亚甲蓝或亮甲酚蓝变得可见。转铁蛋白受体(CD71)的表面表达在网织红细胞上也较高并且并且减少并且它们成熟为红细胞,允许使用抗CD71抗体来富集和分析网织红细胞群(参见,例如,美天旎CD71微珠粒产品插入物号130-046-201)。可替代地,相对于成熟红细胞,肌酸和血红蛋白A1C含量和丙酮酸激酶、天冬氨酸转氨酶、和胆色素原脱氨酶酶活性的分析可以用来评估分离的网织红细胞的性质(参见,例如,(Brun)等人,血液(Blood)76:2397-2403(1990))。例如,相对于成熟红细胞,胆色素原脱氨酶的活性高出近9倍,而血红蛋白A1C含量在网织红细胞中低出近10倍。在一些实施例中,从一种或多种干细胞活体外和/或体内分化适用于产生表达外源抗原的EHC的细胞。在一个实施例中,该一种或多种干细胞是一种或多种造血干细胞。可以进行各种测定来确认所培养的造血干细胞活体外分化成网织红细胞和红细胞,这些测定包括,例如,显微镜检术、血液学、流式细胞术、变形性测量、和血红蛋白分析和功能性质(贾拉塔纳(Giarratana)等人,自然生物技术(NatureBiotech.)23:69-74(2005))。可以使用被,例如,甲苯基亮蓝染色的细胞的显微镜检术评估所培养的造血干细胞的表型。例如,网织红细胞在这些染色条件下表现出核糖体RNA的网状网络,然而红细胞没有染色。还可以针对标准血液学变量使用例如XE2100自动机(希森美康(Sysmex),罗氏诊断公司(RocheDiagnostics))监测去核细胞,这些血液学变量包括红细胞平均容积(MCV;飞升(fL))、平均红细胞血红蛋白浓度(MCHC;%)和平均红细胞血红蛋白(MCH;pg/细胞)。在一些实施例中,针对其物理性质评估表达外源抗原的EHC,例如,尺寸、质量、容积、直径、浮力、密度、和膜性质,例如,黏度、变形性波动、和流动性。在一个实施例中,通过显微镜检术或通过自动化仪器,例如,血液分析仪器,来测量表达外源抗原的EHC的直径。在一个实施例中,表达外源抗原的EHC的直径在约1-20微米之间。在一个实施例中,表达外源抗原的EHC的直径在至少一个维度在约1-20微米之间。在一个实施例中,表达外源抗原的EHC的直径少于约1微米。在一个实施例中,EHC的直径在一个维度大于约20微米。在一个实施例中,表达外源抗原的EHC的直径在约1微米和约20微米之间、在约2微米和约20微米之间、在约3微米和约20微米之间、在约4微米和约20微米之间、在约5微米和约20微米之间、在约6微米和约20微米之间、在约5微米和约15微米之间或在约10微米和约30微米之间。在一个实施例中,使用血液分析仪器测量该表达外源抗原的EHC的平均红细胞容积。在一个实施例中,EHC的红细胞平均容积的体积大于10fL、20fL、30fL、40fL、50fL、60fL、70fL、80fL、90fL、100fL、110fL、120fL、130fL、140fL、150fL、或大于150fL。在一个实施例中,该EHC的红细胞平均容积小于30fL、40fL、50fL、60fL、70fL、80fL、90fL、100fL、110fL、120fL、130fL、140fL、150fL、160fL、170fL、180fL、190fL、200fL、或小于200fL。在一个实施例中,EHC的平均红细胞容积在80-100飞升(fL)之间。在一个实施例中,使用悬浮微通道谐振器或双悬浮微通道谐振器测量表达外源抗原的EHC的平均浮力质量(pg/细胞)(参见,例如,尤恩(Byun)等人,PNAS2013110(19):7580和布赖恩(Bryan)等人,微型实验室(LabChip)201414(3):569)。在一个实施例中,通过在H2O-D2O交换测定中的浮力质量测量表达外源抗原的EHC的干燥密度(参见,例如,费约尔加多(FeijoDelgado)等人,公共科学图书馆期刊(PLOSOne)20138(7):e67590)。在一些实施例中,该表达外源抗原的EHC具有如通过空间光干涉显微镜检术(SLIM)所测量的大于10、20、30、40、50、60、70、80、90、100或大于100mrad的标准差的平均膜变形性波动(参见,例如,(Bhaduri)等人,科技报告(SciReports)2014,4:6211)。在一个实施例中,当用粘度依赖性量子产率荧光团孵育时,通过检测平均荧光来测量表达外源抗原的EHC群的平均膜粘度(参见,例如,海德克(Haidekker)等人,化学与生物学(Chem&Biol)20018(2):123)。在一个实施例中,通过荧光偏振,例如,用BMGLabtechPOLARstarω酶标仪测量表达外源抗原的EHC的膜流动性。例如,为了测量变形性,在培养的第15天,可以将网织红细胞从有核细胞中分离,例如,通过穿过去细胞滤器(例如,LeucolabLCG2,Macopharma),并且随后使用细胞变形计量学进行测定。将去核细胞悬浮于4%聚乙烯吡咯烷酮溶液中并且然后暴露于从60至450mosM增加的渗透梯度。将细胞的激光衍射图案的变化(变形性指数)记录为克分子渗透浓度的函数,来评估细胞膜的动力变形性。在生理学相关的克分子渗透浓度处获得的最大变形性指数与红细胞的平均表面面积相关。在一些实施例中,针对血红蛋白含量分析表达外源抗原的EHC。可以使用血红蛋白的测定来评估分化细胞的表型(贾拉塔纳(Giarratana)等人,自然生物技术(NatureBiotech.)23:69-74(2005))。例如,可以使用伯乐公司变体IIHb分析仪的高效液相层析(HPLC)来评估各种血红蛋白部分的百分比。可以使用连续法用双波长分光光度计测量氧平衡(例如,Hemox分析仪,TCS)。可以使用闪光光解评估血红蛋白的结合性质。在该方法中,在用532nm的10纳秒脉冲光解后,在436nm下分析CO与细胞内血红蛋白四聚体的重新结合。如果需要,本文中所描述的表达外源抗原的EHC可以在制造后进行纯化。许多适合的纯化方法在本领域中是已知的。例如,通过离心、磁导入、辐射、声泳、和化学或物理摘除纯化表达外源抗原的EHC。在一个实施例中,通过用,例如,基质细胞共培养的活体外成熟纯化表达外源抗原的EHC。在一个实施例中,通过化学或酶促处理EHC来纯化表达外源抗原的EHC,例如,通过用去糖基化酶的处理。在一个实施例中,通过使表达外源抗原的EHC失去任何剩余复制能力来纯化表达外源抗原的EHC。在一个实施例中,使表达外源抗原的EHC受到辐射,例如,X射线、γ射线、β粒子、α粒子、中子、质子、元素核、紫外线,以破坏残留的有复制能力的核酸。电离辐射是通过X射线、γ射线、β粒子(高速电子)、α粒子(氦原子的核)、中子、质子及其他重离子,例如氩、氮、碳和其他元素的核传送的能量。X射线和γ射线是类似光的电磁波,但是其能量远大于光能量(其波长更短)。紫外(UV)线是能够破坏细胞的中间能量的辐射,但是紫外线不同于以上提及的电磁辐射形式,因为它不引起原子或分子中的电离(失去电子),而是引起激发(电子能级的变化)。其他形式的辐射--颗粒--是带负电荷的(电子)、带正电荷的(质子,α射线和其他重离子)、电中性的(中子)。辐射诱导的电离可以直接作用于细胞组分分子或间接作用于水分子,产生水衍生的自由基。自由基在非常短的时间内与附近分子反应,导致化学键的断裂或受影响分子的氧化(添加氧原子)。在细胞中的主要作用是DNA断裂。由于DNA由一对互补双链组成,因此可以发生单链或两条链的断裂。然而,据信后者在生物学上更重要。由于DNA分子的双链性质,大多数单链断裂可以正常修复(两条链彼此互补,这样使得完整链可以用作修复其受损的相反链的模板)。然而,在双链断裂的情况下,修复更加困难,并且可能发生断裂端的错误重新接合。这些所谓的错误修复导致了诱导突变,染色体畸变或细胞死亡。DNA区段的缺失是在辐射下存活的细胞中辐射损伤的主要形式。它可能由以下各项引起:(1)在具有两个外端的连接和断裂之间的碎片的损失的DNA分子中两个单独的双链断裂的错配,或(2)在重新连接以修复一个双链断裂前清理断裂端的过程(酶消化核苷酸-DNA的组成分子)。辐射不仅因其成分(电子、质子、中子等)而不同,而且还因其能量而不同。导致沿着其轨道(如中子)的密集电离的辐射称为高线性能量转移(高LET)辐射,一种描述轨道每单位长度释放的平均能量的物理参数。(参见附图。)低LET辐射只沿其轨道稀疏地产生电离,并且因此在细胞内几乎同质。辐射剂量是每单位生物材料的能量的量(例如,每个细胞的离子化数)。因此,高LET辐射比低LET辐射对生物材料更具破坏性--如X和γ射线--因为在相同剂量下,低LET辐射在细胞内更稀疏地诱导相同数量的自由基,而高LET辐射--如中子和α粒子--将其大部分能量转移到细胞的小区域。由来自高LET辐射的密集电离引起的局部DNA损伤比由来自低LET辐射的稀疏电离引起的弥散性DNA损伤更难修复。在一个实施例中,使用大于1、5、10、15、20、25、30、35、40、50、60、70、80、90、100、或大于100kGy的辐射剂量对表达外源抗原的EHC群进行γ辐射。在一个实施例中,使用大于0.1、0.5、1、5、10、15、20、25、30、35、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900、1000、2000、3000、4000、5000、6000、7000、8000、9000、10000、或大于10000mSv的辐射剂量对表达外源抗原的EHC群进行X线辐射。表达外源抗原的EHC群的纯度可以通过测量群体的同质性来评估。在一个实施例中,通过血液分析仪器测量该表达外源抗原的EHC的平均分布宽度。在一个实施例中,表达外源抗原的EHC群具有大于10、100、1000、104、105、106、或大于106的网织红细胞与非网织红细胞比率。表达外源抗原的EHC群的同质性可以通过测量群体的基质细胞含量来评估。在一个实施例中,表达外源抗原的EHC群具有小于1ppb的基质细胞。可替代地或此外,通过测量群体的病毒滴度和/或细菌菌落形成潜力来评估表达外源抗原的EHC群的同质性。在一个施例中,通过体外测定评估表达外源抗原的EHC群的同质性。该体外测定包括本文中描述用于测定EHC一致性的那些,其包括但不限于:扩增速率、数量、蛋白质含量或表达水平、mRNA含量或表达水平、脂质含量、底物的分配、催化活性、或代谢活性。可以使用各种方法分离用于产生表达外源抗原的EHC的成熟红细胞,这些方法像例如,细胞洗涤器、连续流动池分离器、密度梯度分离、荧光激活细胞分选(FACS)、Miltenyi免疫磁性耗竭(MACS)、或这些方法的组合(参见,例如,范德伯格(vanderBerg)等人,临床化学(Clin.Chem.)33:1081-1082(1987);巴尔-兹维(Bar-Zvi)等人,生物化学杂志(J.Biol.Chem.)262:17719-17723(1987);戈德曼(Goodman)等人,实验生物学和医学(Exp.Biol.Med.)232:1470-1476(2007))。可以通过简单离心从全血中分离红细胞(参见,例如,范德伯格(vanderBerg)等人,临床化学(Clin.Chem.)33:1081-1082(1987))。例如,EDTA抗凝的全血可以在4℃下以800×g离心10分钟。除去富含血小板的血浆和血沉棕黄层,并用等渗盐溶液(NaCl,9g/L)洗涤血红细胞三次。可替代地,可以使用利用各种分离介质的密度梯度离心来分离红细胞,这些分离介质像例如:菲可、泛影钠、Histopaque、珀可(Percoll)、微晶纤维素(Sigmacell)、或其组合。例如,将一定体积的Histopaque-1077层叠在等体积的Histopaque-1119的顶部上。将在等体积的等渗盐溶液(NaCl,9g/L)中1:1稀释的EDTA-抗凝的全血层叠在Histopaque的顶部上,并且将样品在室温下以700×g离心30分钟。在这些条件下,粒细胞迁移至1077/1119界面,淋巴细胞,其他单核细胞,并且血小板保留在血浆/1077界面,并且使血红细胞沉淀。将血红细胞用等渗盐溶液洗涤两次。可替代地,可以使用珀可步骤梯度通过离心分离红细胞(参见,例如,巴尔-兹维(Bar-Zvi)等人,生物化学杂志(J.Biol.Chem.)262:17719-17723(1987))。例如,新鲜血液与包含75mM柠檬酸钠和38mM柠檬酸的抗凝血剂溶液混合,并且在Hepes缓冲盐水中简单洗涤这些细胞。通过用α-纤维素和微晶纤维素(1:1)的混合物吸附除去白细胞和血小板。通过在SorvallSS34转子中在2500rpm下通过45%/75%珀可步进梯度离心10分钟,进一步从网织红细胞和残留的白细胞中分离红细胞。当网织红细胞带在45%/75%界面处且剩余白细胞带在0/45%界面处时,在沉淀中回收红细胞。通过在Hepes缓冲盐水中洗涤几次来从红细胞中除去珀可。可用于产生用于分离红细胞的密度梯度的其他材料包括OptiPrepTM,碘克沙醇在水中的60%溶液(来自Axis-Shield,邓迪,苏格兰)。可以从网织红细胞,例如,使用流式细胞术分离出红细胞(参见,例如,戈德曼(Goodman)等人,实验生物学与医学(Exp.Biol.Med.)232:1470-1476(2007))。在这种情况下,将全血离心(550×g,20min,25℃)以从血浆中分离细胞。将细胞沉淀重悬于磷酸盐缓冲盐溶液中,并通过离心(400×g,30分钟,25℃)在菲可帕克(Ficoll-Paque,1.077密度)上进一步分离,以从白细胞分离红细胞。将所得细胞沉淀重悬于补充有10%胎牛血清的RPMI中,并基于大小和粒度在FACS仪器例如BD公司(BectonDickinson)FACSCalibur(BD生物科技公司(BDBiosciences),富兰克林湖,新泽西州,美国)上分选。可以通过免疫磁性耗竭分离红细胞(参见,例如,(Goodman)等人,(2007)实验生物学与医学(Exp.Biol.Med.)232:1470-1476)。在这种情况下,使用具有细胞类型特异性抗体的磁珠来消除非红细胞。例如,使用本文所述的密度梯度,随后进行任何残留的网织红细胞的免疫磁力消耗,从大多数其他血液组分中分离红细胞。在25℃下用人抗体血清预处理细胞20分钟,并且然后用针对网织红细胞特异性抗原(例如CD71和CD36)的抗体进行处理。这些抗体可以直接附接至磁珠或共轭至PE,例如,具有抗PE抗体的磁珠将与之反应。该抗体-磁珠复合物能够例如从红细胞群中选择性提取残留的网状细胞。还可以使用血液成分单采术分离红细胞。血液成分单采术的过程涉及从患者或供体去除全血,使用离心或细胞分选分离血液组分,取出一个或多个分离的部分,以及将剩余组分输回患者或供体中。目前,为此目的使用了许多仪器,例如,来自Baxter公司的Amicus和Alyx仪器(迪尔菲尔德,伊利诺伊州,美国),来自GambroBCT公司的TrimaAccel仪器(莱克伍德,科罗拉多州,美国)和来自Haemonetics公司的MCS+9000仪器(布伦特里,马萨诸塞州,美国)。另外的纯化方法可以是获得适当程度的细胞纯度所必需的。在一些实施例中,从一种或多种网织红细胞活体外和/或体内分化表达外源抗原的EHC。网织红细胞可用于产生表达外源抗原的EHC。网织红细胞是不成熟的血红细胞,并且构成人体中约1%的血红细胞。网织红细胞在骨髓中形成并且成熟。一旦释放到循环中,网织红细胞迅速经历终末分化为成熟红细胞。像成熟红细胞一样,网状细胞不具有细胞核。与成熟红细胞不同,网织红细胞保持进行蛋白质合成的能力。在一些实施例中,将编码外源抗原的重组核酸(例如mRNA)引入网织红细胞中以产生表达外源抗原的EHC。基于随着网织红细胞成熟的细胞密度的差异,可以从外周血中分离出不同年龄的网织红细胞。可以使用通过各种密度梯度的差速离心从外周血中分离网织红细胞。例如,珀可梯度可用于分离网织红细胞(参见,例如,血液(Blood)74:475-481(1989))。通过将珀可(西格玛-奥德里奇公司(Sigma-Aldrich),圣路易斯,密苏里州,美国)稀释至10mM三乙醇胺,117mMNaCl,5mM葡萄糖和1.5mg/ml牛血清白蛋白(BSA)的终浓度,制备密度为1.096和1.058g/ml的无菌等渗珀可溶液。这些溶液具有在295和310mOsm之间的克分子渗透浓度。例如,将五毫升的第一珀可溶液(密度为1.096)添加到无菌的15ml锥形离心管中。例如,将两毫升的第二珀可溶液(密度为1.058)层叠在较高密度的第一珀可溶液上。将二至四毫升全血层叠在管的顶部上。将管在具有甩平式管座的冷冻离心机中以250×g离心30min。网织红细胞和一些白细胞迁移至两个珀可层之间的界面。将界面处的细胞转移到新管中,并用包含5mM葡萄糖、0.03mM叠氮化钠和1mg/mlBSA的磷酸盐缓冲盐水(PBS)洗涤两次。通过在PBS中通过尺寸排阻柱的色谱法除去残留的白细胞。可替代地,可以通过使用免疫磁性分离方法的阳性选择来分离网织红细胞(参见,例如,布朗(Brun)等人,血液(Blood)76:2397-2403(1990))。该方法利用了在成熟前相对于红细胞在网织红细胞表面上表达的大量转铁蛋白受体。用转铁蛋白受体的抗体包被的磁珠可用于从混合的血细胞群中选择性分离网织红细胞。多种哺乳动物物种(包括人)的转铁蛋白受体的抗体可从商业来源获得(例如,亲和生物试剂公司(AffinityBioReagents),戈尔登,科罗拉多州,美国;西格玛-奥德里奇公司,圣路易斯,密苏里州,美国)。可以将转铁蛋白抗体直接连接至磁珠。可替代地,可以通过第二抗体间接将转铁蛋白抗体接连接至磁珠。例如,可以将针对人转铁蛋白的小鼠单克隆抗体10D2(亲和生物试剂公司,戈尔登,科罗拉多州,美国)与包被有绵羊抗小鼠免疫球蛋白G的免疫磁珠(Dynal/英杰(Invitrogen)公司,卡尔斯巴德,加利福尼亚州,美国)混合。然后将免疫磁珠与白细胞耗尽的血红细胞部分一起孵育。在温和混合下,将磁珠和血红细胞在22℃下孵育60-90min,随后使用磁场分离具有附接的网织红细胞的珠子。例如,可以使用溶液(来自英杰公司,卡尔斯巴德,加利福尼亚州,美国)从磁珠除去分离的网织红细胞。可替代地,网织红细胞可以使用本文所述的方法从CD34+造血干细胞的体外生长和成熟中分离。终末分化的有核红细胞可以基于其DNA含量与其他细胞分离。在一个非限制性实施例中,首先用病毒DNA染料标记细胞,例如Hoechst33342(英杰公司)。Hoechst33342是一种细胞渗透性核复染剂,当与双链DNA结合时发射蓝色荧光。培养物中的未分化的前体细胞,巨噬细胞或其他有核细胞通过Hoechst33342染色,而去核红细胞是Hoechst阴性。可以通过使用荧光激活细胞分选仪或其他细胞分选技术将Hoechst阳性细胞从有核红细胞分离。可以通过透析或其他合适的方法从分离的红细胞中除去Hoechst染料。可以通过降低EHC群的核材料含量来纯化表达外源抗原的EHC群。例如,EHC群的摘除率增加,和/或增加或富集去核表达外源抗原的EHC的数量。可以将表达外源抗原的EHC群与小分子一起孵育,例如,肌动蛋白抑制剂,例如,细胞松弛素A、B、C、D、E、F、H、J,并且然后离心除去核材料。可替代地或此外,可以通过逐渐变小的尺寸限制性过滤器以去除核材料来机械操纵表达外源抗原的EHC群。还可以将表达外源抗原的EHC群在纤连蛋白包被的塑料表面上进行孵育,以增加核材料的去除。在一个实施例中,将表达外源抗原的EHC群在具有基质细胞(例如,巨噬细胞)的共培养物种进行孵育,以增加核材料的去除。在一些实施例中,表达外源抗原的EHC群大于5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、99%、99.5%、99.9%,或者大于99.9%去核。在一些实施例中,将表达外源抗原的EHC不与支持细胞(例如与粘附基质层)共培养。在一些实施例中,通过将EHC与外源抗原接触并且将EHC分化,以获得包括外源抗原的去核细胞群,来产生表达外源抗原的EHC群。在没有富集步骤,例如重力分离,磁性或荧光分选,辐射,有核细胞中毒等的情况下获得表达外源抗原的EHC群,以选择去核细胞。在一些实施例中,如通过检测核材料的测定(如本文所描述的那些)所评估的,表达外源抗原的EHC群由大于5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、99%、99.5%、99.9%、或大于99.9%的缺乏核材料的表达外源抗原的EHC构成。在一些实施例中,评估外源抗原(例如由表达外源抗原的EHC展示的外源抗原多肽)的存在,生物活性和/或功能。许多合适的测定是可获得的并且是本领域已知的。在一个实施例中,外源抗原是表达外源抗原的EHC表面上的多肽。外源抗原的存在可以通过测定来评估,这些测定包括但不限于:流式细胞术、蛋白质印迹法、RT-PCR、RNA印迹法、库姆斯重置、质谱法。在一个实施例中,外源抗原是表达外源抗原的EHC内部中的多肽。外源抗原的存在可以通过测定来评估,这些测定包括但不限于:蛋白质印迹法、RT-PCR、RNA印迹法、PCR、DNA印迹法、质谱法。在一个实施例中,该外源抗原是表达外源抗原的EHC的表面上的核酸。外源抗原的存在可以通过测定来评估,这些测定包括但不限于:流式细胞术、用同源荧光探针的流式细胞术、DNA印迹法、RNA印迹法、PCR。在一个实施例中,该外源抗原是表达外源抗原的EHC内部中的核酸。外源抗原的存在可以通过测定来评估,这些测定包括但不限于:DNA印迹法、RNA印迹法、PCR。在一个实施例中,该外源抗原是表达外源抗原的EHC的表面上的小分子。外源抗原的存在可以通过测定来评估,这些测定包括但不限于:流式细胞术、质谱法。在一个实施例中,该外源抗原是表达外源抗原的EHC内部中的小分子。外源抗原的存在可以通过测定来评估,这些测定包括但不限于:质谱法、荧光光谱学。在一个实施例中,外源抗原是表达外源抗原的EHC的膜中的脂质。外源抗原的存在可以通过测定来评估,这些测定包括但不限于:质谱法、流式细胞术、膜的溶解性、荧光偏振、空间光干涉显微镜检术。在一个实施例中,该外源抗原是荧光的或与荧光分子融合或从具有荧光报告蛋白如GFP的重组核酸共表达。在表达外源抗原的EHC中或其上的外源抗原的存在可以通过测定来评估,这些测定包括但不限于:流式细胞术、荧光光谱学、吸收光谱。在一个实施例中,该外源抗原是一种气态分子。在表达外源抗原的EHC中或其上的外源抗原的存在可以通过测定来评估,这些测定包括但不限于:化学发光测定、质谱法。通过流式细胞术,以定量方式,使用校准珠例如可商购获得的细胞计数校准珠来量化个体EHC上的外源抗原的数量,来评估表达外源抗原的EHC中或其上的外源抗原的存在。可替代地或此外,可以通过蛋白印迹法,以定量方式,使用可检测的已知浓度的标准,使用相同检测试剂作为外源抗原来评估表达外源抗原的EHC中或其上的外源抗原的存在,并且以此方式,可以量化个体EHC上外源抗原的数量。在一些实施例中,可以通过相同或不同的方法,同时,以连续方式或平行地评估两种或更多种不同外源抗原的存在。例如,在一个实施例中,可以通过使用对外源抗原具有特异性的抗体的流式细胞术来评估表面上的外源抗原,并且可以通过在流式细胞术中使用不同通道的荧光信号来评估不在荧光表面上的不同外源抗原。在不同的示例中,可以通过流式细胞术评估表面上的外源抗原,并且可以通过蛋白质印迹法评估不在表面上的不同外源抗原。在一个具体实施例中,在细胞来源的终末分化后,外源抗原保留在表达外源抗原的EHC上。例如,表达外源抗原的EHC由培养的EHC产生,并且在细胞终末分化后,通过适合的方法评估外源抗原的表达或存在,例如,通过流式细胞术、蛋白质印迹法、免疫沉淀反应、荧光光谱学、化学发光、DNA印迹法、RNA印迹法、或吸收光谱。在一个具体实施例中,在向受试者给予表达外源抗原的EHC后体内循环后,外源抗原保留在表达外源抗原的EHC上。可以将表达外源抗原的EHC注射到实验用动物或动物模型中,如经静脉内到小鼠中,例如通过尾静脉,或者经静脉内注射到人类中。然后抽血,并且通过适合的测定评估表达外源抗原的EHC上的外源抗原的存在,例如,通过流式细胞术、蛋白质印迹法、免疫沉淀反应、荧光光谱学、化学发光、DNA印迹法、RNA印迹法、或吸收光谱。在一些实施例中,在表达外源抗原的EHC中或其上的外源抗原的生物活性,EHC的总体生物活性,和EHC群的总体活性可以通过体外测定来评估。在一些实施例中,使用模型细胞系快速迭代表达外源抗原的EHC的活性。例如,在模型细胞系中表达合适的外源抗原文库,例如,HEK293T或K562,并且通过合适的测定法评估活性;然后最好的外源抗原候选物,例如,以最高水平表达的外源抗原或在合适的测定中显示最高的活性的外源抗原,表达于,例如,培养的EHC中,以产生表达外源抗原的EHC。在一个实施例中,使用培养的小鼠模型快速迭代表达外源抗原的EHC的活性。例如,在培养的小鼠EHC中表达合适的外源抗原文库;在合适的疾病小鼠模型或用于评估活性的合适的小鼠模型系统中评估活性;然后最好的外源抗原候选物,例如,以最高水平表达的外源抗原或在合适的测定中显示最高的活性的外源抗原,表达于,例如,培养的EHC中,以产生表达外源抗原的EHC。在一些实施例中,外源抗原是酶,并且外源抗原的活性可以通过酶测定来评估,其中检测特定底物分子的消失或检测特定产物分子的出现。这样的测定包括但不限于:比色测定、质谱法、HPLC、荧光测定。例如,a)外源抗原是腺苷脱氨酶(ADA)并且该酶法测定检测腺苷至肌苷的转化;b)该外源抗原是苯丙氨酸羟化酶(PAH)并且该测定检测苯丙氨酸至酪氨酸的转化;c)该外源抗原是苯丙氨酸氨裂解酶(PAL)并且该测定检测苯丙氨酸至反式肉桂酸的转化;d)该外源抗原是胸苷磷酸化酶(TP)并且该测定检测胸苷至胸腺嘧啶和2-脱氧-核糖的转化;e)该外源抗原是嘌呤核苷磷酸化酶(PNP)并且该测定检测肌苷至次黄嘌呤,腺苷至腺嘌呤,和鸟苷至鸟嘌呤的转化;f)该外源抗原是尿黑酸1,2-双加氧酶(HDG)并且该测定检测尿黑酸至马来酰乙酰乙酸的转化;g)该外源抗原是胱硫醚β合酶,并且该测定检测丝氨酸和同型半胱氨酸至胱硫醚的转化;h)该外源抗原是草酸氧化酶,并且该测定检测了草酸盐的氧化。在一些实施例中,在动物模型中评估表达外源抗原的EHC的活性,例如,患有疾病的小鼠模型、和免疫缺陷小鼠、或NSG小鼠,该疾病例如代谢疾病或酶缺陷,或可以证明表达外源抗原的EHC的作用的小鼠,例如,注射了底物并且测量外源抗原介导的转化的产物的小鼠。在一个实施例中,该外源抗原是补体受体1(CR1)多肽,其衍生物或功能片段。能以几种方式评估CR1外源抗原的活性,这些方法包括,例如,通过CR1外源抗原的免疫复合物的特异性捕获,免疫复合物到巨噬细胞的有效转移,或来自小鼠的免疫复合物的体内清除。可以通过一个或多个过程评估过表达CR1外源抗原的EHC的功能性:捕获包括CR1外源抗原的EHC表面上的免疫复合物,释放免疫复合物至巨噬细胞同时保留包括CR1外源抗原的EHC,以及包括CR1外源抗原的EHC的正常循环。这三个参数可以在体外测定。本领域描述了免疫复合物捕获测定,例如,乌丁(Oudin)等人,免疫学杂志(JImmunol)2000和斯契费尔利(Schifferli)等人,免疫学杂志1991。例如,将标记的免疫复合物与表达天然CR1或CR1外源抗原多肽或其片段的EHC一起孵育,并且通过流式细胞术测定由EHC捕获的免疫复合物的数目。本领域描述了巨噬细胞转移测定,例如,库恩(Kuhn)等人,免疫学杂志1998。例如,将荷载到表达天然CR1或CR1外源抗原多肽或其片段的红细胞上的标记的免疫复合物与巨噬细胞一起孵育。免疫复合物从红细胞表面向巨噬细胞的转移以及巨噬细胞对红细胞的消耗或保留可以通过流式细胞术进行测量。通过分析EHC形态和变形性可以预测正常循环。表达天然CR1或CR1外源抗原多肽或其片段的EHC的形态可以通过眼睛使用标准显微技术来评估,例如,通过贾拉塔纳(Giarratana)等人,血液,2011和瑞皮克(Repik)等人,临床和实验免疫学(ClinExpImmunol)2005所描述的。表达天然CR1或CR1外源抗原多肽或其片段的EHC的变形性可以通过细胞变形计量学来评估,该细胞变形计量学又称为激光辅助光学旋转细胞分析(LORCA),例如,如贾拉塔纳等人,血液,2011所描述的。例如,将表达外源CR1抗原的EHC(包括CR1多肽外源抗原的EHC)与免疫复合物(如,体外产生的免疫复合物或患者衍生的免疫复合物)一起孵育。例如,通过流式细胞术使用免疫复合物中的荧光标记或通过流式细胞术使用针对免疫复合物的元件的第二检测剂,评估由CR1外源抗原对免疫复合物的捕获。在一个实施例中,首先将表达外源CR1抗原的EHC与免疫复合物孵育,然后与巨噬细胞孵育,这些巨噬细胞如原代巨噬细胞、原代单核细胞、培养的巨噬细胞、培养的单核细胞、U937细胞、PMA-激活的U937细胞、AA9细胞、RAW264.7细胞、J774细胞、THP1细胞、KG-1细胞、NR8383细胞、MV-4-11细胞、3D4/31细胞、MD细胞、Fcwf-4细胞、DH82细胞。例如,针对由表达外源CR1抗原的EHC转移的免疫复合物的存在,通过流式细胞术或射线照相术测定巨噬细胞。捕获的免疫复合物从培养的EHC到巨噬细胞的转移是本领域中的标准测定,参见,例如,瑞皮克(Repik)等人,2005,临床和实验免疫学,140:230;李(Li)等人,2010,传染免疫(InfectionImmunity),78(7):3129。在一个实施例中,在动物模型中评估表达外源CR1抗原的EHC的活性。例如,可以使用合适的小鼠模型,如免疫缺陷小鼠或NSG小鼠。小鼠疾病模型可以是例如免疫复合物疾病,例如狼疮。小鼠模型包括NZBWF1/J、MRL/MpJ、MRL/MpJ-Fas(lpr)、Smn.C3-Fasl/J、NZM2410/Aeg、129S4-Cd48、Cg-Sle1、NZM-Sle1Sle2Sle3/LmoJ、和BXSB.129P2。可替代地或此外,可以将疾病表型引入小鼠,例如通过注射免疫复合物。表达外源CR1抗原的EHC可以注射到任何合适的小鼠(或其他动物模型)中以测试EHC的一种或多种生物效应,例如,通过表达外源CR1抗原的EHC清除注射的免疫复合物。在一个实施例中,该外源抗原是补体调节分子或具有补体调节活性。外源抗原的这种活性可以通过体外和体内测定来评估。例如,外源抗原的活性可以通过测量体外补体激活测定中的降低来评估,这些测定例如:测量补体介导的敏化的绵羊红细胞的裂解的CH50测定或测量替代途径补体介导的非敏化的兔红细胞的裂解的AH50测定。可替代地,外源抗原的活性可以通过检测重组补体组分的切割或切割的不存在(可能暴露或不暴露新表位)来评估,该组分已经与外源抗原一起孵育,其包括但不限于,例如:将重组C2切割成C2a和C2b,将因子B切割成因子Ba和因子Bb,将因子C3b切割成iC3bH和iC3bL,将C3bBb切割成C3b和Bb,将C4bBb切割成C4b和Bb,或将因子C4b切割成iC4bH和iC4bL。合适的重组补体成分的切割或切割的不存在可以通过本领域已知的蛋白质分析方法来评估,这些方法包括但不限于,例如:色谱、凝胶电泳、ELISA、和蛋白质印迹法。针对外源抗原活性的适合的体内测定包括将表达外源抗原的EHC注射到动物中,例如小鼠,并且通过组织学染色检查补体因子的沉积,例如膜攻击复合物。在一个实施例中,外源抗原能够结合或捕获靶标,并且可以通过在体外或体内检测外源抗原上捕获的靶标来评估外源抗原的活性。在一个实施例中,将表达外源抗原的EHC与体外靶标一起孵育,并且使用体外测定检测外源抗原对靶标的捕获,这些体外测定包括但不限于,例如:流式细胞术、免疫组织化学、磁分离、射线照相术、集落形成测定、显微镜检术。在一个实施例中,将表达外源抗原的EHC与体外靶标一起孵育,并且使用体外共培养测定检测外源抗原对靶标的捕获,这些体外共培养测定包括但不限于,例如:表达调理的外源抗原的EHC的巨噬细胞消耗测定,T细胞激活测定,B细胞刺激测定,肥大细胞脱粒测定,感染性潜在测定。在一个实施例中,将表达外源抗原的EHC与体外靶标一起孵育,并且使用体外测定来测量捕获的靶标的释放或解离速率,这些体外测定包括但不限于,例如:流式细胞术、免疫组织化学、磁分离、射线照相术、集落形成测定、显微镜检术。可以在体内测定中测定表达外源抗原的EHC对靶标的捕获,例如,在动物中,其包括患有疾病的小鼠模型(其中靶标天然存在于小鼠中)。合适的疾病包括细菌感染,病毒感染,真菌感染,免疫复合物疾病,自抗体疾病,高脂血症,高血糖症。在其他小鼠模型中,将靶标经外部给予小鼠,例如通过注射或通过喂食。在这些测定中,通过针对靶标的减少或保留检测动物(例如,血浆、组织),或通过从动物(例如,从血液、从血浆、从组织)分离或收集表达外源抗原的EHC并且使用体外测定(这些体外测定包括但不限于,例如:流式细胞术、免疫组织化学、磁分离、射线照相术、集落形成测定、显微镜检术)测定外源抗原上靶标的存在,来测定由表达外源抗原的EHC对靶标的捕获。在一些实施例中,该外源抗原能够结合或捕获靶标,并显着增加体内靶标的清除,或基本上降低循环中靶标的浓度。表达外源抗原的EHC上的外源抗原的活性可以通过检测体外或体内靶标的增强的清除来评估。在一个实施例中,将表达外源抗原的EHC与体外靶标一起孵育,并且使用体外测定检测外源抗原对靶标的捕获,这些体外测定包括但不限于,例如:流式细胞术、免疫组织化学、磁分离、射线照相术、集落形成测定、显微镜检术。随后,将表达外源抗原的EHC在共培养测定中与已知促进清除的细胞例如巨噬细胞或单核细胞一起孵育,并且通过以下各项评估靶标和表达外源抗原的EHC的清除,例如,流式细胞术、免疫组织化学、磁分离、射线照相术、集落形成测定、显微镜检术。在一个实施例中,将表达外源抗原的EHC与体外靶标一起孵育,并且使用体外测定检测外源抗原对靶标的捕获,这些体外测定包括但不限于,例如:流式细胞术、免疫组织化学、磁分离、射线照相术、集落形成测定、显微镜检术。随后,将表达外源抗原的EHC在模拟体内EHC的清除机制的物理系统中孵育,该清除机制例如人工脾,微通道,填充柱,树脂,组织外植体,离心机,并且通过以下各项评估靶标和表达外源抗原的EHC的清除,例如,流式细胞术、免疫组织化学、磁分离、射线照相术、集落形成测定、显微镜检术。在一个实施例中,在体内测定中,例如,在动物中,测定由表达外源抗原的EHC对靶标的清除,这些动物包括例如:患有疾病(这些疾病例如细菌感染,病毒感染,真菌感染,免疫复合物疾病,自抗体疾病,高脂血症,高血糖症)的小鼠模型(其中靶标天然存在于小鼠中),或者例如:小鼠模型(其中将靶标经外部给予小鼠,例如通过注射或通过喂食)。在这些测定中,通过针对靶标的减少检测动物(例如,血浆、组织),或通过从动物(例如,从血液、从血浆、从组织)分离或收集表达外源抗原的EHC并且使用体外测定(这些体外测定包括但不限于,例如:流式细胞术、免疫组织化学、磁分离、射线照相术、集落形成测定、显微镜检术)测定外源抗原上靶标的存在,来测定由表达外源抗原的EHC对靶标的清除。在一些实施例中,表达外源抗原的EHC能够将合适的外源抗原递送至特定的亚细胞区室,例如溶酶体。例如,可以将外源抗原递送至靶细胞(例如,巨噬细胞)的溶酶体区室。通过对应于荧光外源抗原或荧光外源抗原检测剂的点状斑点的显微镜检术和检测来评估外源抗原向靶细胞的溶酶体区室的成功递送。可替代地或此外,通过荧光外源抗原检测剂与用于已知溶酶体标志物的荧光检测剂(例如,lysotracker,LAMP-1)的显微镜检术和共定位来评估外源抗原向靶细胞的溶酶体区室的成功递送。在一些实施例中,外源抗原是可以降解有毒组分的酶,这些有毒组分在细胞的溶酶体中积聚,该细胞表现出有溶酶体贮积病的基因型或表现型,或者衍生自患有溶酶体贮积病的患者。例如,外源抗原能够降解在细胞中积累的毒性物质并拯救细胞表型,例如,预防细胞死亡。可以针对EHC不能复制,EHC通过脉管系统安全地循环的能力,和缺乏EHC的免疫原性,评估表达外源抗原的EHC群。在一些实施例中,通过使用合适的体外或体内测定测量EHC群的复制潜力来评估表达外源抗原的EHC群的安全性。例如,针对表达外源抗原的EHC基本上不能自我复制的测试包括:a)当注射到免疫受损小鼠中时基本上不能形成肿瘤;b)当在软琼脂中培养时基本上不能形成菌落;c)在胸苷掺入测定中基本上不能掺入胸苷;d)在用编码荧光报道分子的DNA转染时基本上缺乏阳性信号,例如,小于10%、1%、0.1%、0.01%、0.001%、1ppm、100ppb、10ppb、1ppb、100ppt、10ppt、1ppt、或小于1ppt阳性信号;e)在用核染料染色时基本上缺乏阳性信号,例如,小于10%、1%、0.1%、0.01%;和0.001%、1ppm、100ppb、10ppb、1ppb、100ppt、10ppt、1ppt、或小于1ppt阳性信号;f)在用恶性血液病的细胞标志物(例如,CKIT、CD34、EpCam)染色时基本上缺乏阳性信号,例如,小于10%、1%、0.1%、0.01%、0.001%、1ppm、100ppb、10ppb、1ppb、100ppt、10ppt、1ppt、或小于1ppt阳性信号。在某些实施例中,提供了表达外源抗原的EHC,其不包含大量的复制核酸。在某些实施例中,通过测量给予的EHC在体内(在受试者的循环系统中)循环而不引起实质性血管闭塞或诱导凝血级联的能力来评估表达外源抗原的EHC群的安全性。任选地,可以评估表达外源抗原的EHC的循环药代动力学。在一个实施例中,通过经静脉内将EHC注射到动物(例如小鼠)中来评估表达外源抗原的EHC的循环药代动力学。小鼠可以是NSG(nod-SCID-γ)免疫缺陷小鼠。在注射EHC前,使小鼠耗尽巨噬细胞,例如,通过腹膜内注射人血红细胞,或通过静脉内注射氯膦酸盐脂质体。可以用荧光染料,例如CFSE,标记表达外源抗原的EHC。在注射EHC后,抽取血液并且通过以下各项评估剩余的表达外源抗原的EHC的数量,例如:流式细胞术、蛋白质印迹法,并且从这些数据推断表达外源抗原的EHC的清除率。可以将人血红细胞注射到与表达外源抗原的EHC相同的动物模型中,并且比较EHC和人血红细胞的清除率。在一个实施例中,用体外测定评估由表达外源抗原的EHC激活的凝血级联的风险。在一个实施例中,将表达外源抗原的EHC与人血一起孵育,并且在高岭土、带负电荷的磷脂和钙的存在下,通过测量凝结所需的时间(激活部分凝血激酶时间(aPTT)测试)来评估凝血级联激活,参见,例如,埃克斯纳和里卡德(ExnerandRickard),生物医学(Biomedicine)197727(2):62,或者在促凝血酶原激酶和钙的存在下,通过测量凝结所需的时间(凝血酶原时间(PT)测试)来评估凝血级联激活,参见,例如,杰奎斯和邓禄普(JaquesandDunlop)1945,美国生理学杂志(AmJPhysiol)145:67。aPTT测试的正常范围约为25-38秒。PT测试的正常范围约为10-12秒。在一个实施例中,通过将EHC经静脉内注射到动物中并且评估凝血级联的激活来评估由表达外源抗原的EHC所诱导的任何不良事件。通过抽血并且通过例如蛋白印迹法或ELISA评估血浆中的凝血级联组分水平来评估凝血级联诱导的水平。凝血级联组分通常是纤维蛋白原分解产物,例如纤维蛋白肽A和纤维蛋白肽B。例如,通过抽血并且通过血小板激活测定评估血浆中凝血活性的水平来评估凝血级联诱导的水平,例如,将血浆与血小板一起孵育并且通过流式细胞术评估血小板的激活,例如,通过染色激活的标记物,例如,通过染色PAC-1、CD62p、或CD40L。在一个实施例中,通过将EHC经静脉内注射到动物中并且评估炎症途径的激活来评估由表达外源抗原的EHC所诱导的任何不良事件。可以通过抽血并且通过例如蛋白印迹法或ELISA评估血浆中的炎性细胞因子水平来评估炎症的水平。在一个实施例中,该炎症细胞因子是干扰素γ,肿瘤坏死因子α和IL-12片段p70。在一个实施例中,通过将EHC经静脉内注射到动物中并且评估组织状态的激活来评估由表达外源抗原的EHC所诱导的任何不良事件,这些组织是例如:肝、脾、心、肺、脑、皮肤、肾。该组织状态可以通过肉眼尸检、组织解剖、组织学染色、和通过显微镜成像来评估。在一个实施例中,表达外源抗原的EHC在体内循环而不引起显着血管闭塞或凝固级联的激活的能力是通过测量EHC的变形性来评估的。使用体外测定评估表达外源抗原的EHC的变形性。例如,该测定是渗透脆性测定。表达外源抗原的EHC的机械脆性可以通过在Couette型剪切系统中测量响应于剪切应力的结构完整性来评估。在一个实施例中,使用血细胞计数器评估表达外源抗原的EHC的变形性。在一个实施例中,通过在定义的压力下通过激光衍射使用激光辅助光学旋转细胞分析仪(LORCA)仪器测量伸长指数来评估表达外源抗原的EHC的变形性。在一个实施例中,通过在微流体装置中在固定压力下通过限定尺寸的一系列微米级收缩来测量相变时间,来评估表达外源抗原的EHC的变形性。在一个实施例中,通过在微流体装置中在固定压力下通过限定尺寸的一系列微米级收缩来测量存活率,来评估表达外源抗原的EHC的变形性。微流体装置可以选自以下之一,其包括但不限于:具有微米级收缩的聚二甲基硅氧烷(PDMS)芯片(例如,霍尔茨(Hoelzle)等人,可视化实验杂志(JVisExp)201491:e51474);具有漏斗形收缩的芯片(例如,(Guo)等人,芯片实验室(LabChip)201212:1143);具有支柱的PDMS芯片(例如,张(Zhang)等人,PNAS2012109(46):18707);或体外人工脾微珠填充柱(纪尧姆迪普莱纳(GuillaumeDePlaine)等人,血液,2011,117(8))。在一个实施例中,表达外源抗原的EHC在体内循环而不引起显着血管闭塞或凝固级联的激活的能力是通过测量EHC的血管闭塞来评估的。可以使用体外测定评估表达外源抗原的EHC的血管闭塞。例如,使用离体测定评估表达外源抗原的EHC的血管闭塞。将表达外源抗原的EHC与参比人血红细胞以1:1的比例进行孵育,并且与使用Rh-错配的血液的参比测定相比,通过光学显微镜法评估多细胞玫瑰花结的诱导。通过在流动条件下测量EHC与人血管内皮细胞的粘附来评估表达外源抗原的EHC的血管闭塞,参见,例如,考尔(Kaul)DK,芬尼根(Finnegan)E,和巴拉比诺(Barabino)GA(2009),微循环(Microcirculation)16(1):97-111。可替代地或此外,通过在大鼠血管内皮的离体灌注测定中测量外周阻力单位(PRU)增加来评估血管闭塞,参见,例如,考尔(Kaul),法夫里(Fabry)和纳高(Nagel),PNAS1989,86:3356。此外,通过活体镜检法评估血管闭塞,参见,例如,泽娜迪(Zennadi)等人,2007,血液,110(7):2708。血管闭塞还可以通过测量EHC体外渐变高度流动室的流速和粘附来评估,参见,例如,泽娜迪等人,2004,血液,104(12):3774。在一些实施例中,通过使用合适的体外或体内测定测量EHC群的免疫原性来评估表达外源抗原的EHC群的安全性。例如,表达外源抗原的EHC群a)在使用来自预期受体受试者的血清的库姆斯试验中并不诱导凝集或者b)在使用合并的人血清的库姆斯试验中不诱导凝集。在一个实施例中,表达外源抗原的EHC群衍生自祖细胞,该祖细胞已经针对主要的血型抗原进行了基因分型并与接受者的血型抗原基因型进行了匹配。在一个实施例中,表达外源抗原的EHC群包括外源抗原或其他外源蛋白,该外源抗原或其他外源蛋白通过计算机模拟T细胞表位预测算法具有小于10%、1%、0.1%、0.01%、0.001%、或小于0.001%预测的T细胞反应性。在一个实施例中,表达外源抗原的EHC群包括外源抗原或其他外源蛋白,该外源抗原或其他外源蛋白在体外T细胞激活测定(例如,Antitope公司EpiScreen测定)中具有小于10%、1%、0.1%、0.01%、0.001%、或小于0.001%反应性。例如,可以将来源于红细胞的表达外源抗原的EHC离心并重新悬浮于合适的溶液(例如,标准AS-3溶液)中,用于输注到对其有需要的受试者中。在一些实施例中,待输注的表达外源抗原的EHC具有与受体相同的ABO类型,以使输注相关的免疫反应的风险最小化。还可以预处理表达外源抗原的EHC,以除去血型特异性抗原或以其他方式降低抗原性。适合于此目的方法包括但不限于:美国专利申请公开号20010006772和20030207247中所描述的那些。治疗组合物提供了包含EHC的组合物,这些EHC具有有效水平的感兴趣的外源抗原。这类组合物包含许多EHC,例如,1x103个细胞、或1x104、1x105、1x106、1x107、1x108、1x109、1x1010、1x1011、1x1012、或大于1x1012个细胞。例如,本发明的EHC可以作为堆积的红血细胞在盐溶液中以10%、20%、30%、40%、50%、60%、70%、80%、90%或大于90%质量/体积比的浓度(%m/v)进行给予。向患者给予的时间可以在从10分钟至4小时、或更久的范围内。本发明的培养的EHC可以储存在合适的缓冲液中,例如,FDA批准的抗凝血剂保存液,如抗凝血剂柠檬酸盐-右旋糖A(ACD-A)、柠檬酸盐-磷酸盐右旋糖(CPD)、柠檬酸磷酸盐-右旋糖-右旋糖(CP2D)、或柠檬酸盐-磷酸盐-右旋糖-腺嘌呤(CPDA-1)。组合物可储存长达21天。可替代地,本发明的培养的EHC可以储存在批准的添加剂溶液中,例如,AS-1(Adsol)、AS-3(Nutricel)、AS-5(Optisol)、或AS-7(SOLX)。可替代地,本发明的培养的EHC可以储存在甘油低温防护溶液中。这些组合物可以冷冻并储存长达10年。冷冻的细胞可以通过连续的洗涤步骤解冻和脱甘油化,例如在使用前用0.9%的氯化钠。考虑到人类和鼠红细胞的寿命分别为120和50天(参见,例如,坎多尔沃(Khandelwal)等人,输注(Transfusion)2007),刺激人工红细胞衰老以增加抗原呈递细胞的吞噬作用可能是有利的。从循环中去除红细胞的最重要的以及生理的机制是在暴露RBC细胞表面上的新抗原位点之后免疫介导(辛格(Singer)等人,PNAS1986),例如分别用钙离子载体或BS3化学处理人工诱导的磷脂酰丝氨酸外化(斯克罗伊特(Schroit)等人,生物化学杂志(JBiolChem)1985)或带3蛋白聚集(凯(Kay),PNAS1975;图里尼(Turrini)等人,生物化学杂志1991)。本发明的培养的EHC可以用吞噬作用诱导剂,例如,钙离子载体或BS3来处理。许多本发明的培养的EHC可以包括已经用吞噬作用诱导剂(例如,钙离子载体或BS3)处理了不同时间长度的EHC,这样使得当向受试者给予时,许多本发明的培养的EHC以不同的速率被吞噬,例如,在一天或若干天过程中连续进行或不是作为大丸剂。本文提供了包括许多培养的EHC的组合物和药物组合物,这些EHC包括感兴趣的外源抗原。组合物和药物组合物可以包括适当的储存缓冲液的溶液,例如,抗凝血柠檬酸-右旋糖A。该包括许多培养的EHC(这些EHC包括感兴趣的外源抗原)的组合物和药物组合物可以另外包括批准的添加剂,例如Adsol。该包括许多培养的EHC(这些EHC包括感兴趣的外源抗原)的组合物和药物组合物可以另外包括用于冷冻保存的甘油低温防护溶液。本文提供了包括感兴趣外源抗原的EHC,该外源抗原选自表F、表G、表H、表I和表J的抗原的一种或多种,例如,髓磷脂碱性蛋白、蛋白脂质蛋白、髓磷脂少突胶质细胞糖蛋白、胰β细胞抗原、胰岛素、鞭毛蛋白、谷蛋白、2S白蛋白、透明质酸酶、因子VIII、因子IX、和抗-TNFa、腺苷脱氨酶、L-天冬酰胺酶、拉布立酶、抗胸腺细胞球蛋白、L-精氨酸酶、L-蛋氨酸酶、前胰岛素、胰岛素原、胰岛素、GAD65、GAD67、IA-2、ΙΑ-2β、甲状腺球蛋白、甲状腺过氧化物酶、促甲状腺激素受体、髓磷脂少突胶质细胞糖蛋白、蛋白脂质蛋白、胶原II、胶原IV、乙酰胆碱受体、基质金属蛋白1和3、分子伴侣热休克蛋白47、原纤维蛋白-1、PDGF受体a、PDGF受体β、核蛋白SS-A、伴花生球蛋白(Arah1)、过敏原II(Arah2)、花生凝集素(Arah6)、α-乳白蛋白(ALA)、乳运铁蛋白、谷蛋白、低分子量谷蛋白、α-麦醇溶蛋白、γ-麦醇溶蛋白、大麦醇溶蛋白、黑麦碱、燕麦蛋白及其组合。能以组合物或药物组合物提供包括感兴趣的外源抗原的多种EHC。本文提供了编码表F、表G、表H、表I和表J的一种或多种抗原的表达载体,如髓磷脂碱性蛋白、蛋白脂质蛋白、髓磷脂少突胶质细胞糖蛋白、胰β细胞抗原、胰岛素、鞭毛蛋白、谷蛋白、Arah2、2S白蛋白、透明质酸酶、因子VIII、因子IX、和抗TNFa,任选地融合至表C的内源红细胞蛋白之一。本文提供了包括表达外源抗原的EHC的药物组合物,这些EHC适用于向受试者给予。这些药物组合物通常包括表达外源抗原的EHC群和以一种适用于向受试者给予的形式的药学上可接受的载体。药学上可接受的载体部分地通过所给予的具体组合物、连同通过给予该组合物所使用的具体方法来确定。因此,存在多种多样的药物组合物的适合的配制品,这些药物组合物包括表达外源抗原的EHC群。(参见,例如,雷明顿药学大全(Remington’sPharmaceuticalSciences),麦克出版公司(MackPublishingCo.),伊斯顿,宾夕法尼亚州,第18版,(1990))。药物组合物通常配制为无菌的,基本上等渗的并完全符合美国食品和药物管理局的所有良好生产规范(GMP)规定。药学上可接受的赋形剂包括通常安全,无毒和所需的赋形剂,包括兽医用途以及人类药物使用可接受的赋形剂。这样的赋形剂可以是固体、液体、半固体或气体(在气雾组合物的情况下)。载体或稀释剂的实例包括但不限于:水、盐水、林格氏溶液、右旋糖溶液和5%人血清白蛋白。用于药物活性物质的此类介质和化合物的用途是本领域中熟知的。除非与本文描述的表达外源抗原的EHC不相容,否则任何常规介质或化合物均可以考虑在组合物中的用途。补充治疗剂也可以并入这些组合物中。典型地,将药物组合物配制成与其预期的给予途径相容。通过以下途径给予表达外源抗原的EHC:静脉内、口服、皮下、动脉内、皮内、经皮、直肠、颅内、腹膜内、鼻内;肌内途径或作为吸入剂。表达外源抗原的EHC可任选与其他治疗剂联合给予,所述治疗剂至少部分有效地治疗表达外源抗原的EHC预期针对的疾病、障碍或病症。用于肠胃外、皮内、或皮下应用的溶液或悬浮液可以包括以下组分:无菌稀释剂,如注射用水、盐溶液、非挥发油、聚乙二醇、甘油、丙二醇或其他合成溶剂;抗菌化合物,如苯甲醇或对羟基苯甲酸甲酯;抗氧化剂,如抗坏血酸或亚硫酸氢钠;螯合化合物如乙二胺四乙酸(EDTA);缓冲剂,如乙酸盐、柠檬酸盐或磷酸盐;以及用于调节张力的化合物,如氯化钠或葡萄糖。可用酸或碱调节PH值,例如盐酸或氢氧化钠。胃肠外制剂可以被封装在由玻璃或塑料制成的安瓿、一次性注射器或多剂量小瓶中。适合于可注射使用的药物组合物包括无菌水溶液(在水溶性的情况下)或分散体以及用于临时制备无菌可注射溶液或分散液液的无菌粉末。对于静脉内的给予,适当的载体包括生理盐水、抑菌水、CremophorELTM(BASF,Parsippany,N.J.)或磷酸酯缓冲盐水(PBS)。在所有情况下,该组合物必须是无菌的并且应该具有达到容易注射的程度的流动性。此类药物形式在制造和储存条件下必须稳定并且必须被保存以防诸如细菌和真菌的微生物污染作用。载体可以是溶剂或分散介质,包含例如水、乙醇、多元醇(例如,甘油、丙二醇和液体聚乙二醇,等等),和其合适的混合物。恰当的流动性可(例如)通过应用衣料如卵磷脂来维持,在分散体的情况下通过维持所需的颗粒大小来维持,以及通过应用表面活性剂来维持。防止微生物的作用可以通过不同的抗细菌以及抗真菌化合物,例如对羟苯甲酸酯、三氯叔丁醇、苯酚、抗坏血酸、硫柳汞等来实现。在许多情况下,优选在组合物中纳入等渗化合物,例如糖、多元醇如甘露醇、山梨醇、氯化钠。可注射组合物的延长吸收可以通过在组合物中包含一种延缓吸收的化合物,例如单硬脂酸铝和明胶来实现。如所希望的,可以通过将表达外源抗原的EHC以有效的量和在适合的溶剂中与本文列举的成分的一种或组合掺在一起来制备无菌可注射溶液。通常,通过将表达外源抗原的EHC并入包含碱性分散介质和任何所希望的其他成分的无菌载体中来制备分散体。就用于制备无菌可注射溶液的无菌粉末而言,制备方法为真空干燥和冷冻干燥技术,这些方法产生活性成分的粉末以及来自其先前的无菌过滤溶液的任何另外的所需成分。表达外源抗原的EHC能以贮库注射或植入制剂的形式给予,其能以允许表达外源抗原的EHC、它们的外源抗原和/或它们的任选有效负载的持续或脉冲释放的这种方式进行配制。口服的组合物通常包括一种惰性稀释剂或一种可食用的载体。它们可以被封装在明胶胶囊中或被压成片剂。出于口服治疗性给予目的,表达外源抗原的EHC可以随赋形剂一起掺入并且以片剂、药锭剂、或胶囊剂的形式使用。可以使用用以口腔清洗的流体载体来制备口服组合物,其中流体载体中的化合物经口服、漱、咳出或咽下。可以包括药学上相容的粘合化合物和/或佐剂材料作为组合物的一部分。片剂、丸剂、胶囊剂、锭剂等可以包含以下成分或具有类似性质的化合物中的任一种:结合物,如微晶纤维素、黄芪胶或明胶;赋形剂,如淀粉或乳糖,崩解化合物,如海藻酸、Primogel、或玉米淀粉;润滑剂,如硬脂酸镁或氢化植物油;助流剂,如胶体二氧化硅;甜味化合物,如蔗糖或糖精;或调味化合物,如薄荷、水杨酸甲酯或橙味调味品。对于通过吸入给予,表达外源抗原的EHC从加压容器或分配器(其中包含合适的推进剂,例如气体(如二氧化碳))、或喷雾器以气溶胶喷雾形式递送。包括表达外源抗原的EHC的组合物的全身性给予还可以通过经粘膜的或经皮的方式。对于经粘膜或经皮肤给予,在该配制品中使用适合有待渗透的障碍的渗透剂。这样的渗透剂通常是本
技术领域:
所熟知的,并且对于经粘膜给予包括,例如洗涤剂、胆汁盐和夫西地酸衍生物。经黏膜给予可通过使用鼻喷剂或栓剂来完成。对于经皮肤给予,如本领域中公知的,将修饰的血红细胞配制成软膏剂、油膏剂、凝胶剂、或乳膏。表达外源抗原的EHC还可以用于直肠递送的栓剂(例如使用常规的栓剂基质,如可可脂和其他甘油酯)或保留灌肠剂的形式制备为药物组合物。在一些实施例中,用载体来制备表达外源抗原的EHC,其将降低表达外源抗原的EHC从受试者体内消除的速率。例如,控释的配制品是适合的,包括植入物和微胶囊递送系统。可使用生物可降解、生物相容的聚合物,如乙烯醋酸乙烯酯、聚酸酐、聚乙醇酸、胶原、聚原酸酯及聚乳酸。用于制备此类配制品的方法对本领域的普通技术人员而言应是显而易见的。这些材料可以从阿尔扎(AlzaCorporation)公司和诺维信生物有限公司(NovaPharmaceuticals.Inc)购得。在一个实施例中,将包括表达外源抗原的EHC的药物组合物经静脉内给予将从药物组合物受益的受试者。在其他实施例中,将该组合物给予淋巴系统,例如,通过淋巴管注射或通过结节内注射(参见,例如,森蒂(Senti)等人,2008PNAS105(46):17908),或者通过肌内注射,通过皮下给予,通过直接注射到胸腺中或到肝脏中。在一个实施例中,将包括表达外源抗原的EHC的药物组合物以液体悬浮液给予。在一个实施例中,将药物组合物以凝结的配制品给予,该凝结的配制品在给予后能够形成贮库,并且在一个优选的实施例中,缓慢释放表达外源抗原的EHC到循环中,或在一个优选的实施例中,以贮库形式保留。在一个实施例中,使用能够维持表达外源抗原的EHC的活力的方法和缓冲液组合物储存包括表达外源抗原的EHC的药物组合物。例如,在储存前保持厌氧状态的脱氧作用,pH的操作,代谢前体的补充,渗透平衡的操纵,悬浮介质的体积的增加和/或通过添加保护性分子而减少氧化应激可用于维持表达外源抗原的EHC的生存力。使用这些策略的组合的若干研究已经报道了红细胞生存力的维持,允许延长存储超过6周(参见,例如,吉田(Yoshida)和舍甫科普蕾丝(Shevkoplyas),输注(BloodTransfus)20108:220)。可以使用药学上可接受的载体或赋形剂来递送本文中所描述的表达外源抗原的EHC。赋形剂是指用作稀释剂或载体的惰性物质。一般,将药学上可接受的载体与化合物一起使用,以便使该化合物可用于疗法或作为产品。通常,对于任何物质,药学上可接受的载体是与底物组合以递送至受试者的材料。常规的药物载体,水性基质,粉末或油性基质,增稠剂等可以是必要的或希望的。在一些情况下,载体对于递送是必需的,例如,以溶解用于液体递送的不溶性化合物;用于控制底物的pH以保持其活性的缓冲液;或防止储存容器中的底物损失的稀释剂。然而,在其他情况下,载体为了方便起见是,例如,更方便给予的液体。本文所述化合物的药学上可接受的盐可以根据本领域技术人员已知的方法合成。通常,药学上可接受的组合物是高度纯化的,不含污染物,是生物相容的并且无毒的,以及适于给予受试者。如果水是载体的组分,那么水是高度纯化的,并且加工成没有污染物的,例如内毒素。药学上可接受的载体可以是乳糖、右旋糖、蔗糖、山梨醇、甘露醇、淀粉、阿拉伯树胶、磷酸钙、藻酸盐、明胶、硅酸钙、微晶纤维素、聚乙烯吡咯烷酮、纤维素、水、糖浆、甲基纤维素、甲基羟基苯甲酸酯、丙基羟基苯甲酸酯、滑石、硬脂酸镁和/或矿物油,但不限于此。该药物组合物可以进一步包括滑润剂、润湿剂、增甜剂、增味剂、乳化剂、悬浮剂、和/或防腐剂。提供了包含表达外源抗原的EHC的药物组合物,这些EHC具有有效水平的外源抗原。这类组合物包含许多表达外源抗原的EHC,例如,1x103个EHC、或1x104、1x105、1x106、1x107、1x108、1x109、1x1010、1x1011、1x1012、或大于1x1012个EHC。在具体实例中,从EHC产生的表达外源抗原的EHC可以作为堆积的红血细胞在盐溶液中以10%、20%、30%、40%、50%、60%、70%、80%、90%或大于90%质量/体积比的浓度(%m/v)进行给予。向患者给予的时间可以在从10分钟至4小时、或更久的范围内。在具体实例中,从EHC产生的表达外源抗原的EHC可以储存在合适的缓冲液中,例如,FDA批准的抗凝血剂保存液,如抗凝血剂柠檬酸盐-右旋糖A(ACD-A)、柠檬酸盐-磷酸盐右旋糖(CPD)、柠檬酸磷酸盐-右旋糖-右旋糖(CP2D)、或柠檬酸盐-磷酸盐-右旋糖-腺嘌呤(CPDA-1)。组合物可储存长达21天。可替代地,从EHC产生的表达外源抗原的EHC可以储存在批准的添加剂溶液中,例如,AS-1(Adsol)、AS-3(Nutricel)、AS-5(Optisol)、或AS-7(SOLX)。可替代地,从EHC产生的表达外源抗原的EHC可以储存在甘油低温防护溶液中。这些组合物可以冷冻并储存长达10年。冷冻的细胞可以通过连续的洗涤步骤解冻和脱甘油化,例如在使用前用0.9%的氯化钠。本文提供了包括许多培养的EHC的组合物和药物组合物,这些EHC包括外源抗原。组合物和药物组合物可以包括适当的储存缓冲液的溶液,例如,抗凝血柠檬酸-右旋糖A。该包括许多培养的EHC(这些EHC包括外源抗原)的组合物和药物组合物可以另外包括批准的添加剂,例如Adsol。该包括许多培养的EHC(这些EHC包括外源抗原)的组合物和药物组合物可以另外包括用于冷冻保存的甘油低温防护溶液。在一个实施例中,表达外源抗原的EHC能够与另一种表达外源抗原的EHC形成多复合物凝集体,例如,二聚体、三聚体、多聚体。在一个实施例中,该表达外源抗原的EHC能够与循环系统的组分(例如,红细胞、网织红细胞、血小板、巨噬细胞、淋巴细胞、T细胞、B细胞、肥大细胞)形成多复合物凝集体,例如,二聚体、三聚体、多聚体。表达源抗原的EHC及其药物组合物的给予剂量和频率可以由主治医生基于各种因素来确定,这些因素如疾病的严重性,患者的年龄,性别和饮食,任何炎症的严重性,给予时间和其他临床因素。在一个实施例中,以最低限度有效的剂量开始静脉内给予,并且剂量在预选的时间过程中增加直到观察到积极效果。随后,剂量的增量增加被限制到产生相应效果增加的水平,同时考虑可能出现的任何不利影响。适合剂量的非限制性实例可以,例如,从1x103至1x1014、从1×103至1×107、从1×105至1×106、从1×107至1×1011、从1×108至1×109、从1×1010至1×1014、从1×1011至1×1013、或从5×1011至5×1012个表达外源抗原的EHC范围内。具体的实例包括约1×103、2×103、3×103、4×103、5×103、6×103、7×103、8×103、9×103、1×104、2×104、3×104、4×104、5×104、6×104、7×104、8×104、9×104、1×105、2×105、3×105、4×105、5×105、6×105、7×105、8×105、9×105、1×106、2×106、3×106、4×106、5×106、6×106、7×106、8×106、9×106、1×107、2×107、3×107、4×107、5×107、6×107、7×107、8×107、9×107、1×108、2×108、3×108、4×108、5×108、6×108、7×108、8×108、9×108、1×109、2×109、3×109、4×109、5×109、6×109、7×109、8×109、9×109、1×1010、2×1010、3×1010、4×1010、5×1010、6×1010、7×1010、8×1010、9×1010、1×1011、2×1011、3×1011、4×1011、5×1011、6×1011、7×1011、8×1011、9×1011、1×1012,或更多个表达外源抗原的EHC。每种剂量的表达外源抗原的EHC可以间隔给予,如每日一次、每周一次、每周两次、每月一次或每月两次。每种EHC可表达一定范围的抗原分子,例如,从约100至10^7,或从约10^3至10^6。具体实例包括约1000、3000、5000、1x10^4、3x10^4、5x10^4、1x10^5、3x10^5、5x10^5、1x10^6、3x10^6、5x10^6、1x10^7、或更多个外源抗原分子/EHC。“基于EHC的比例剂量”是相对于循环实体的天然发生量,作为剂量所给予的表达外源抗原的EHC的数目。循环实体可以是细胞,例如,红细胞、网织红细胞、或淋巴细胞,或靶标,例如抗原、抗体、病毒、毒素、细胞因子等。这些单位定义为表达外源抗原的EHC/循环实体,即SCMRC/CE。该剂量单位可以包括10-7、10-6、10-5、10-4、10-3、10-2、10-1、1、10、102、103、104、105、106、107、108、109。本文所述的药物组合物包括表达外源抗原的EHC和任选药物活性剂或治疗剂。该治疗剂可以是生物试剂,小分子试剂或核酸试剂。提供了包含包括本文所述的表达外源抗原的EHC的药物组合物的剂型。在一些实施例中,该剂型被配制成用于静脉内注射的液体悬浮液。提供了医疗装置,这些医疗装置包括容纳包括本文描述的表达外源抗原的EHC的药物组合物的容器和用于将药物组合物静脉内注射到受试者的施用器。提供了医疗试剂盒,这些医疗试剂盒包括包含本文描述的表达外源抗原的EHC的药物组合物和用于将药物组合物静脉内注射到受试者的医疗装置。表达外源抗原的EHC的药学上可接受的悬浮液优选包装在约10至约250ml的体积中。包装可以是适于输注的注射器或IV袋。进行悬浮液的给予,例如,通过静脉内或动脉内注射,任选使用来自IV袋或诸如此类的滴注。给予通常经静脉内在手臂中或通过中央导管来进行。对于超过50ml的给予,优选使用滴注。疾病的治疗提供了诱导免疫耐受性的方法。这些方法包括向对诱导免疫耐受性有需要的受试者以足以在该受试者中诱导免疫耐受性的量和/或给予频率给予红细胞的药物组合物,该红细胞包括本文提供的感兴趣的外源抗原。本发明的药物组合物提供了包括可用于促进或增强免疫耐受性的感兴趣的外源抗原的红细胞。免疫耐受性可以用来治疗、预防、或减少与免疫激活有关的疾病、障碍、或病症。免疫激活疾病包括自身免疫性疾病,像例如,多发性硬化症、1型糖尿病、类风湿性关节炎、和膜性肾炎、以及表F中所列出的那些。免疫激活疾病还包括炎性疾病,像例如,克罗恩病、溃疡性结肠炎、乳糜泻、或其他自发性炎性肠病、和表G中所列出的那些。免疫激活疾病还包括过敏性疾病,像例如,哮喘、花生过敏、贝类过敏、花粉过敏、牛奶蛋白过敏、昆虫叮咬过敏、和乳胶过敏、以及表H中所列出的那些。免疫激活疾病还包括响应于治疗性蛋白的免疫激活(其减轻了该治疗性蛋白的功效),该治疗性蛋白被给予来治疗主要病症,像例如,血友病A中的凝血因子VIII、血友病B中的凝血因子IX、类风湿性关节炎及其他炎性疾病中的抗肿瘤坏死因子α(TNFa)抗体、戈谢病中的葡糖脑苷脂酶、或急性成淋巴细胞白血病(ALL)中的天冬酰胺酶,和表I、表J、表5、和表7中所列出的那些。还提供了用于治疗免疫激活疾病的方法。这些方法包括向对诱导治疗有需要的受试者以足以治疗免疫激活疾病的量给予红细胞的药物组合物,该红细胞包括本文提供的感兴趣的外源抗原。例如,患有或疑似患有免疫激活疾病例如自身免疫性疾病,炎性疾病或过敏性疾病的受试者将从所提供的治疗方法受益。在一些实施例中,患者患有自身免疫性疾病或病症或自抗体介导的疾病或病症,其中患者的免疫系统对内源性分子具有活性,例如,蛋白抗原,这样使得免疫系统攻击内源分子,诱导炎症,损伤组织,以及其他方式引起自身免疫或自抗体疾病或病症的症状。该免疫应答可以由结合内源性分子的抗体驱动,或者它可以由攻击表达内源性分子的细胞的过度活化T细胞驱动,或者它可能由其他免疫细胞(如调节T细胞、NK细胞、NKT细胞、或B细胞)驱动。在这些实施例中,抗原蛋白或其片段可以在本发明的去核造血细胞上表达。当向患有疾病或病症的患者给予一次或多次时,这些细胞群将足以诱导对抗原蛋白的耐受性,使得其不再诱导免疫系统的激活,并且因此将治疗或改善潜在的疾病或病症的症状。例如,患有获得性血栓性血小板减少性紫癜(TTP)的患者具有异常的自抗体介导的疾病,其中产生针对内源性ADAMTS13蛋白的抗体,使其在进行其血管性血友病因子切割活性方面无效,这导致在整个脉管系统中形成微血栓和随后的血小板减少。在此实施例中,该ADAMTS13抗原在去核造血细胞上表达,并且向患有TTP的患者以有效诱导对ADAMTS13的耐受性的量给予,从而减少循环中抑制性抗ADAMTS13自抗体的量,并恢复身体裂解血管性血友病因子的能力,从而减少疾病的症状。在一个优选的实施例中,只有ADAMTS13的抗原片段在去核造血细胞上表达。在一个优选的实施例中,全长ADAMTS13在去核造血细胞上表达。在一个优选的实施例中,该全长ADAMTS13在去核造血细胞上表达,并且具有酶促活性,使得给予的细胞产物能够诱导耐受性并且还治疗性地裂解血管性血友病因子。在另一个实例中,患有非典型溶血性贫血综合征(aHUS)的患者对内源性蛋白补体因子H(CFH)具有异常的自抗体应答,阻止CFH执行其补体调节功能。其结果是,补体过度激活发生在导致血管内溶血的脉管系统中。在此实施例中,该CFH抗原在去核造血细胞上表达,并且向患有aHUS的患者以有效诱导对CFH的耐受性的量给予,从而减少循环中抑制性抗CFH自抗体的量,并恢复身体抑制补体的能力,从而减少疾病的症状。在一个优选的实施例中,只有CFH的抗原片段在去核造血细胞上表达。在一个优选的实施例中,全长CFH在去核造血细胞上表达。在一个优选的实施例中,该全长CFH在去核造血细胞上表达,并且具有治疗活性,使得给予的细胞产物能够诱导耐受性并且还治疗性地促进补体调节。在另一个实例中,患有多发性硬化症(MS)的患者对覆盖神经元的多肽髓磷脂具有自身免疫应答。其结果是,T细胞攻击髓磷脂,并且所产生的炎症引起神经纤维的脱髓鞘,并且削弱了沿着神经发送电信号的能力,导致多发性硬化症的症状。在该实施例中,髓磷脂抗原在去核造血细胞上表达,并且以有效诱导对髓磷脂抗原的耐受性的量给予患有MS的患者,从而减少抗髓磷脂免疫应答并且恢复身体向有髓神经纤维发送电脉冲的能力,从而减少疾病的症状。在一个优选的实施例中,只有髓磷脂的一个或多个抗原片段在去核造血细胞上表达。在一个优选的实施例中,全长髓磷脂蛋白在去核造血细胞上表达。在另一个实施例中,患有1型糖尿病(T1D)的患者对胰腺的β胰岛细胞具有自身免疫应答。其结果是,T细胞杀死β胰岛细胞,减少或消除胰腺产生和分泌胰岛素的能力,这导致T1D的症状和病理。在该实施例中,β细胞抗原在去核造血细胞上表达,并且以有效诱导对β细胞抗原的耐受性的量给予患有T1D的患者,从而减少抗β细胞免疫应答并且恢复胰腺产生和分泌胰岛素的能力,从而减少疾病的症状。在一个优选的实施例中,只有β细胞抗原的一个或多个抗原片段在去核造血细胞上表达。在一个优选的实施例中,全长β细胞抗原蛋白在去核造血细胞上表达。还提供了响应于治疗性蛋白治疗方案减少或减轻免疫激活的方法。这些方法包括向需要响应于治疗性蛋白治疗方案减少或减轻免疫激活的受试者给予包括本文提供的感兴趣的外源抗原的红细胞的药物组合物,其量响应于治疗性蛋白治疗方案足以减少或减轻响应中的免疫激活。在一些实施例中,患者患有疾病或病症,为此可以给予治疗性蛋白以治疗或改善该疾病或病症的症状,但是治疗性蛋白是免疫原性的,这样使得患者引发针对治疗性蛋白的免疫应答,使得其在治疗或改善原始疾病方面不再有效。例如,该免疫原性治疗蛋白可以衍生自非人来源,例如,牛、猪或非人灵长类动物,或衍生自非哺乳动物来源,例如,细菌、真菌或植物衍生的,或者该免疫原性治疗性蛋白可以衍生自人类来源,但是重复暴露和给予可能足以诱导免疫原性。该免疫应答可能由抗体驱动,这些抗体结合至免疫原性治疗蛋白并且抑制其功能(中和抗体)或者这些抗体结合至免疫原性治疗蛋白并且触发其通过其他免疫细胞的清除(调理抗体)。在这些实施例中,免疫原性治疗蛋白或其抗原片段可以在本发明的去核造血细胞上表达。当向患有疾病的患者给予一次或多次时,这些细胞群将足以诱导对免疫原性治疗蛋白的耐受性,使得其不再被免疫系统中和或调理。在一个优选的实施例中,在本发明的去核造血细胞表面上表达的免疫原性治疗蛋白在循环中的细胞上具有治疗活性,使得当向患者给予时,细胞和蛋白的组合物能够诱导耐受性并且治疗或改善潜在疾病或病症的症状。在另一个优选的实施例中,在本发明的去核造血细胞表面上表达的免疫原性治疗蛋白的抗原片段在循环中的细胞上具有治疗活性,使得当向患者给予时,细胞和蛋白的组合物能够诱导耐受性,但给予免疫原性治疗蛋白的单独配制品以治疗或改善潜在疾病或病症的症状。例如,患有血友病A的患者需要输注凝血因子VIII(FVIII)以恢复适当的凝血。然而,许多患者形成针对FVIII的中和抗体,尽管它是人源的,这使得治疗无效并且导致危及生命的出血风险。在一个实施例中,将表达外源性FVIII的去核造血细胞给予患有血友病A的患者,使得(1)循环活性FVIII的水平恢复至改善症状和预防严重不受控制的出血所必需的水平,并且使得(2)对FVIII诱导耐受性。在另一个实例中,患有类风湿性关节炎的患者需要注射抗TNFa抗体以减少与该疾病相关的炎症。然而,患者可能形成针对抗TNFa抗体的中和抗体,这使得治疗性抗体无效。在这种情况下,患者通常遭受症状恶化,并且不得不增加抗TNFα抗体的剂量,或者切换到具有不同氨基酸编码序列的不同抗TNFα抗体。在此实施例中,将表达抗TNFα的抗原性片段的去核造血细胞给予患有类风湿性关节炎的患者,该患者已经形成了针对抗TNFα抗体的中和抗体。该组合物以足以诱导对抗TNFα抗体的耐受性的剂量给予,允许有效给予抗TNFα以降低循环TNFα水平,并因此减少患者中类风湿性关节炎的症状。在另一个实例中,用聚乙二醇化苯丙氨酸氨裂解酶(PAL)(非人类酶)治疗患有苯丙酮尿症(PKU)的患者。患者形成针对PAL的调理和中和抗体,其在给予治疗性蛋白时也引起过敏反应。这种免疫应答不仅使PAL无效,而且还威胁患者的健康。在一个实施例中,将表达外源PAL的去核造血细胞以足以诱导对PAL耐受性的量给予患有PKU的患者。在一个优选的实施例中,细胞表达的PAL在细胞上是具有活性的,并且该组合物能够降低苯丙氨酸的循环水平并且治疗或改善苯丙酮尿症的症状以及防止对PAL的危险的免疫反应。在另一个优选的实施例中,PAL的抗原性片段在去核造血细胞上表达,并且该细胞表达的片段不具有治疗活性,因此向患者给予单独的PAL配制品以治疗或改善苯丙酮尿症的症状。可以在给予PAL的治疗配制品之前给予诱导耐受性细胞组合物,或者可以在给予PAL的治疗配制品的同时给予诱导耐受性的细胞组合物。在一些实施例中,患者患有过敏性疾病,例如,对动物皮屑、黑胡桃、巴西坚果、腰果、栗子、尘螨、蛋、英国核桃、鱼、榛子、昆虫毒液、乳胶、牛奶、霉菌、花生、花粉、草、贝类、大豆、树坚果、或小麦过敏。患有过敏的患者可以在与过敏原的抗原片段接触时产生免疫应答,例如通过饮食、皮肤接触、注射或环境暴露。免疫应答可涉及IgE抗体,敏化的肥大细胞,脱颗粒,组胺释放和过敏反应,以及典型免疫细胞如T细胞、B细胞、树突细胞、T调节细胞、NK细胞、嗜中性粒细胞、和NKT细胞。过敏反应可能引起不适或者它可能严重到导致死亡,并且因此需要患者以及他或她的家人和看护人的持续警惕。在这些实施例中,抗原蛋白或其片段可以在本发明的去核造血细胞上表达。当向患有过敏性疾病或病症的患者给予一次或多次时,这些细胞群将足以诱导对抗原蛋白的耐受性,使得当暴露时其不再诱导免疫系统的激活,并且因此将治疗或改善潜在的过敏性疾病或病症的症状。在一个实例中,患有花生过敏的患者在接触花生抗原AraH1后具有免疫应答。在该实施例中,AraH1在去核造血细胞上表达,并且以有效诱导对AraH1的耐受性的量给予患有花生过敏的患者,从而减少抗过敏疫应答并且恢复个人安全地消耗花生的能力,从而减少疾病的症状。在一个优选的实施例中,只有AraH1的一个或多个抗原片段在去核造血细胞上表达。在一个优选的实施例中,全长AraH1蛋白在去核造血细胞上表达。本发明的某些方面涉及EHC,这些EHC包括被人白细胞抗原(HLA)错配介导的疾病中免疫细胞识别的抗原,这些人白细胞抗原(HLA)错配介导的疾病如,例如,移植物抗宿主疾病或器官移植排斥。在一些实施例中,患者患有人白细胞抗原(HLA)不匹配的疾病或病症,其中免疫细胞被组织上的HLA抗原被激活并攻击组织。这通常发生在来自不完全匹配并导致移植排斥的医学状况的供体的器官或组织的同种异体移植后,其中患者的免疫系统攻击外源组织或器官并导致移植的器官或组织死亡。另一种常见的HLA不匹配病症是移植物抗宿主疾病(GVHD),其中患者已经从不是完美匹配的供体接受了同种异体骨髓移植,并且其中移植的免疫细胞(移植物)变得激活并且攻击受体器官(宿主),其被认为是外源的,造成宿主组织和器官的损害,并导致包括死亡的严重后果。HLA-错配免疫激活通常由T细胞介导,但是还可以涉及T调节细胞、NK细胞、NKT细胞、B细胞、抗体、树突状细胞、单核细胞、巨噬细胞和嗜中性粒细胞。在这些实施例中,抗原HLA分子或其片段可以在本发明的去核造血细胞上表达。当向患有HLA错配疾病或病症的患者给予一次或多次时,这些细胞群将足以诱导对抗原HLA错配的耐受性,使得其不再诱导免疫系统的激活,并且因此将治疗或改善潜在的HLA错配疾病或病症的症状,例如,移植器官的存活或患者的存活。在一个优选的实施例中,在细胞表面上表达的HLA分子还包含荷载到HLA分子中的肽。包括用于本文所述方法的外源抗原的红细胞可以是自体衍生的,即来自同一受试者,或可以是同种异体衍生的,即来自不同的细胞供体。该药物组合物可以例如通过静脉内输注或肌内注射给予受试者。其他实施例应当理解,本发明并非限于具体的方法、试剂、化合物组合物或生物系统,当然它们可以发生变化。还应该理解的是,本文使用的术语仅是为了对具体的方面进行说明的目的,并不旨在进行限制。在本说明书中讨论的所有特征能以任何组合来组合。在本说明书中讨论的每个特征可以由用于相同,等同或类似目的替代特征替换。因此,除非另有说明,所讨论的每个特征仅仅是等效或类似特征的通用系列的示例。在一些实施例中,包括CR1外源抗原的表达外源抗原的EHC不在小鼠中产生和/或不由小鼠类红细胞产生。在一些实施例中,包括CR1外源抗原的表达外源抗原的EHC不在实验动物中产生和/或不由衍生自实验动物的类红细胞产生。在一些实施例中,表达外源抗原的EHC由巨核细胞和血小板产生。在一些实施例中,表达外源抗原的EHC是从类红细胞产生的,例如,红细胞或网织红细胞。在一些实施例中,表达外源抗原的EHC不由嗜中性粒细胞,嗜酸性粒细胞或嗜碱性粒细胞产生。在一些实施例中,表达外源抗原的EHC不由单核细胞或巨噬细胞产生。在一些实施例中,表达外源抗原的EHC不由CD34+Thy-1+造血干细胞或富集于CD34+Lin-或CD34+Thy-1+Lin-细胞中的细胞群产生。在一些实施例中,表达外源抗原的EHC不包括含HIV共同受体的细胞外结构域的外源抗原。在一些实施例中,表达外源抗原的EHC不包括能够结合病毒的外源抗原。在一些实施例中,表达外源抗原的EHC不包括含CD4的外源抗原。在一些实施例中,表达外源抗原的EHC不包括含HIV共同受体的外源抗原。在一些实施例中,表达外源抗原的EHC不包括外源抗原,该外源抗原包括CXCR4、CCR5、CCR1、CCR2、CCR3、CCR4、CCR8、CXCR1、CXCR2、CXCR3、CXCR6、GPR15、APJ、CMKLR1、或CX3CR1或其组合。在一些实施例中,表达外源抗原的EHC不包含编码腺苷脱氨酶抗原的外源核酸。在一些实施例中,表达外源抗原的EHC不包括含腺苷脱氨酶(ADA)的外源抗原。在一些实施例中,表达外源抗原的EHC不包括编码癌基因的外源核酸。在一些实施例中,表达外源抗原的EHC不包括含癌基因的外源抗原。在一些实施例中,表达外源抗原的EHC不包含编码cdx1、cdx2、或cdx4的外源核酸。在一些实施例中,表达外源抗原的EHC不包括含cdx1、cdx2、或cdx4、或其任何组合的外源抗原。在一些实施例中,表达外源抗原的EHC不包括外源抗原,该外源抗原包括含配体结合域的嵌合多肽。在一些实施例中,表达外源抗原的EHC不包括外源抗原,该外源抗原包括能够结合配体的S结构域。在一些实施例中,表达外源抗原的EHC不包括外源抗原,该外源抗原包括CD3ζ、CD3η、IL-2受体、IL-3受体、IL-4受体、IL-7受体、IL-11受体、IL-13受体、GM-CSF受体、LIF受体、CNTF受体、抑癌蛋白M受体、TGF-β受体、EGF受体、ATR2/neu、HER2/neu、HER3/c-erbB-3、Xmrk、胰岛素受体、IGF-1受体、IRR、PDGF受体、CSF-1受体、c-kit、STK-1/flk-2、FGF受体、flg、bek、NGF受体、Ror1和Ror2或其组合。在一些实施例中,表达外源抗原的EHC不包括外源抗原,该外源抗原包括人乳头瘤病毒的E6或E7基因。在一些实施例中,表达外源抗原的EHC不包括外源抗原,该外源抗原包括肿瘤抗原。在一些实施例中,表达外源抗原的EHC不包括外源抗原,该外源抗原包括葡糖脑苷脂酶。在一些实施例中,表达外源抗原的EHC不包括外源抗原,该外源抗原包括天冬酰胺酶。在一些实施例中,表达外源抗原的EHC不包括外源抗原,该外源抗原包括精氨酸脱亚氨基酶。在一些实施例中,表达外源抗原的EHC不包括融合分子,该融合分子能够促进将表达外源抗原的EHC融合至靶细胞,该靶细胞i)不同于该外源抗原和/或ii)独立于该外源抗原起作用,其中该外源抗原能够与靶标相互作用。在一些实施例中,表达外源抗原的EHC不包括外源抗原,该外源抗原包括合胞素-1。在一些实施例中,表达外源抗原的EHC不包括光敏合成化合物,例如,可以被光子或可淬灭化合物激活的化合物。在一些实施例中,表达外源抗原的EHC不包括能够产生可检测应答的可激活的分子检测剂。在一些实施例中,表达外源抗原的EHC不包括诊断化合物。在一些实施例中,表达外源抗原的EHC不包括病毒或细菌。在一些实施例中,表达外源抗原的EHC不由自体CD34+细胞产生或不包括自体CD34+细胞。在一些实施例中,使用从本文所述的类红细胞产生的表达外源抗原的EHC的治疗和预防方法不包括,例如,通过与红细胞相关的检测剂检测体内类红细胞的步骤。在一些实施例中,表达外源抗原的EHC不由人类供体多能造血干细胞产生。在一些实施例中,表达外源抗原的EHC群在生物反应器中不扩增。在一些实施例中,表达外源抗原的EHC群在扩增和/或分化后不包括单一种类的分化人血细胞。在一些实施例中,表达外源抗原的EHC是不分化的、成熟的人血细胞。在一些实施例中,表达外源抗原的EHC不由衍生自万能供血者的血细胞产生,例如,血型O,Rh因子阴性。在一些实施例中,不使用表达外源ADA多肽抗原的EHC来治疗严重的联合免疫缺陷(ADA-SCID)。在一些实施例中,表达外源抗原的EHC的扩增和分化的方法不包括在包括骨髓增生受体(mpl)配体的介质中培养表达外源抗原的EHC。在一些实施例中,表达外源抗原的EHC不包括有效负载,该有效负载包括合成的三磷酸化核苷类似物。在一些实施例中,表达外源抗原的EHC不包括有效负载,该有效负载包括2’,3’-双脱氧胞苷-5’-三磷酸酯(ddCTP)和/或3’-叠氮-3’-脱氧胸苷-5’-三磷酸酯(AZT-TP)。在一些实施例中,表达外源抗原的EHC不包括有效负载,该有效负载包括双膦酸酯。在一些实施例中,表达外源抗原的EHC是通过将类红细胞与外源抗原和任选地有效负载接触产生,无需裂解和再密封细胞以掺入外源抗原和/或有效负载。在一些实施例中,表达外源抗原的EHC是通过将类红细胞与外源抗原和任选地有效负载接触产生,其中接触不包括低渗透析。在一些实施例中,表达外源抗原的EHC是通过将类红细胞与外源抗原和任选地有效负载接触产生,其中接触不包括将外源抗原和/或有效负载加载到类红细胞中或其上。在一些实施例中,在不是待接触的类红细胞的实体中产生外源抗原和/或从不包括待接触的类红细胞的样品中分离外源抗原。例如,针对多肽外源抗原,合适的实体包括细胞系、体外表达系统、细菌表达系统等。在一些实施例中,由EHC表达的外源抗原多肽存在于EHC的表面上,但不与EHC的表面非共价结合。在一些实施例中,抗原与EHC表面的非共价附接不由抗体,抗体片段,抗体样多肽或非抗体多肽结合支架介导。在一些实施例中,外源抗原与EHC表面的非共价附接不针对类红细胞抗原,如带3(CD233)、水通道蛋白-1、Glut-1、Kidd抗原、RhAg/Rh50(CD241)、Rh(CD240)、Rh30CE(CD240CE)、Rh30D(CD240D)、Kx、血型糖蛋白B(CD235b)、血型糖蛋白C(CD235c)、血型糖蛋白D(CD235d)、凯尔(CD238)、Duffy/DARCi(CD234)、CRl(CD35)、DAF(CD55)、红细胞糖苷酯、CD44、ICAM-4(CD242)、Lu/B-CAM(CD239)、XGl/XG2(CD99)、EMMPRIN/neur.helin(CD147)、JMH、glycosyltransferase、Cartwright、Dombrock、C4A/CAB、Scianna、MER2、s番茄素、BA-l(CD24)、GPIV(CD36)、CD108、CD139、dH抗原(CD173)、或另一种红细胞结合部分。在一些实施例中,该外源抗原多肽不是由EHC分离产生的并且然后共轭至EHC。在一些实施例中,该外源抗原多肽不是酶促共轭的,例如,通过自催化异肽键形成反应,例如,通过转肽酶、分选酶、和/或异肽酶进行。在一些实施例中,该外源抗原多肽不使用分选酶进行酶促共轭。在一些实施例中,该外源抗原多肽没有,例如,通过交联剂如碳化二亚胺(包括分选酶1-乙基-3-(3-二甲基氨丙基)碳化二亚胺(EDC))进行化学共轭。在一个实施例中,该外源抗原不是由EHC分离产生的并且然后由EHC封装。在一个实施例中,该外源抗原的封装不是在外源抗原存在下由EHC的低渗透析介导的。本发明所属领域的技术人员可从前文描述和相关附图中获益进而很容易想出这些发明的许多改进形式和其他实施例。因此,应当理解本发明不限于公开的具体实施例,改进和其他实施例意欲包括于附加的权利要求范围内。尽管其中使用特殊术语,它们仅以通用和描述性意义而非限制目的使用。如在本说明书和所附权利要求书中所使用的,单数形式“一个/一种(a/an)”、以及“该(the)”包括复数指代,除非上下文另外清楚地指示。替代方案(例如,“或”)的使用应当被理解为意指替代方案中的一个、两个或其任何组合。当指可测量的值例如量、持续时间等时,本文使用的术语“约”意在包括与规定值的±20%或±10%,更优选±5%的变化,甚至更优选±1%,并且还更优选±0.1%的偏差,因为这样的变化适于执行所披露的方法。如本文所用,任何浓度范围,百分比范围,比率范围或整数范围应理解为包括所述范围内的任何整数的值,并且在适当时,包括其分数(例如整数的十分之一和百分之一),除非另有说明。本文所用的“包括(Comprise、comprising、comprises)”和“包括(comprisedof)”与“包括(include、including、includes)”或“包含(contain、containing、contains)”是同义,并且是包容性或开放性术语,其指定以下内容的存在,例如组分,并且不排除或不阻止本领域中已知或本文披露的另外的未列举的组分、特征、元素、成员、步骤的存在。在此使用的,术语“如”、“例如”等旨在表示示例性实施例,并不是限制本披露的范围。除非另外指明,本文所用的所有科学技术术语具有如本发明所属领域的普通技术人员通常理解的相同意义。虽然任何类似或等同于本文所描述的方法和材料的方法与材料均可用于本发明的测试的实践中,本文描述了优选的材料和方法。出于所有目的,所有在本说明书中引用的公开文件以及专利申请都通过引用以其全文结合在此,如同出于所有目的,各个单独的公开文件或专利申请被确切地并且单独地指明为通过引用而结合。仅提供在此讨论的这些公开在本申请的提交日期之前的披露内容。本文任何内容均不得解释为承认本文描述的发明人由于在先发明或由于其他任何原因而无权优先于这样的披露。定义“给予(Administration、administering)”及其变体意指将组合物,如表达外源抗原的EHC或试剂引入受试者,并且包括组合物或试剂的同时和顺序引入。将组合物或试剂引入受试者是通过任何合适的途径,包括经口服、经肺、经鼻内、经肠胃外(经静脉内、经肌内、经腹膜内、经皮下)、经直肠、经淋巴内合肌、或经局部。给予包括自我给予以及由另一个人进行给予。合适的给予途径允许组合物或试剂发挥其预期功能。例如,如果合适的途径是静脉内的,则通过将组合物或试剂引入受试者的静脉中来给予组合物。给予可以通过任何合适的途径进行。“锚”或“锚结构域”或“A结构域”用于指外源抗原多肽中的部分,包括与EHC的细胞膜接触的融合或嵌合外源抗原多肽。外源抗原多肽可以通过以下各项与脂质细胞膜层相互作用:磷脂尾插入、共价结合至脂质层成分、离子键、氢键、或跨越一个或多个脂质细胞膜层的单通道或多通道跨膜多肽结构域。在此使用的,术语“抗体”涵盖无论是天然还是部分或全部合成产生的免疫球蛋白,及其片段。该术语还涵盖具有与免疫球蛋白结合结构域同源的结合结构域的任何蛋白。这些蛋白可以衍生自天然来源,或部分或全部经合成产生。“抗体”还包括多肽,该多肽包括来自特异性结合和识别抗原的免疫球蛋白基因或其片段的框架区。术语抗体的使用意在包括完整抗体,多克隆,单克隆和重组抗体,其片段,并且还包括单链抗体,人源化抗体;鼠抗体;嵌合的,小鼠-人,小鼠-灵长类动物,灵长类动物-人单克隆抗体,抗独特型抗体,抗体片段,例如scFv,(scFv)2,Fab,Fab'和F(ab')2,F(ab1)2,Fv,dAb和Fd片段,双抗体和抗体相关多肽。抗体包括双特异性抗体和多特异性抗体,只要它们显示所需的生物活性或功能。本文使用的术语“抗原结合片段”是指完整免疫球蛋白的片段,以及多肽的任何部分,该多肽包括具有特异性结合抗原的能力的抗原结合区。例如,抗原结合片段可以是F(ab')2片段,Fab'片段,Fab片段,Fv片段或scFv片段,但不限于此。Fab片段具有一个抗原结合位点,并且包含轻链和重链的可变区,轻链的恒定区和重链的第一恒定区CH1。Fab’片段与Fab片段的不同之处在于Fab’片段另外包括重链的铰链区,包括在重链CH1区的C末端处至少一个半胱氨酸残基。产生F(ab’)2片段,由此Fab’片段的半胱氨酸残基通过铰链区的二硫键连接。Fv片段是仅具有重链可变区和轻链可变区的最小抗体片段,并且用于产生Fv片段的重组技术是本领域熟知的。双链Fv片段可以具有一种结构,其中重链可变区通过非共价键与轻链可变区连接。单链Fv(scFv)片段通常可以具有与双链Fv片段中一样的二聚体结构,其中重链可变区通过肽接头与轻链可变区共价结合,或者重链和轻链可变区在其C末端彼此直接连接。可以使用蛋白酶获得抗原结合片段(例如,用木瓜蛋白酶消化完整抗体以获得Fab片段,并用胃蛋白酶消化以获得F(ab’)2片段),并且可以通过基因重组技术进行制备。dAb片段由VH结构域组成。单链抗体分子可包括具有许多个体分子的聚合物,例如二聚体、三聚体或其他聚合物。“施用器”是指用于连接到受试者的任何装置。这包括,例如,针、插管、导管和输液管。当用于描述多个化合物或分子之间的关系时,“与...相关”涵盖例如外源抗原和靶标之间或表达外源抗原的EHC和靶标之间的任何相互作用。这包括酶相互作用,离子结合,共价结合,非共价结合,氢键,伦敦力,范德华力,疏水相互作用,亲脂相互作用,磁相互作用,静电相互作用等。当用于描述靶标、实体、化合物、试剂或分子与疾病、障碍、病症、症状或表型之间的关系时,“与...相关”是可以合理地在它们之间形成的任何关系,其包括因果关系、或统计显着的关系、经验建立的关系、暗示的关系,无论是否是疾病,障碍,病症,症状或表型的病因。“自身免疫性疾病”通常是受试者的免疫系统攻击身体自身细胞,引起组织破坏的病症。自身免疫障碍可以使用血液测试、脑脊液分析,肌电图(测量肌肉功能)和脑的磁共振成像来诊断,但是对于自抗体(或自体抗体)在血液中的抗体测试是特别有用的。通常,IgG类抗体与自身免疫性疾病相关。“结合”描述化合物或分子之间的相互作用,例如在外源抗原和靶标之间或在表达外源抗原的EHC和靶标之间,其通过共价结合或非共价结合发生,包括离子结合、静电相互作用、氢键、伦敦力、范德华力、疏水相互作用、亲脂相互作用等。“多肽的生物活性”是指由多肽(如外源抗原多肽)引起的任何分子活性或表型(例如,结合,信号转导,催化等)。在此使用的,术语“生物样品”是指从受试者分离的任何类型的生物来源的材料,包括例如DNA,RNA,脂质,碳水化合物和蛋白质。术语“生物样品”包括从受试者分离的组织,细胞和生物体液。生物样品包括例如但不限于:全血、血浆、血清、精液、唾液、眼泪、尿、粪便材料、汗液、颊、皮肤、脑脊液、骨髓、胆汁、毛发、肌肉活检、器官组织或本领域普通技术人员已知的生物来源的其他材料。生物样品可以从,例如,内脏活检或从癌症中获得。生物样品可以从用于诊断或研究的受试者获得,或者可以从健康受试者获得,作为对照或用于基础研究。本文使用的“清除率”通过测量以下各项的量或浓度来计算,例如随着时间保留在受试者的循环系统中的外源抗原、靶外源抗原、或表达外源抗原的EHC。例如,在第一样品中所检测的1%、2%、3%、4%、5%、10%、15%、20%、30%、40%、50%、60%、70%、80%、90%、95%、或99%靶标仍可以在第二样品中检测到,该第二样品在1小时、5小时、10小时、24小时、2天、3天、4天、5天、6天、7天、2周、3周、4周、2个月、3个月、4个月、5个月、6个月、7个月、8个月、9个月、10个月、11个月、12个月、2年、3年、4年或5年后取得。清除率可以替代地表示为:每单位时间(例如,每天)的实体数目(例如,靶标/外源抗原)。清除率的增加是大于未处理或健康的合适对照受试者中所显示速率的速率。清除率的降低是小于未处理或健康的合适对照受试者中所显示速率的速率。增加或减少可以是1%、2%、3%、4%、5%、10%、15%、20%、30%、40%、50%、60%、70%、80%、100%、150%、200%、500%、1000%或可以是1.1倍、1.2倍、1.3倍、1.4倍、1.5倍、2倍、3倍、4倍、5倍、10倍、20倍、50倍、100倍、500倍或1000倍。在此使用的,“切割”是破坏靶标(如多肽或核酸)中存在的键合相互作用以产生两个或更多个实体的过程,所述实体可在切割后彼此分离。分离可以涉及例如破坏离子键,共价键,极性共价键,非极性共价键或金属键。当切割应用于多肽靶标时,切割可以涉及破坏一个或多个肽键。当切割应用于小分子靶标时,切割可以涉及破坏一个或多个碳或硫键。当切割应用于核苷酸序列时,切割可以涉及破坏一个或多个磷酸二酯键。当切割应用于微生物例如细菌、真菌或病毒时,切割可以涉及膜或衣壳结构的裂解。切割可以通过酶(例如催化活性外源抗原多肽)进行。外源抗原可以包括,例如,外切核酸酶、内切核酸酶或蛋白酶活性。在此使用的“受试者的循环系统”涵盖全血和任选的人类中的淋巴系统占据的空间,包括血浆和所有循环的细胞和分子,并且分布在所有组织的动脉,静脉,毛细血管和淋巴管中。“循环浓度”是在定义为循环系统的空间中靶标,例如,细胞、多肽(如抗体、致病抗原等)、治疗剂、小分子、代谢物或其他实体、外源抗原或表达外源抗原的EHC的浓度。在某些实施例中,浓度可以定义为给定体积中的游离(未结合)实体的数量。在其他实施例中,可以将浓度定义为给定体积中的实体的总数。本文使用的术语“互补决定区(CDR)”是指在免疫球蛋白的重链或轻链的可变区中发现的氨基酸序列。CDR决定了抗体的特异性,并且可以提供用于结合抗原的特异性表位的接触残基。重链和轻链可以分别包括三个CDR(CDRH1、CDRH2、和CDRH3,以及CDRL1、CDRL2、和CDRL3)。具有比CDR更高度保守的氨基酸序列的四个框架区,分离VH或VL中的CDR区。在此使用的“复合物”包括两个或更多个实体的结合。复合物可以包括一种或多种多肽,核酸,脂质,碳水化合物,无机化合物,有机化合物等。复合物可以是功能性(多单元多肽)或非功能性(例如,聚集体或沉淀物),并且可以具有有益或有害性质(例如,免疫复合物)。复合物可以是天然发生的或可以是人造的或合成的。合成的复合物包括更高有序的实体,例如亚细胞结构和细胞(如果它们包括合成的化合物或分子)。对于氨基酸序列,熟练的技术人员应当认识到,对一种核酸、肽、多肽或蛋白质序列进行的单个取代、删除或添加(这在经编码的序列中改变、增加或删去一个单一氨基酸或小百分数的氨基酸)是一种“保守修饰的变体”,其中该改变导致一种氨基酸被一种化学上相似的氨基酸取代。在此使用的术语“保守性氨基酸取代”通过在以下组中的每一个内的氨基酸中的取代来说明:(1)甘氨酸、丙氨酸、缬氨酸、亮氨酸和异亮氨酸,(2)苯丙氨酸酪氨酸和色氨酸,(3)丝氨酸和苏氨酸,(4)天冬氨酸和谷氨酸,(5)谷氨酰胺和天冬酰胺,和(6)赖氨酸、精氨酸和组氨酸。在治疗的疾病,障碍或病症的症状的上下文中,“减少”是指与作为症状表现的疾病或病症相关的可测量或可传递的参数的减少。可测量的参数的实例是受试者体温的降低,取自受试者的样品中靶标浓度的降低,炎症强度或发炎区域的大小的减小,浸润细胞数目的减少,与疾病、障碍或病症相关的发作次数的减少,器官大小增加/减少,体重增加/减少等。可传递的参数的实例是,例如,受试者自身对健康和生活质量的评估。例如,对于自抗体介导的疾病,减少可以定量为以下参数之一或其组合:减轻的炎症、减少的突发、降低的疲劳、减少的血液凝固、减少的肿胀、增加的能量或增加的毛发生长等等。可以定量的参数是适于评估正在治疗的特定疾病,障碍或病症的那些参数。在治疗的疾病,障碍或病症的症状的上下文中,延迟是指使用治疗显着延长否则会加重的可控的健康状况。“降解”定义为靶直接或间接减少、失活、分解、解构、裂解、溶解、破坏、减弱、损伤、削弱、变质,缩小或分裂的过程。“不同的多肽来源”是指生物体或物种,编码该多肽的遗传序列,该多肽或其部分源自该生物体或物种。在某些实施例中,包括不同多肽来源的多肽的融合体可以包括由人腺苷脱氨酶的遗传序列和来自紫色色杆菌的苯丙氨酸羟化酶的遗传序列编码的外源抗原多肽。“结构域”是多肽的一部分,例如外源抗原多肽,其通常具有3维结构,并且可以表现出不同的活性,功能,例如催化、酶促、结构作用,或结合功能。“富集的细胞群”是指基本上由特定感兴趣的细胞构成的细胞群。在富集群中,群体中50%或更多的细胞是感兴趣的细胞,例如,在细胞中的50%、60%、70%,通常80%、85%、90%,更通常92%、95%、96%、97%、98%或99%,有时多达100%细胞。从复杂混合物或细胞的异源培养物中分离感兴趣的细胞可以通过本领域已知的任何方便的手段进行,例如通过亲和分离技术,如使用包被有亲和试剂的磁珠的磁分离,亲和层析,或用附接到固体基质的亲和试剂的“淘选”,例如,平板,或其他方便的技术。提供精确分离的其他技术包括荧光激活细胞分选器,其可以具有不同程度的复杂性,例如多色通道,低角度和钝角光散射检测通道,阻抗通道等。通过使用与死细胞相关的染料,可以针对死细胞来选择细胞。可以采用对所希望细胞的生存力不过度有害的任何技术。“去核”是通过失活或去除核来使细胞呈现非复制状态。“表位”包括抗体或其他配体或结合分子结合的抗原上的任何区段。表位可以由例如氛基酸或糖侧链分子的化学活性的表面集团组成,并且通常具有特异性的三维结构特征以及特异性的电荷特征。在一些实施例中,外源抗原包括特异性表位。在一些实施例中,靶标包括特异性表位。在此使用的“类红细胞”包括有核血红细胞,血红细胞前体和去核血红细胞以及表A1中列出的那些。例如,这些类红细胞是脐带血干细胞、CD34+细胞、造血干细胞(HSC)、脾集落形成(CFU-S)细胞、髓共同祖细胞(CMP)、胚细胞集落形成细胞、爆发集落形成单位红细胞(BFU-E)、巨核细胞红祖细胞(MEP)、红细胞菌落形成单位(CFU-E))、网织红细胞、红细胞、诱导性多能干细胞(iPSC)、间充质干细胞(MSC)、多染性成红细胞、晚正成红细胞(orthochromaticnormoblast)、或其组合。在一些实施例中,这些类红细胞是永生的或永生化的细胞。例如,永生化的成红细胞可以通过CD34+造血祖细胞的逆转录病毒转导产生,以表达Oct4、Sox2、Klf4、cMyc,并且抑制TP53(例如,如黄(Huang)等人,分子治疗(MolTher)2013,电子版先于印刷版9月3日中所述)。此外,这些细胞可以旨在用于自体使用或提供同种异体输注的来源。类红细胞可以与外源抗原接触以产生表达外源抗原的EHC。包含外源抗原的类红细胞是表达外源抗原的EHC的一个实例。在一些实施例中,培养类红细胞。在一些实施例中,红系祖细胞与外源抗原接触以产生表达外源抗原的EHC。在此使用的,术语“凝血细胞”是指干细胞巨核细胞-血小板谱系的细胞,其包括例如,巨核细胞和血小板,或通过血小板生成素诱导分化的细胞,或表达与该谱系相关的表面标志物的细胞,例如,CD41(GPIIb/IIIa)、CD42a(GPIX)、CD42b(GPIb)、和CD61(avb3,玻连蛋白受体)、PAC-1(激活的IIb/IIIa)、CD62P(P-选择素)、CD31(PECAM)和CD63。在此使用的,术语“去核造血细胞”(EHC)是指造血细胞,人类的或非人类的,其是或已经如本文所定义的被去核的。该定义包括如本文定义的“类红细胞”和“凝血细胞”。在此使用的,术语“赋形剂”是指添加到药物组合物中以进一步促进化合物的给予的惰性物质。赋形剂的实例包括但不限于:碳酸钙、磷酸钙、各种糖类和淀粉、纤维素衍生物、明胶、植物油、抗凝剂和聚乙二醇。本文所用的术语“外源的”意指由在细胞没有内天然发现的碳水化合物,多糖,脂质,寡核苷酸或多肽产生的细胞组分或功能,或者对于细胞内源的细胞组分或功能的增强或操作,包括,例如,包括外源多肽抗原和内源蛋白或其功能片段的融合蛋白。包括外源抗原多肽的外源抗原是“外源的”或“异源的”,因此它可以不天然发生,例如包括不同多肽或物种来源的结构域的融合体或嵌合体,它可以不天然发生于天然发生的细胞(如未修饰的红细胞或血小板)中,它可能不以与天然发生的多肽相同的方式起作用,或者它可能不以外源抗原多肽发生的量天然发生,例如,在实施例中,其中与未修饰细胞中天然发生的多肽的表达相比,外源抗原是过表达的。在一些实施例中,多肽外源抗原是从外源核酸表达。在一些实施例中,将外源抗原从来源中分离并且荷载到或共轭至表达外源抗原的EHC。当在核酸的上下文中使用时,术语“外源的”包括转基因和重组核酸。在此使用的,术语“表达(expression或expressing)”是指产生多肽(例如外源抗原多肽)的过程,包括转录和翻译。表达可以,例如,通过许多方法增加,这些方法包括:增加编码多肽的基因的数目,增加基因的转录(例如通过将基因置于组成型启动子的控制下),增加基因的翻译,敲除竞争性基因或这些的组合和/或其他方法。术语“表达(expression或expressing)”还包括包含外源多肽的EHC,该外源多肽同时由EHC主动表达,但是已停止的活性表达(定义为转录和翻译)。例如,外源抗原多肽在去核事件之前由EHC主动表达(即,转录和翻译),并且抗原多肽在去核后被EHC保留,但不再活性表达,例如,因为缺乏编码核酸。例如,该EHC可以包括由外源核酸编码的外源抗原多肽。在去核期间,该外源抗原多肽被EHC保留,而外源核酸不被保留,这种EHC被称为“表达抗原(antigen-expressing或expressingantigen)”,即使在抗原多肽的活性表达(转录和翻译)被有效地终止和/或EHC不包含大量的复制核酸的情况下。“功能性”外源抗原或表达外源抗原的EHC表现出期望的或特定的活性或特征,包括酶促、催化或代谢活性,结构完整性,免疫原性互补性,靶结合和正确定位或能够促进期望的或特定的效应或表型。“融合体或嵌合体”是多肽序列或相应的编码核苷酸序列,其衍生自在自然界中没有一起发现的两个或更多个序列的组合。这可以是衍生自相同基因组内的单独基因或衍生自明显不同物种基因组的异源基因的单独序列的组合。“遗传物质”是指具有能够编码基因的腺嘌呤,胸腺嘧啶,尿嘧啶,胞嘧啶和鸟嘌呤的核苷酸序列的核酸分子。本文使用的术语“重链”应理解为包括全长重链,该全长重链包括具有确定抗原特异性的氨基酸序列的可变区(VH),和具有三个恒定结构域(CH1、CH2和CH3)的恒定区,及其片段。此外,本文使用的术语“轻链”应理解为包括全长轻链,该全长轻链包括具有确定抗原特异性的氨基酸序列的可变区(VL),和的恒定区(CL),及其片段。术语“同源物”表示多肽,包括在其一级,二级或三级结构中的相应位置处具有相同或保守残基的外源抗原多肽。功能同源物包括表现出相似功能和/或特异性(例如,针对特定靶标)的外源抗原和其他多肽。天然发生的完整抗体或免疫球蛋白包括四种多肽:两条全长轻链和两条全长重链,其中每条轻链通过二硫键与重链连接。每条重链具有恒定区和可变区。类似地,每条轻链具有恒定区和可变区。有五种重链类(同种型):伽马(γ)、缪(μ)、阿尔法(α)、德尔塔(δ)、或艾普西隆(ε),以及另外地若干亚类伽马1(γ1)、伽马2(γ2)、伽马3(γ3)、伽马4(γ4)、阿尔法1(α1)、和阿尔法2(α2)。该轻链恒定区可以是卡帕(κ)或拉姆达(λ)类型。可变区在抗体之间在序列上不同,并且用于给定抗体对其特定抗原的结合和特异性。如本文所用,术语“增加”,“增强”,“刺激”和/或“诱导”(和类似术语)通常是指相对于自然、预期或平均值或相对于对照条件,直接或间接改进或增加浓度、水平、功能、活性或行为。如本文所用,术语“抑制”,“压制”,“降低”,“干扰”和/或“减少”(和类似术语)通常是指相对于自然、预期或平均值或相对于对照条件,直接或间接减少浓度、水平、功能、活性或行为。如本文所用的“文库”包括具有不同核酸序列的核酸分子(例如,DNA,RNA)的集合,克隆的遗传多样性集合,不同多肽的集合,细胞(如EHC)的多样集合,等等。如本文所用的,“哺乳动物受试者”包括所有哺乳动物,包括但不限于:人、家养动物(例如狗,猫等),农场动物(例如牛,绵羊,猪,马等)和实验室动物(例如猴,大鼠,小鼠,兔,豚鼠等)。术语“个体”、“受试者”、“宿主”、和“患者”在本文中可互换使用,并且是指希望诊断、治疗治疗的任何哺乳动物受试者,特别是人。本文所述的方法适用于人类疗法和兽医应用。在一些实施例中,该受试者是哺乳动物,并且在其他实施例中,该受试者是人。“医疗装置”是指用于递送一定剂量的表达外源抗原的EHC和/或治疗剂的任何装置,设备或机器。这包括容器、瓶子、小瓶、注射器、袋子、药筒、盒子、药盒(magazines)、圆筒或小罐。“医疗试剂盒”是指包括医疗装置或施用器、适当剂量的表达外源抗原的EHC(该表达外源抗原的EHC任选地包括治疗剂)、以及相关标记和说明书的包装单元。如本文所用,术语“调节(modulate、modulating)”、“修饰”和/或“调节剂”通常指通过增加或减少来改变的能力,例如直接或间接促进/刺激/上调或干扰/抑制/下调特定浓度、水平、表达、功能或行为,例如作为拮抗剂或激动剂。在一些情况下,调节剂可以相对于对照,或相对于通常预期的活性的平均水平或相对于活性的对照水平增加和/或降低某一浓度、水平、活性或功能。本文使用的“膜”是将内部空间与外部空间分隔开的边界层,其包括一种或多种生物化合物,通常是脂质,和任选的多肽。膜可以是脂双层。在某些实施例中,膜包括磷脂酰胆碱,鞘磷脂,溶血磷脂酰胆碱,磷脂酰乙醇胺,磷脂酰丝氨酸,磷脂酰肌醇或磷脂酸中的一种或多种。在一些实施例中,膜包括一种或多种多肽,例如锚蛋白和辅酶Q10。包括在膜的定义中的是细胞膜,这些细胞膜包括,例如,磷脂双层和细胞膜相关多肽。短语“核酸分子”是指脱氧核糖核苷酸或核糖核苷酸碱基的单链或双链聚合物。它包括染色体DNA和自我复制质粒、载体、mRNA、tRNA、siRNA等,其可以是重组的,并且当将核酸引入细胞中时可以从其中表达外源多肽。直系同源物定义为通过物种形成从共同祖先基因演化的不同物种中的基因。术语“药学上可接受的”及其语法变体是指能够给予受试者或其上的组合物、载体、稀释剂和试剂,而不产生达到将阻止给予组合物的程度的不希望的生理作用。例如,“药学上可接受的赋形剂”包括可用于制备通常安全,无毒和所需的药物组合物的赋形剂,并且包括兽医用途以及人类药物用途可接受的赋形剂。这样的赋形剂可以是固体、液体、半固体或气体(在气雾组合物的情况下)。本文使用的,术语“药学上可接受的载体”包括任一种标准的可药用载体,例如磷酸缓冲盐溶液、水、乳剂,如油/水或水/油,以及多种类型的润湿剂。该术语还包括由美国联邦政府的管理机构批准的或在美国药典中列出的用于动物(包括人)的任何试剂,以及不会对受试者造成显着刺激并且不消除所给予的化合物的生物活性和性质的任何载体或稀释剂。一些试剂可以作为“药学上可接受的盐”给予,例如,由无机和有机酸制备的药学上可接受的盐。从无机酸衍生的盐包括盐酸、氢溴酸、硫酸,硝酸、磷酸等。从有机酸衍生的盐包括乙酸、丙酸、乙醇酸、丙酮酸、草酸、苹果酸、丙二酸、琥珀酸、马来酸、富马酸、酒石酸、柠檬酸、苯甲酸、肉桂酸、扁桃酸、甲磺酸、乙磺酸、对甲苯磺酸、水杨酸等。盐也可以由无机和有机碱制备。从无机碱衍生的盐仅通过举例的方式包括钠、钾、锂、铵、钙和镁盐。从有机碱衍生的盐包括但不限于:伯胺,仲胺和叔胺的盐。本领域任何普通技术人员将知道如何选择适当的药学上可接受的载体,其药学上可接受的盐用于实施本发明而无需过多的实验。如本文所用,术语“药物组合物”是指一种或多种本文所述的化合物,例如表达外源抗原的EHC,其混合或掺和或悬浮于一种或多种其他化学组分中,例如生理学可接受的载体和赋形剂。药物组合物的一个目的是促进化合物向受试者的给予。某些实施例提供具有与所需功能或活性相关的序列的多种多肽分子,例如外源抗原多肽。多肽是指氨基酸残基的链,其不考虑翻译后修饰(例如,磷酸化或糖基化)和/或与另外的多肽复合,与核酸和/或碳水化合物、或其他分子合成为多亚基复合物。因此,蛋白多糖在本文中也称为多肽。在某些实施例中,表达外源抗原的EHC包括多肽外源抗原。在某些实施例中,表达外源抗原的EHC包括任选膜相关的一种或多种非外源抗原多肽。术语“药物活性剂”或“药物试剂”定义为当给予受试者时对受试者具有可测量的或可传递的作用的任何化合物,例如小分子药物或生物制剂(例如,多肽药物或核酸药物),例如,它减轻或减少疾病,障碍或病症的症状。在一些实施例中,可以在递送表达外源抗原的EHC之前,与其组合,或之后给予药物试剂。在一些实施例中,药物活性剂与表达外源抗原的EHC发挥协同治疗效果。在一些实施例中,药物活性剂与表达外源抗原的EHC发挥附加治疗效果。“启动子”定义为可引导可操作地连接的核酸转录的一系列核酸控制序列。启动子包括靠近转录起始位点的必需的核酸序列。启动子也任选地包括远端增强子和阻遏物元件。“组成型”启动子是一种在大多数环境和发育条件下有活性的启动子。“诱导型”启动子是指在环境调节或发育调节的情况下活化的启动子。术语“操控连接的”启动子是指核酸表达调控序列(如启动子、转录因子结合位点)和第二核酸序列之间具有功能上的相关性,其中表达调控序列指导第二序列相应的核酸序列的转录。本文所用的“外源抗原”是能够与靶标相互作用,例如与靶标缔合或结合至靶标的实体。外源抗原可以包括多肽,或者可以基本上由多肽组成。在一些实施例中,该外源抗原包括多肽、碳水化合物、核酸、脂质、小分子、或其组合。在外源抗原是天然发生的化合物或分子的实施例中,抗原是“外源的”,在意义上,就其在EHC中的存在而言,它是外源或异源化合物或分子。在其他实施例中,抗原是“外源的”,在意义上,它是人造化合物或分子,如融合体或嵌合体、非天然发生的多肽、碳水化合物、核酸、脂质或其组合,或人造小分子或其他治疗剂。例如,外源抗原可以包括包含S结构域、A结构域和U结构域中的一种或多种的融合体或嵌合体。该S结构域是暴露于EHC周围环境的表面结构域,例如受试者的循环系统。该A结构域是将S结构域附接到EHC的细胞膜的锚定结构域。该U结构域面向EHC的未暴露侧或位于EHC内(即,在其胞内空间中)。不论任何结构域,外源抗原可以位于表达外源抗原的EHC的表面上,或者可以位于EHC内。外源抗原可以与表达外源抗原的EHC的膜缔合,例如,外源抗原锚定在膜中,共轭或以其他方式结合到膜上。在一些实施例中,外源抗原可以通过化学或酶促共轭与表达外源抗原的EHC的膜共轭。在其他实施例中,该外源抗原不与膜共轭。在一些实施例中,该外源抗原不与表达外源抗原的EHC的膜缔合,并且位于EHC的膜包封的胞内空间内。在一些实施例中,位于EHC的胞内空间内的外源抗原基本上不扩散出EHC和/或可以不渗透膜。在其他实施例中,外源抗原可以基本上扩散出EHC和/或可以渗透膜。在一些实施例中,将外源抗原加载,例如引入或放入EHC中。加载的外源抗原不是通过表达外源抗原的EHC生物合成的。适合荷载的外源抗原可以例如在基于细胞的表达系统中产生,从生物样品中分离,化学或酶促合成,并且然后荷载到EHC中或其上。在一些实施例中,该外源抗原可以在荷载后通过外表达源抗原的EHC进一步修饰。在其他实施例中,该外源抗原在荷载后不被修饰。在一些实施例中,该外源抗原多肽不加载到EHC上或其中。在一些实施例中,制备外源抗原,例如通过表达外源抗原的EHC进行生物合成。通常,外源抗原多肽由来自引入EHC的外源核酸分子(例如,DNA或mRNA)的表达外源抗原的EHC表达。当抗原在EHC上表达时,该外源抗原可具有保留的生物学功能。该外源抗原可以结合和/或螯合靶标。可替代地或此外,该外源抗原可以对靶标表现出催化活性,例如,外源抗原可以转化或修饰该靶标,或者可以降解该靶标。然后可以任选地从外源抗原中释放产物。表达外源抗原的EHC的“驻留期(Residency)”是指其在生理位置中其花费的时间段。表达外源抗原的EHC的特定位置可以在其寿命中改变,并且“驻留期”适用于在各种环境中所花费的时间段,这些环境包括血管循环、外周组织、毛细血管、消化系统、肺系统、鼻组织、表皮表面和间质组织。在具体实施例中,表达外源抗原的EHC驻留于受试者的循环系统中。“复制核酸”是指脱氧核糖核酸(DNA),其能够被专用于增加DNA拷贝数量的酶复制。通常,DNA复制导致从一个原始DNA分子产生两个相同的复制品。DNA复制包括通过与模板链匹配的DNA聚合酶将核苷酸并入生长的DNA链中,经一次一个产生磷酸二酯键。“螯合”被定义为靶标的隔绝、封闭、分离、分开、隐藏、绝缘或隔离,并防止其与其环境自由相互作用。如本文所用,“特异性结合”或“特异性相互作用”描述了两个实体(例如,具有外源抗原的靶标,如具有抗原的抗体,具有配体的受体,具有底物的酶,具有抗生物素蛋白的生物素等)之间的任何相互作用,其是可饱和的,常常是可逆的并且因此具有竞争性,因为这些术语是化学和生物化学领域的普通技术人员所理解的。例如,当结合对的一个成员具有带电荷、极性或疏水部分的形状和分布的位点时,发生涉及生物分子(例如,蛋白质、肽和核酸)的特异性结合,使得同源配体与该位点的相互作用特征在于有利的能量学(即,结合的负自由能)。相互作用的特异性可以测量或表达为结合常数(Kd)。Kd可以在从mM范围至fM范围的范围内,包括pM范围,μM范围和nM范围。典型的Kd值低于约10-6M,低于约10-7M,低于约10-8M,并且在一些实施例中低于约10-9M。如本文所用,术语“基本上地”或“基本上的”是指,例如,特定空间中实体的存在、水平或浓度,一个实体对另一个实体的作用,或治疗效果。例如,如果相对于基线,增加是2倍、3倍、4倍、5倍、10倍、50倍、100倍或1000倍,那么实体的活性、水平或浓度显着增加。如果相对于基线,增加是5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、100%、200%、或500%,那么实体的活性、水平或浓度也显着增加。如果实体可以通过本领域已知的方法来检测,那么实体可以基本上存在于特定空间中。如果实体以低于本领域已知的测定和方法的检测限的水平存在,那么实体可以基本上不存在于特定空间中。在一些实施例中,如果实体几乎不可检测,但仅以非功能量或不引起或改变表型的微量,那么该实体可以基本上不存在于特定空间中。在其他实施例中,如果实体存在并且只能在构成群体的少数成分中检测到,例如小于10%、9%、8%、7%、6%、5%、4%、3%2%或小于1%、0.5%、0.1%的群体组分,那么该实体可以基本上不存在于特定空间中。例如,当去核时外源核酸可能不被保留,细胞呈现为不可复制,并且去核细胞不能继续表达由外源核酸编码的外源抗原多肽。细胞继续显着翻译外源多肽的能力的丧失“有效终止”蛋白质表达。在某些实施例中,表达外源抗原的EHC基本上不能自我复制,例如核酸的复制。例如,如果在掺入测定中,与标记的核苷(如胸苷)接触,表达外源抗原的EHC基本上不掺入核苷。在一些实施例中,该表达外源抗原的EHC不包含大量的自我复制核酸。术语多核苷酸或核酸序列的“基本一致性”意指包括具有至少25%序列一致性的序列的多核苷酸。可替代地,百分比一致性可以是从25%至100%的任何整数。更优选的实施例包括与使用本文描述的过程的参照序列相比,至少25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、或99%;优选地使用标准参数的BLAST。“合成的”是指一种化合物或分子,其是人造和非天然发生的,或者如果它是天然发生的,则放置在其不会自然存在的环境或位置中,或者如果它在环境或位置中天然存在,则处于纯净状态,或以它在环境或位置中不会自然存在的量、浓度或数量存在。合成实体可以是任选地从其天然状态,外源核酸,外源(异源)外源抗原等经化学或酶促修饰的分离或纯化的化合物。如本文所定义的合成化合物或分子在任何实体中的存在使得整个实体是“合成的”。例如,包括外源抗原的细胞是合成的细胞。本文所用的“靶表”是能够与外源抗原相互作用,例如与外源抗原缔合或结合外源抗原的实体。“靶标”包括但不限于:多肽(例如,抗体或抗体相关多肽、补体成分、淀粉样蛋白、病原体、毒素、朊病毒),分子(例如,代谢物、类固醇、激素、碳水化合物;寡糖;化学品;多糖、DNA;RNA;脂质、氨基酸、元素、毒素或病原体),复合物(例如,免疫复合物)或细胞(例如,癌细胞、巨噬细胞、细菌、真菌、病毒或寄生虫)。靶标旨在通过本文提供的方法进行检测、诊断、损伤,破坏或改变(例如,功能补充)。特定目标可以是游离的或者是与受试者循环系统中其他实体缔合的。本文使用的“靶标自抗体”是与自身免疫性疾病相关的自抗体。可以使用抗体结合测试来检测和分析此类自抗体,这些测试涉及将受试者的抗体接触受试者自身组织,通常为甲状腺、胃、肝和肾组织的样品。与“自身”组织(包括自身抗原)结合的抗体指示出自身免疫障碍。“转基因”或“外源核酸”是指引入EHC的外源或天然核苷酸序列。转基因和外源核酸在本文中可互换使用,并且涵盖重组核酸。如本文所用,“治疗(treat、treating、和/或treatment)”是用于获得有益的或所期望的临床结果,药理学和/或生理学效应的方法,例如减轻症状,预防或消除所述症状,并且是指特定疾病,障碍或病症的治疗性治疗和预防疾病的或预防性治疗。有益的或所期望的临床结果,药理学和/或生理学效应包括但不限于:预防疾病、障碍或病症避免在可能易患疾病、障碍或病症但尚未经历或表现出疾病(预防疾病治疗)症状的受试者中发生,减轻疾病、障碍或病症的症状,缩小疾病、障碍或病症的程度,疾病、障碍或病症的稳定(即不恶化),防止疾病、障碍或病症的传播,延迟或减缓疾病、障碍或病症进展,改善或缓解疾病、障碍或病症,及其组合,以及与如果不接受治疗的预期存活相比延长存活。“治疗剂”或“治疗性分子”包括当以有效量存在时对有需要的受试者产生所希望的治疗效果、药理学和/或生理学作用的化合物或分子。术语“治疗有效量”或“有效量”是向受试者所给予的试剂足以实现有益或所希望的临床结果,药理学和/或生理效应的量。有效量能够以一次或多次给予来给予。有效量通常足以减轻,改善,稳定,逆转,减慢或延迟疾病状态的进展。因此,有效量是指试剂的量或特定量的试剂的给予频率,其足以合理地实现所希望的治疗和/或预防疾病效果。例如,它可以包括导致与正在治疗的疾病或病症相关的症状的预防、治疗或减少,例如,与自身免疫应答相关的疾病或医学病症,过度活性免疫激活,或需要免疫耐受性的抑制性抗体产生,或与靶标多肽相关的疾病或医学病症。给予受试者的治疗组合物的量将取决于疾病的类型和严重程度以及取决于个体的特征,例如一般健康、病理状况、饮食、年龄、性别、体重和对药物的耐受性。它还将取决于疾病的程度、严重程度和类型。此外,有效量将取决于所使用的配制和给予方法,例如给予时间、给予途径、排泄速度和反应敏感性。本领域技术人员将能够根据这些和其他因素确定合适的剂量。组合物还可以与一种或多种另外的治疗化合物组合给予。对于成人来说,药物组合物的所希望的剂量可以在约0.001至100mg/kg的范围内。在一个实施例中,以最低限度有效的剂量开始静脉内给予,并且剂量在预选的时间过程中增加直到观察到积极效果。随后,剂量的增量增加被限制到产生相应效果增加的水平,同时考虑可能出现的任何不利影响。合适剂量的非限制性实例可以在,例如,从1×1010至1×1014、从1×1011至1×1013、或从5×1011至5×1012个本发明的表达外源抗原的EHC的范围内。具体实例包括约5×1010、6×1010、7×1010、8×1010、9×1010、1×1011、2×1011、3×1011、4×1011、5×1011、6×1011、7×1011、8×1011、9×1011、1×1012、或更多个本发明的表达外源抗原的EHC。每种剂量的表达外源抗原的EHC可以间隔给予,如每日一次、每周一次、每周两次、每月一次或每月两次。“未结合”是指外源抗原能够与之相互作用的靶标的状态。未结合的靶标不与另一个实体或外源抗原缔合。未结合的外源抗原不与另一个实体或靶标缔合。一旦靶标与外源抗原或另一个实体缔合,则认为其是“结合的”。未结合的靶标包括循环中靶标的可溶形式。结合的靶标包括嵌入的循环或外周组织中的实体的,与循环或外周组织中的实体缔合、连接或以其他方式相互作用的靶标。靶标可与之相互作用的实体包括循环细胞、外周内皮组织、免疫复合物、糖脂、微生物、免疫球蛋白、血清白蛋白、凝血因子、脂蛋白和电解质。“变体”是通过一个或多个氨基酸取代、缺失、插入或其他修饰而与原始蛋白质不同的多肽。这些修饰不显着改变原始蛋白质的生物活性。在许多情况下,变体保留原始蛋白质的生物活性的至少10%、20%、30%、40%、50%、60%、70%、80%、90%、95%、或100%。变体的生物学活性也可以高于原始蛋白质的生物学活性。变体可以是天然发生的,如通过等位基因变异和多态性,或有意地工程化。变体的氨基酸序列与原始蛋白质的氨基酸序列基本相同。在许多实施例中,变体与原始蛋白质具有至少50%、60%、70%、80%、85%、90%、95%、99%或更多的全局序列一致性或相似性。序列一致性或相似性可以使用本领域已知的各种方法来确定,如基本局部比对工具(BLAST),点阵分析或动态规划方法。在一个实例中,通过使用遗传学计算机组(GCG)程序GAP(尼德曼-翁施算法)确定序列一致性或相似性。变体和原始蛋白质的氨基酸序列可以在一个或多个区域中基本相同,但在其他区域中是不同的。如本文所用,术语“载体”是核酸分子,优选自主复制核酸分子,其将插入的核酸分子(如转基因或外源核酸)转移和/或复制到宿主细胞中和/或其之间。它包括基因组中插入有重组DNA片段并用于将重组DNA或转基因导入EHC中的质粒或病毒染色体。实例以说明的方式而不是以限制的方式提供以下实例。实例1:培养具有异源基因表达的类红细胞通过使用Mini-MACS柱(美天旎公司(MiltenyiBiotec);94%+/-3%纯度)的超磁性微珠选择从外周血中分离CD34细胞。基于补充有稳定的谷氨酰胺、330ug/mL全人转铁蛋白、10ug/mL重组人胰岛素、2IU/mL肝素和5%溶剂/洗涤剂病毒灭活血浆的IMDM,在红细胞分化介质(EDM)中培养细胞。扩展过程包括3个步骤。在第一步(第0天至第7天)中,在1uM氢化可的松、100ng/mLSCF、5ng/mLIL-3和3IU/mLEpo的存在下,在EDM中培养10^4/mLCD34细胞。在第4天,将1体积的细胞培养物稀释于4体积的包含CSF、IL-3、Epo和氢化可的松的新鲜介质中。在第二步(第7天至第11天)中,将细胞以10^5/mL重新悬浮于补充有SCF和Epo的EDM中。在第三步(第11天至第18天)中,在仅补充有Epo的EDM中培养细胞。在第11天和第15天分别将细胞计数调节至7.5x10^5到1x10^6和5-10x10^6细胞/mL。超过第18天,包含Epo的培养基每周更新两次。在37℃下在5%CO2的空气中保持培养物。将感兴趣的抗原的编码序列置于红细胞特异性启动子的控制下,例如,GATA-1,并用聚腺苷酸尾终止,参见,例如,瑞皮克(Repik)等人,临床和实验免疫学(ClinExpImmunol)2005,140:230。该序列在慢病毒载体中编码(例如,EF1,系统生物科学有限公司(SystemBiosciences,Inc.))。载体通过标准方法从293T细胞产生。在培养的第1-4天期间,将慢病毒载体转导入人造血祖细胞,例如,CD34+细胞或永生化的成红细胞或iPS细胞,例如如常(Chang)等人,自然生物技术(NatBiotechnol)2006,24:1017所述。如上所述进行后续的扩增和分化阶段。实例2:通过低渗/高渗循环将蛋白质荷载到类红细胞中将抗原(在这种情况下为OVA)以0.5或5mg/ml的终浓度添加到RBC悬浮液中,并且血细胞比容(Hct)为70%。在低渗透析过程(50mOsmol/kg)后,通过在37℃下添加高渗溶液(1900mOsmol/kg)30分钟来重新密封RBC。用0.9%NaCl+0.2%葡萄糖溶液洗涤荷载OVA的RBC四次,在4℃下以1000×g离心10分钟,并用血浆或缓冲液调节至Hct为50%。实例3:用异双功能交联剂进行细胞表面标记通过用5mMTCEP孵育来制备具有暴露的还原性硫醇基基团的抗原(抗原-SH)。在共轭之前通过尺寸排阻色谱法除去还原剂。将细胞用马来酰亚胺-PEG-NHS交联剂(例如,SM(PEG)x(赛墨科技公司(ThermoScientific)))孵育30分钟。NHS基团与细胞表面抗原上的游离胺基反应。反应的细胞在PBS中洗涤以除去过量的交联剂。将抗原-SH溶液引入细胞加交联剂溶液中,并允许马来酰亚胺-SH反应进行30分钟。将细胞沉淀并洗涤以除去未结合的抗原。实例4:用分选酶进行细胞表面标记将包含分选酶A受体序列(LPXTG)的表达表面蛋白质的细胞与100uM分选酶A在由50mMTris-Cl(pH7.5)、150mMNaCl、1mMCaCl2组成的反应缓冲液中在37℃下孵育4小时。引入10x摩尔过量的亲核试剂,GGG-抗原,并且允许反应在室温下进行过夜。反应后,通过沉淀和洗涤细胞除去过量的分选酶和未反应的亲核试剂。实例5:用点击化学进行细胞表面标记根据制造商的说明书(例如,格伦研究公司(GlenResearch)),将细胞与炔-NHS酯反应以将炔基团与细胞表面上暴露的伯胺共轭。通过沉淀和洗涤细胞除去过量的炔-NHS酯。根据制造商的说明书(例如,赛墨科技公司(ThermoScientific)),将抗原与叠氮化物-NHS酯反应,以将叠氮基团与抗原上暴露的伯胺共轭。通过尺寸排阻色谱法除去过量的叠氮化物-NHS酯。炔细胞和叠氮化物抗原在室温下通过铜催化的环加成进行反应。实例6:小鼠中T细胞增殖的测量根据制造商的说明书,使用CD8磁珠阴性选择试剂盒(美天旎生物技术公司)分离来自OTI(CD45.2+)小鼠脾脏的CD8+T细胞。将新鲜分离的CD8+OTI细胞重新悬浮于PBS中,并在室温下用1μMCFSE(英杰公司)标记6分钟,并且用等体积的具有10%(体积/体积)的FBS(Gibco公司)的伊斯科夫改良的达尔伯克培养基(Iscove’smodifiedDulbecco’smedium)(IMDM)淬灭反应1min。将细胞洗涤,计数,并且在注射前重悬于纯IMDM中。注射总计3×106个CFSE标记的CD8+OTI细胞,静脉内注射进入接受者CD45.1+小鼠的尾静脉中。对于短期增殖研究,在过继转移后24小时注射包括OVA的类红细胞。在抗原给予后5天收获脾细胞,并通过流式细胞术染色以进行分析。实例7:小鼠中的OVA抗原激发模型如上所述,将总计3×105个CFSE标记的OTICD8+T细胞注射到CD45.1+受体小鼠中。在过继转移后1和6天,小鼠静脉给予100μL包括OVA的类红细胞到尾静脉中。在过继转移后15天,用皮内注射到每个后腿垫的25μL盐水中的5μg的OVA和25ng的超纯大肠杆菌LPS(英杰公司)激发小鼠(Hock法:总剂量为10μg的OVA和50ng的LPS)。抗原特异性B和T细胞的定量和血清抗体滴度描述如下。实施例8:小鼠中抗原特异性B和T细胞的定量如上所述,在OVA激发后4天杀死小鼠,并且分离脾脏和引流淋巴结细胞用于再刺激。对于细胞内细胞因子的流式细胞术分析,在1mg/mLOVA或1μg/mLSIINFEKL肽(Genscript)的存在下再刺激细胞3h。添加布雷菲德菌素-A(5μg/mL;西格玛),并在染色和流式细胞术分析前重新开始再刺激3h。对于分泌因子的ELISA测量,在100μg/mLOVA或1μg/mLSIINFEKL肽的存在下再刺激细胞4天。旋转细胞,并且根据制造商的说明书,收集介质用于使用IFN-γ和IL-10Ready-Set-Go试剂盒(eBioscience公司)的ELISA分析。实例9:循环抗体滴度的定量通过在OVA包被的板上以不同稀释度孵育小鼠血清,随后与山羊抗小鼠IgG-HRP进行最终孵育来检测OVA特异性血清IgG(南方生物技术(SouthernBiotech))。实例10:细胞外SpyTag-SpyCatcher耐受性诱导合成包含凯尔和SpyTag的编码序列的表达盒,并将其插入慢病毒载体EF1中。用载体转化CD34+细胞群。通过FACS定量凯尔-SpyTag融合蛋白的表达。然后将细胞外表达凯尔-SpyTag的细胞置于与SpyCatcher序列和cMyc标签融合的Arah(1-6)肽的溶液中。孵育后,使用FAC分选细胞以定量凯尔-SpyTag与SpyCatcher-ArahX-cMyc的共价异肽共轭。细胞也被低渗裂解,并通过蛋白质印迹法定量凯尔-SpyTag-SpyCatcher-ArahX-cMyc的存在。实例11:基因装配编码以下基因的DNA作为cDNA购自Dharmacon(GE生命科学(GELifeSciences))或通过DNA2.0和Genscript从头合成:血型糖蛋白A(UniprotIDP02724),凯尔(UniprotIDP23276),针对乙型肝炎表面抗原的抗体scFv(博斯(Bose)等人,2003,分子免疫学(MolImmunol)40(9):617,GenBankIDAJ549501.1),腺苷脱氨酶(UniprotIDP00813),来自紫色色杆菌的苯丙氨酸羟化酶(GenBankIDAF146711.1),补体受体1(UniprotIDP17927),CD46(GenBank:BAA12224.1),CD55(UniprotIDP08174),CD59(UniprotIDP13987),绿色荧光蛋白(UniprotIDP42212),胸苷磷酸化酶(UniprotIDP19971),葡糖脑苷脂酶(UniprotIDP04062),β2糖蛋白1(UniprotIDP02749),磷脂酶α2受体(UniprotIDQ13018),胶原α-3(IV)(UniprotIDQ01955),血清淀粉样蛋白P(UniprotIDP02743),脂蛋白脂肪酶(UniprotIDP06858),天冬酰胺酶(UniprotIDP00805),因子IX(UniprotIDF2RM35),ADAMTS13(UniprotIDQ76LX8)。1.单基因克隆(CR1)通过本领域已知的标准分子生物学方法将基因组装到表达载体中。补体受体1(CR1)基因由商业供应商(DNA2.0)合成并且在标准克隆载体(pJ系列)中提供。通过聚合酶链反应(PCR)使用具有非同源末端序列的寡核苷酸从pJ载体扩增基因,以制备用于插入哺乳动物表达载体(系统生物科学公司(SystemBiosciences),pM系列):上游寡核苷酸由与上游pM插入位点同源的25nt和与CR1的起始同源的25nt组成;下游寡核苷酸由与下游pM插入位点同源的25nt和与CR1的末端同源的25nt组成。扩增产物通过凝胶电泳(凯杰公司(Qiagen))纯化。使用与上游和下游插入位点同源的尾对尾寡核苷酸通过PCR将pM载体线性化,并通过PCR纯化(凯杰公司)纯化。通过吉普森(Gibson)装配将CR1扩增子连接到线性化的pM载体中,详细描述于吉普森2011,酶学方法(MethodsEnzymology),第498卷,第394页。通过桑格测序确认序列。2.两个基因的融合(凯尔scFv膜)凯尔的基因作为cDNA购买,并在标准克隆载体(pJ系列)中提供。通过商业供应商(DNA2.0)合成乙型肝炎表面抗原特异性抗体scFv的基因(scFv,描述于博斯2003,分子免疫学(MolecularImmunology)40:617),并在标准克隆载体(pJ系列)中提供。通过聚合酶链反应(PCR)使用具有非同源末端序列的寡核苷酸从pJ载体扩增基因,以制备用于插入哺乳动物表达载体(系统生物科学(SystemBiosciences),pM系列)中。用上游寡核苷酸和下游寡核苷酸扩增凯尔,该上游寡核苷酸由与上游pM插入位点同源的25nt和与凯尔的5’末端同源的25nt组成,该下游寡核苷酸由与scFv的5’末端同源的25nt和与凯尔的3’末端同源的25nt组成。用上游寡核苷酸和下游寡核苷酸扩增scFv,该上游寡核苷酸由与凯尔插入位点的3’末端同源的25nt和与scFv的5’末端同源的25nt组成,该下游寡核苷酸由与下游pM插入位点同源的25nt和与scFv的3’末端同源的25nt组成。扩增产物通过凝胶电泳(凯杰公司)纯化。使用与上游和下游插入位点同源的尾对尾寡核苷酸通过PCR将pM载体线性化,并通过PCR纯化(凯杰公司)纯化。通过一步法吉普森装配将凯尔和scFv扩增子连接到线性化的pM载体中,该一步法吉普森装配详细描述于吉普森,2011,酶学方法(MethodsEnzymology),第498卷,第394页。通过桑格测序确认序列。3.基因之间的接头装配(凯尔-scfv)凯尔的基因作为cDNA购买,并在标准克隆载体(pJ系列)中提供。通过商业供应商(DNA2.0)合成乙型肝炎表面抗原特异性抗体scFv的基因(scFv,描述于博斯2003,分子免疫学(MolecularImmunology)40:617),并在标准克隆载体(pJ系列)中提供。通过聚合酶链反应(PCR)使用具有非同源末端序列的寡核苷酸从pJ载体扩增基因,以制备用于插入哺乳动物表达载体(系统生物科学(SystemBiosciences),pM系列)中。用上游寡核苷酸和下游寡核苷酸扩增凯尔,该上游寡核苷酸由与上游pM插入位点同源的25nt和与凯尔的5’末端同源的25nt组成,该下游寡核苷酸由与scFv的5’末端同源的25nt、编码(GlyGlyGlySer)x2间隔区的24nt和与凯尔的3’末端同源的25nt组成。用上游寡核苷酸和下游寡核苷酸扩增scFv,该上游寡核苷酸由与凯尔插入位点的3’末端同源的25nt、编码(GlyGlyGlySer)x2间隔区的24nt和与scFv的5’末端同源的25nt组成,该下游寡核苷酸由与下游pM插入位点同源的25nt和与scFv的3’末端同源的25nt组成。扩增产物通过凝胶电泳(凯杰公司)纯化。使用与上游和下游插入位点同源的尾对尾寡核苷酸通过PCR将pM载体线性化,并通过PCR纯化(凯杰公司)纯化。通过一步法吉普森装配将凯尔和scFv扩增子连接到线性化的pM载体中,该一步法吉普森装配详细描述于吉普森,2011,酶学方法(MethodsEnzymology),第498卷,第394页。通过桑格测序确认序列。4.表位标签附着(凯尔-scFv)凯尔的基因作为cDNA购买,并在标准克隆载体(pJ系列)中提供。通过商业供应商(DNA2.0)合成乙型肝炎表面抗原特异性抗体scFv的基因(scFv,描述于博斯2003,分子免疫学(MolecularImmunology)40:617),并在标准克隆载体(pJ系列)中提供。通过聚合酶链反应(PCR)使用具有非同源末端序列的寡核苷酸从pJ载体扩增基因,以制备用于插入哺乳动物表达载体(系统生物科学(SystemBiosciences),pM系列)中。用上游寡核苷酸和下游寡核苷酸扩增凯尔,该上游寡核苷酸由与上游pM插入位点同源的25nt和与凯尔的5’末端同源的25nt组成,该下游寡核苷酸由与scFv的5’末端同源的25nt、编码(GlyGlyGlySer)x2间隔区的24nt和与凯尔的3’末端同源的25nt组成。用上游寡核苷酸和下游寡核苷酸扩增scFv,该上游寡核苷酸由与凯尔插入位点的3’末端同源的25nt、编码(GlyGlyGlySer)x2间隔区的24nt和与scFv的5’末端同源的25nt组成,该下游寡核苷酸由与下游pM插入位点同源的25nt、编码HA表位标签的27nt序列tacccctatgacgtgcccgactatgcc(Seq.IDNo.8)、和与scFv的3’末端同源的25nt组成。扩增产物通过凝胶电泳(凯杰公司)纯化。使用与上游和下游插入位点同源的尾对尾寡核苷酸通过PCR将pM载体线性化。下游引物另外包含编码HA表位标签的27nt序列tacccctatgacgtgcccgactatgcc(Seq.IDNo.8)。通过PCR纯化(凯杰公司)纯化线性化载体。通过一步法吉普森装配将凯尔和scFv扩增子连接到线性化的pM载体中,该一步法吉普森装配详细描述于吉普森,2011,酶学方法(MethodsEnzymology),第498卷,第394页。通过桑格测序确认序列。5.两个基因的融合(报告基因装配)(GPA-HA)补体受体1(CR1)和绿色荧光蛋白(GFP)基因由商业供应商(DNA2.0)合成并且在标准克隆载体(pJ系列)中提供。通过聚合酶链反应(PCR)使用具有非同源末端序列的寡核苷酸从pJ载体扩增CR1基因,以制备用于插入哺乳动物表达载体(系统生物科学公司(SystemBiosciences),pM系列):上游寡核苷酸由与上游pM插入位点同源的25nt和与CR1的起始同源的25nt组成;下游寡核苷酸由与病毒衍生的T2A序列gagggcagaggaagtcttctaacatgcggtgacgtggaggsgsstcccggccct(Seq.IDNo.7)同源的54nt组成。通过聚合酶链反应(PCR)使用具有非同源末端序列的寡核苷酸从pJ载体扩增GFP基因,以制备用于插入哺乳动物表达载体(系统生物科学公司(SystemBiosciences),pM系列):上游寡核苷酸由与病毒衍生的T2A序列gagggcagaggaagtcttctaacatgcggtgacgtggaggsgsstcccggccct(Seq.IDNo.7)同源的54nt和与GFP的起始同源的25nt组成;下游寡核苷酸由与下游pM插入位点同源的25nt和与GFP的末端同源的25nt组成。扩增产物通过凝胶电泳(凯杰公司)纯化。使用与上游和下游插入位点同源的尾对尾寡核苷酸通过PCR将pM载体线性化,并通过PCR纯化(凯杰公司)纯化。通过吉普森装配将CR1和GFP扩增子连接到一起并且连接到线性化的pM载体中,详细描述于吉普森2011,酶学方法(MethodsEnzymology),第498卷,第394页。通过桑格测序确认序列。实例12:mRNA装配使用标准分子生物学方法将感兴趣的基因克隆到pSP64载体(普洛麦格公司(Promega))的多克隆位点中。用EcoRI(NEB)消化载体以产生包含SP6启动子,感兴趣的基因和30个核苷酸长的多聚腺苷酸尾的线性化dsDNA载体。根据制造商的说明书,通过与SP6RNA聚合酶(普洛麦格公司)反应合成mRNA,包括5’帽类似物(ARCA)的推荐浓度以合成加帽的mRNA转录物。然后用DNA酶处理反应混合物以消化模板载体(来自普洛麦格公司的核糖核酸探针),并使用EZNAMicroEluteRNAClean-Up试剂盒(Omega公司)纯化mRNA。实例13:细胞培养1.人血红细胞(RBC)通过使用Mini-MACS柱(美天旎公司(MiltenyiBiotec);94%+/-3%纯度)的超磁性微珠选择从外周血中分离CD34细胞。基于补充有稳定的谷氨酰胺、330μg/mL全人转铁蛋白、10μg/mL重组人胰岛素、2IU/mL肝素和5%溶剂/洗涤剂病毒灭活血浆的IMDM,在红细胞分化介质(EDM)中培养细胞。扩展过程包括3个步骤。在第一步(第0天至第7天)中,在1μM氢化可的松、100ng/mLSCF、5ng/mLIL-3和3IU/mLEPO的存在下,在EDM中培养10^4/mLCD34细胞。在第4天,将1体积的细胞培养物稀释于4体积的包含CSF、IL-3、EPO和氢化可的松的新鲜介质中。在第二步(第7天至第11天)中,将细胞以10^5/mL重新悬浮于补充有SCF和EPO的EDM中。在第三步(第11天至第18天)中,在仅补充有EPO的EDM中培养细胞。在第11天和第15天分别将细胞计数调节至7.5x10^5到1x10^6和5-10x10^6细胞/mL。超过第18天,包含EPO的培养基每周更新两次。在37℃下在5%CO2的空气中保持培养物。2.小鼠血红细胞从小鼠胎儿肝脏祖红细胞培养小鼠类红细胞的方法是本领域已知的,参见,例如,史(Shi)等人,2014,PNAS2014111(28):10131。从胎儿肝脏中分离小鼠祖红细胞。胎儿肝脏从查士睿华实验室购买。将肝脏置于冰上的1mlPBS中。上下吸取以获得单细胞悬浮液溶液并通过70um粗滤器(BD福尔肯公司,35-2235)。用1mlPBS冲洗该网。合并流通(1ml/胚胎)。在1.5kRPM下将细胞沉淀5min,用红细胞裂解缓冲液(来自干细胞的氯化铵溶液)重新悬浮,并在冰上孵育10min。在1.5kRPM下将细胞沉淀5min,除去裂解缓冲液,并用10mlPBS-2%FBS重新悬浮。以50ul/小鼠添加chromPure大鼠IgG(杰克逊免疫研究(JacksonImmunoResearch),#012-000-003),并在4℃下孵育5min。以1ul/1*10^6个细胞添加生物素化的抗小鼠TER119(BDPharmingen公司,#553672),并在4℃下孵育15min。以2ul/1*10^6个细胞添加Ms谱系组(飞世尔科技公司(FisherScientific)(赛默飞世尔科技公司(ThermoFisherScientific))#BDB559971)至细胞并在4℃下孵育15min。用10倍体积的PBS洗涤一次,并在4度下以1.5kRPM旋转细胞5min。添加链霉抗生物素蛋白颗粒Plus-DM(磁珠)(BDPharmingen公司,#557812)(5ul/1*10^6个细胞),并在4℃下孵育30min。在磁性支架上制备2-4个FACS管。将2毫升细胞等分到每个管(总计4ml),并小心地从管中取出细胞,并放入另一个管,另一方面避免磁条珠的破坏。重复同样的程序,并且取Ter119阴性和谱系阴性细胞到一个新的管。浓缩这些细胞,并用50-100ulPBS(2%FBS)重新悬浮细胞。将纯化的红细胞祖细胞在包括以下各项的分化培养基中培养(针对40mL):IMDM:29ml、FBS(干细胞):6ml(最终15%)、在IMDM中的10%BSA(干细胞):4ml(终浓度1%)、10mg/ml全转铁蛋白:2000ul(终浓度:500ug/ml)、100*L-谷氨酰胺:400ul、100*青霉素链霉素:400ul、10U/ulEpo:2ul(终浓度:0.5U/ml)、10mg/ml胰岛素:40ul(终浓度:10ug/ml)。于24孔板中在37℃下在分化培养基中培养2*10^5个细胞/ml。在44-48小时的总培养后,进行分析,例如通过如本文进行的流式细胞术。使用(Hoechst着色剂)门控出去核血红细胞,用于分化谱分析。成功的培养将产生16倍的增长。3.血小板捐赠的CD34+细胞从弗雷德哈钦森癌症研究中心(FredHutchinsonCancerResearchCenter)获得。将富含CD34+的细胞以2-4×10^4个细胞/mL置于无血清培养基中,并在第4天通过添加等体积的介质进行介质更新。在第6天,计数和分析细胞:洗涤1.5×10^5个细胞,并置于1mL的补充有由TPO30ng/mL、SCF1ng/mL、白细胞介素(IL)-67.5ng/mL和IL-913.5ng/mL组成的细胞因子混合物的相同介质中,以诱导巨核细胞分化。在第10天,用新鲜介质替换悬浮培养物的1/2-1/4。所有细胞因子购自派普泰克公司。将培养物在潮湿气氛(10%CO2)中在39℃下孵育培养最初6天,并且在37℃下孵育最后8天。用血细胞计数器计数活的有核细胞(0.4%台盼蓝;英杰公司,伯灵顿,安大略省,加拿大)。根据制造商的说明书(干细胞技术公司(StemCellTechnologies),温哥华,不列颠哥伦比亚,加拿大),使用用于骨髓CPC的MethoCultH4436和用于集落形成单位巨核细胞(CFU-Mk)的MegaCult-C测定克隆形成祖细胞(CPC)。为了评估分化,通过流式细胞术使用FACS-Calibur(BD公司)用针对CD61mCD42b、CD41、CD61和CD49b的抗体对细胞染色。针对细胞周期分析,用磷酸盐缓冲盐水(PBS)冲洗细胞,用甲醛2%(西格玛,圣路易斯,密苏里州,美国)固定5min,并用0.1%的TritonX-100(伯乐公司,赫库斯,加利福尼亚州,美国)进行透性化。遵循制造商的说明书(BD生物科技公司),然后用mAb-Ki-67-FITC(BD生物科技公司,圣何塞,加利福尼亚州,美国)标记细胞,洗涤并重新悬浮于0.5mLPBS-1%胎牛血清(FBS)-0.01%叠氮化物7-氨基-放线菌素D(7-AAD)中。实例14:细胞分离1.原代RBC使用无菌技术在包含低分子量肝素,达肝素钠(9单位/mL血液)的管中收集全血。将血液以5000xg离心5分钟,在除去血浆和血沉棕黄层(两者都可以保留用于以后使用)后,在冷(4℃)磷酸盐缓冲盐水(PBS)中伴随离心洗涤红细胞两次。将所得血红细胞群体在4℃下储存在CPDA-1抗凝血剂或甘油溶液中,用于长期保存。2.原代血小板在适当的IRB方案下,从健康个体收集全血(40ml)于3.8%的柠檬酸钠中(1:9柠檬酸盐对血液体积/体积)。将血液以200g离心15分钟以分离富含血小板的血浆(PRP)。然后在1μM前列腺素I2存在下,在改良的Tyrode’s缓冲液(包含138mMNaCl、5.5mM右旋糖、12mMNaHCO3、0.8mMCaCl2、0.4mMMgCl2、2.9mMKCl2、0.36mMNa2HPO4和20mMHepes(pH7.4))中洗涤血小板,并重新悬浮于相同的缓冲液中。实例15:原代或培养的细胞的辐射可以进行表达外源抗原的EHC的群体的辐射以确保它们不能复制。此类方案类似于本领域已知的用于辐射细胞例如原代血红细胞的方案。简而言之,取一个单位(350ml)的全血并分成两份等分试样,每份175ml,从而将10个这样的单位分成20份。通过自含γ细胞辐射器(GammaCell1000,Theratronics公司)使来自每个血液单位的一份等分试样(175ml)经受25Gy,并且不超过50Gy的γ辐射。然后将血液在4℃下在常规血液储存条件下储存。在采样点成色剂(汾沃公司(Fenwal),美国)的帮助下,在第0、7、14和21天,从这10个辐射的和10个未辐射的血袋进行取样。进行细胞增殖的测试,这些测试包括胸苷掺入测定以定量有任何丝分裂潜力。通过吸光光谱测定上清液的游离血红蛋白,通过比色测定(Pierce)测定游离乳酸脱氢酶,以评估细胞裂解的水平。实例16:类红细胞的去核在补充有100单位/ml青霉素和100单位/ml链霉素的IMDM介质中,在涂有胶原的12-mm直径盖玻片上,使红细胞生长至半细胞覆盖(1至4X10^4个细胞/cm2)。胶原蛋白是必要的,以防止所有细胞在离心过程中从盖玻片上脱落。使细胞在相同介质或具有10%小牛血清的Dulbecco改良的Eagle介质中在盖玻片上生长成单层(5X104个细胞/cm2)。不必用胶原包被细胞盖玻片。为了去除细胞核,将盖玻片倒置(细胞侧向下),并置于15mlCorex离心管的底部,该离心管包含具有10g细胞松弛素B/ml的2-5ml的介质。将具有盖玻片的离心管立即放入SorvallRc2离心机中,该SorvallRC2离心机已经通过旋转(SS34)转子使头部在适当位置以10,000rpm(温度调节器设置在37℃-39℃)旋转约1hr而被加热至37℃。离心的时间长度和速度是成功去核的关键因素。将细胞以9000rpm在37℃±20℃下旋转1hr,并将细胞在37℃±20℃下以6500rpm旋转50min。离心后,从离心机中去除盖玻片,并将细胞侧向上置于包含3ml无细胞松弛素B的介质的35-mm(福尔肯公司)组织培养皿(Biolquest公司)中。在370下30-60分钟内,细胞形态正常,并且90%-99%缺乏细胞核。通过用胰蛋白酶-EDTA(大岛生物公司(GrandIslandBiologicalCo.))处理从盖玻片除去有核细胞,并将细胞悬浮于正常介质中。然后将有核细胞以小滴复制在保持在35-mm组织培养皿中的22-mm2盖玻片上,并且置于培养箱中。在复制后的时间间隔处,将盖玻片安装在载玻片(12)上,并且用Zeiss相位差、偏振光和Nomarski光学器件对去核进行观察。实例17:细胞接触1.核酸-转染将感兴趣的核酸放大,以提供约5ug核酸/10^5个待荷载的EHC(例如细胞,例如红细胞、血小板或造血前体细胞)。将核酸在Opti-MEM介质(生命科技公司(LifeTechnologies))中以1ug至50μL介质的比例稀释。然后将稀释的核酸与转染试剂(针对DNA的Trans-IT、针对mRNA的Trans-ITmRNA、针对siRNA的Trans-ITsiRNA、MirusBio)以1:1体积比混合,并且允许在室温下形成复合物持续5分钟。将核酸复合物添加细胞中持续12-24小时。任选地,在这段时间后,可以用新鲜介质更换该介质,使得转染试剂不再存在。2.核酸-病毒转导将感兴趣的基因用来自系统生物科学公司的MSCV启动子序列克隆到慢病毒载体pCDH的多克隆位点中。慢病毒在293T细胞中通过用脂质体转染细胞产生。在转染前一天,将5x10^6个293T细胞(Lenti-X293T细胞系,Clontech目录号#632180)置于P10培养皿中。细胞融合应该在70%左右。每个构建体转染一个板。将20μl(10μg)pPACKH1(系统生物科学公司)质粒混合物+2μglenti构建体+20μlPlus试剂(生命科技公司,目录号11514-015)在400μlOptimem中组合,并在室温下孵育15min。将30μl的LF2000(生命科技公司,目录号11668-019)稀释到400μlOptimem中,逐滴添加DNA混合物中,并且室温下孵育15min。将DNA混合物添加到细胞中(细胞在9mlOptimem中)。将细胞孵育6小时,并且然后将介质更换为DMEM/10%FBS。转染后48小时通过以1,500rpm离心5分钟收集病毒上清液。收集上清液并以1ml等分试样在-80℃下冷冻。将靶细胞在本文所述的培养方法的第3-7天转导。将5x10^5个培养的细胞置于24孔板中的500μL包含20μg/mL聚凝胺的介质中。针对每种病毒,在一式三份的孔中转导细胞。将病毒上清液添加到另一500μL介质中,并且通过吸移混合样品。感染通过旋转接种,以2000rpm在室温下旋转平板90分钟来实现。在旋转接种后,将细胞在37℃下孵育过夜,并且第二天添加1mL具有合适细胞因子的新鲜IMDM介质。3.核酸-阳离子聚合物可以从多个商业供应商(例如,IDT-DNA,CoralvilleIa)购买编码感兴趣的转基因并包括上游启动子序列和下游多聚腺苷酸尾的mRNA。使用RNAIMax(英杰公司,卡尔斯巴德,加利福尼亚州)或TRANSIT-mRNA(MimsBio公司,麦迪逊,威斯康星州)阳离子脂质递送载体进行RNA转染。首先将RNA和试剂在Opti-MEM基础介质(英杰公司,卡尔斯巴德,加利福尼亚州)中稀释。将100ng/uLRNA稀释5倍,并将5μL的RNAIMax/μg的RNA稀释10倍。在将它们分配到培养基中之前,合并稀释的组分并在室温下孵育15分钟。针对TRANSIT-mRNA转染,将100ng/uLRNA在Opti-MEM中稀释10倍,并且添加BOOST试剂(以2μL/μg的RNA的浓度),添加TRANSIT-mRNA(以2μL/μg的RNA的浓度),并且然后在室温下孵育2分钟后将RNA-脂质复合物递送至培养基。RNA转染在NutristemxenofreehES介质(剑桥,麻萨诸塞州)或Opti-MEM加2%FBS中进行。可以使用各种已知的方法,如荧光标记或报道蛋白,如绿色荧光蛋白(GFP),监测mRNA转录物向宿主细胞的成功引入。修饰的mRNA的成功转染也可以通过,例如,蛋白质印迹法和免疫细胞化学测量靶标多肽的蛋白表达水平来确定。针对遵循类似的RNA-脂质复合物比率的大体积放大到多升(5-10,000L)培养形式,可以遵循类似的方法。4.核酸-电穿孔可以从多个商业供应商(例如,IDT-DNA,CoralvilleIa)购买编码感兴趣的转基因并包括上游启动子序列和下游多聚腺苷酸尾的mRNA。通过用mRNA转录物转染红细胞谱系细胞并通过定量RT-PCR使用设计用于特异性检测外源转录物的引物来测量转染效率来优化电穿孔参数。针对某些细胞制剂,将150uF电容器放入悬浮于具有2mm间隙的标准电穿孔比色皿中的50μl的Opti-MEM(英杰公司,卡尔斯巴德,加利福尼亚州)中的2.5×10^6个细胞中,足以重复递送超过10,000个拷贝的修饰的mRNA转录物/细胞,如使用标准曲线方法测定的,同时保持高活力(>70%)。细胞密度可以从1×10^6个细胞/50μl至2.5×10^6个细胞/500的密度变化,并且需要从110V至145V来转染细胞,并且在每个细胞的转录物拷贝中测量到相似的效率。可以类似于与用上述约束(李(Li)等人,2002;金恩(Geng)等人,2010)描述的方法类似的大体积流电穿孔策略进行大的多升(5-10,000L)电穿孔。5.多肽-脂质体细胞,包括原代末期分化细胞,例如红细胞,可以在其表面及其细胞质中荷载外源蛋白质。蛋白质的荷载可以使用脂质体进行。在氮气下将有机溶剂中的脂质(Pro-Ject试剂,Pierce)在玻璃闪烁管中干燥成薄膜。每10^5个细胞使用约2uL脂质。根据制造商的说明书,用Dylight-650(Pierce)标记多克隆小鼠IgG(艾博抗公司(Abcam))。将0.1mg/mL的在PBS中的蛋白质溶液添加到干燥的脂质混合物中。将溶液移液若干次,在室温下孵育5分钟,然后剧烈涡旋以产生包封的脂质体。添加无血清介质以使每10^5个细胞总体积为500uL。然后将脂质体混合物与细胞在37℃下孵育3-4小时。图1显示了用脂质体将外源蛋白质(在这种情况下是荧光标记的IgG)荷载到原代红细胞中。通过流式细胞术测量荷载。荷载是剂量依赖性的,因为0.06%的细胞在没有脂质体的情况下是具有荧光的,约60%的细胞在低脂质体剂量下是具有荧光的,并且约85%的细胞在高脂质体剂量下是具有荧光的。图1中的数据强烈证明外源蛋白质可以用脂质体荷载到类红细胞中。6.多肽-机械破碎可以使用包含1μm、2μm、3μm、4μm、5μm、10μm宽的通道的微流体装置来荷载细胞,所述通道瞬时穿过细胞,使得当细胞被压迫通过系统时允许有效负载进入。硅基装置是使用光刻和深反应离子蚀刻技术在麻省理工学院微加工设备处制造的。在该过程中,用六甲基二硅氮烷处理450-μm厚的6”硅晶片,在3,000rpm下用光致抗蚀剂(OCG934;富士胶片)旋涂60秒,通过具有收缩通道设计的铬掩模暴露于UV光(EV1;EVG),并在AZ405(AZ电子材料)溶液显影100秒。在90℃下烘焙20min后,通过深反应离子蚀刻(SPTS技术公司(SPTSTechnologies))将晶片蚀刻至所需的深度(通常为15μm)。使用包含出入孔图案的不同掩模并使用更厚的光致抗蚀剂AZ9260(AZ电子材料公司(AZElectronicMaterials)),在晶片的对侧(即,不包含蚀刻的通道的一侧)上重复该过程。然后在将晶片阳极键合到Pyrex晶片并切割成单个器件之前,使用湿氧化来生长100-200nm的氧化硅。在每次实验之前,目视检查装置并安装到具有入口和出口储存器的保持器上(全部在室内设计并且由Firstcut公司生产)。这些储存器与使用Buna-NO形环(McMaster-Carr)的装置相连接以提供适当的密封。入口储存器连接到使用特氟隆管的自制压力调节器系统以提供必要的驱动力以推动材料通过装置。首先将红细胞群体悬浮于与所需递送材料混合的所需递送缓冲液[生长介质、PBS或补充有3%FBS和1%F-68普朗尼克(西格玛)的PBS]中,并且置于装置的入口储存器中。该储存器连接到由调节器控制的压缩空气管线,并且所选择的压力(0-70psi)用于驱动流体通过该装置。然后从出口贮存器收集处理的细胞。在处理之后,将细胞在递送溶液中在室温下孵育5-20min,以在进行任何进一步处理之前确保孔封闭。为了递送荧光标记的苯丙氨酸氨羟化酶(PAH),如上所述进行实验,使得递送缓冲液包含0.1-0.3mg/mLPAH。GFP敲低测量为,相对于未处理对照,细胞群的平均荧光强度的降低百分比。7.多肽-表面共轭用Traut试剂(2-亚氨基硫烷HCl,Pierce)处理细胞表面以硫醇化伯胺。Traut试剂用EDTA溶解在Tris缓冲液(pH8)中以防止巯基的氧化。使用约1pmolTraut试剂处理10^6个细胞。在室温下将Traut试剂与细胞孵育1小时。通过离心和洗涤细胞除去过量或未反应的试剂。可用的巯基的数目可以使用Ellman试剂来测量。同时,根据制造商的说明书,用胺对巯基交联剂,如SMCC(Pierce)处理合适的外源抗原多肽。通过脱盐除去过量的交联试剂。然后将马来酰亚胺官能化的蛋白与硫醇化的细胞一起孵育若干小时。通过离心和洗涤从共轭的细胞中分离未反应的蛋白质。8.多肽-非共价表面附着在哺乳动物表达载体(Genlantis)中,将针对乙型肝炎表面抗原的抗体scFv的基因(scFv,描述于博斯2003,分子免疫学,40:617)融合至6-组氨酸亲和标签并且融合至编码结合小鼠血型糖蛋白A的多肽序列的基因,HWMVLPWLPGTLDGGSGCRG。通过使用标准方法瞬时转染HEK-293T细胞产生完全融合蛋白,并根据制造商的说明书在Ni-NTA亲和树脂(Pierce)上进行纯化。将纯化的融合蛋白与>100nM浓度的小鼠红细胞一起孵育,以允许肽与血型糖蛋白A快速平衡和结合。9.多肽-脂质插入膜根据制造商的方案,将Traut试剂(赛默飞世尔公司)用于在包含胺的合适的外源抗原多肽分子上产生巯基基团。将反应混合物在振荡器上在室温(RT)下孵育1h,并且根据制造商的说明书通过旋转脱盐柱(Zeba,MWCO7K,赛墨科技公司)洗涤以除去未反应的Traut试剂。基于制造商的方案,使用Ellman试剂(Pierce)定量修饰的多肽上巯基基团的产生。将DSPE-PEG3400-mal(在PBS中的1×10^-3M,4μL,脂质:多肽=1:1的摩尔比)(所有脂质购自AvantiPolarLipids并且在氩气下在-20℃下作为氯仿溶液储存)添加到脱盐的多肽溶液中并在室温下在振荡器上孵育。1小时后,使用离心过滤装置(Microcon,密理博公司)在4℃下以14000g将样品溶液过滤15min,以除去小分子并且悬浮于600μLPBS中(1mg/mL多肽)。将200μL全血悬浮在1000μLPBS中并且以1500g旋转30秒,重复四次。最后,将RBC悬浮于800μLPBS中。通过将上述RBC悬浮液和各种量的DSPE-PEG-多肽溶液(1mg/mL)混合,然后在37℃下孵育15-30min,制备RBC/DSPE-PEG-多肽的共轭。将混合物在室温下保持5min,然后在PBS中洗涤三次,并且重新悬浮至5×10^8/mL的最终RBC浓度。使用自动化细胞计数器(Countess,英杰公司)测量细胞浓度。10.多肽-低渗荷载合适的外源抗原多肽(在这种情况下为小鼠IgG)购自艾博抗公司,并且以0.25mg/mL添加到等渗溶液中的RBC悬浮液中,血细胞比容(Hct)为70%。将悬浮液在250mL包含10mM磷酸钠(pH7.4)、10mM碳酸氢钠和20mM葡萄糖的低渗溶液中进行透析,在4℃下以15rpm搅拌1小时。然后通过添加1/10体积的包含5mM腺嘌呤、100mM肌苷、100mM丙酮酸钠、100mM磷酸钠、100mM葡萄糖、12%(w/v)NaCl的再封闭溶液(pH7.4)来等渗重新密封细胞。然后将细胞在37℃下孵育30分钟。11.多肽-细胞穿透肽鱼精蛋白共轭的多肽的制造是本领域已知的,参见,例如,权(Kwon)等人,2009,控制释放期刊(JContrRel)139(3):182。将在50mMHEPES缓冲液(pH8)中的5mg/ml低分子量鱼精蛋白(LMWP)与异型双功能交联剂3-(2-吡啶基二硫代)丙酸N-羟基琥珀酰亚胺(SPDP,西格玛-奥德里奇公司)以1:10的摩尔比在DMSO中混合,并且在室温下振荡1h。然后将反应混合物用50mM二硫苏糖醇(DTT,西格玛-奥德里奇公司)处理,并且在肝素亲和柱上通过HPLC纯化硫醇化的LMWP。通过超滤收集产物,冻干,并在-20℃下储存直至进一步使用。为了共轭,将5mg/ml合适的外源抗原多肽与磷酸盐缓冲液中的SPDP(乙醇中的40μl的0.1MSPDP至1ml蛋白质溶液)混合,并在室温下搅拌1h。通过FPLC用0.1M磷酸盐缓冲液(pH7.4)通过快速脱盐和缓冲液交换除去未反应的SPDP。然后将活化的多肽与10倍摩尔过量的上述制备的LMWP-SH在4℃下共轭24h。通过离子交换色谱法使用肝素亲和柱,然后进行五轮离心过滤(分子量截留值:5,000Da)来分离LMWP-多肽轭合物。浓缩合并的LMWP-多肽共轭物,并且通过MALDI-TOF质谱法测定共轭度。针对吸收实验,将新鲜的绵羊红细胞(安倍医疗公司(MPBiomedicals),梭伦,俄亥俄州)以5×10^8个细胞/ml的密度悬浮于Hank平衡盐溶液(HBSS)中,并且然后与0.5mg/ml的LMWP-多肽轭合物的溶液在室温下在温和振荡下孵育30min。然后用HBSS洗涤RBC并储存在2℃-8℃下。12.多肽-化学渗透性将3×10^8个RBC与200μM的林格氏溶液中的氯丙嗪(西格玛奥德里奇)预孵育30min。然后,将合适的外源抗原多肽添加到林格氏溶液(1至4μM)至400μl的终体积,并在温和搅拌下在室温下孵育30min。孵育后,将细胞洗涤两次,重新悬浮于林格氏溶液中并且收集用于分析。13.多肽-酶促共轭与分选酶的细胞表面酶促共轭是本领域已知的,参见,例如史等人PNAS2014111(28):10131。为了用多肽标记GPAN末端,将30uL的500uM金黄色葡萄球菌分选酶和在C末端具有LPETGG的1mM多肽在50mMTris(pH7.5)、150mMNaCl中在冰上预孵育15分钟,并且添加到在DMEM中的5x10^7RBC中。在偶尔温和混合的情况下,将分选酶和细胞混合物在冰上孵育30min,然后在4℃下以500xg旋转2min,以除去缓冲液/DMEM,然后用1mL冰冷的PBS洗涤三次。实例18:多肽存在的评估1.荧光转基因如本文所述,培养类红细胞。如本文所述,通过吉普森装配构建编码在C末端具有HA标签的融合于具有介入性病毒T2A肽的GFP的血型糖蛋白A的转基因。如本文所述,通过慢病毒转导将转基因导入类红细胞。转导后两天,收集细胞,在PBS缓冲液中洗涤,并在流式细胞术(Attune,生命科技公司)上分析。将转导效率评估为群体中GFP阳性细胞的百分比。2.细胞表面蛋白针对细胞表面蛋白,可以早在转染后2天通过流式细胞术用对蛋白质或共表达表位标签具有特异性的抗体检测蛋白质表达水平。如本文所述,培养类红细胞。通过如本文所述的吉普森装配构建编码在N末端具有HA标签的血型糖蛋白A的转基因。如本文所述,通过慢病毒转导将转基因导入类红细胞。转染后两天,收集细胞,在PBS缓冲液中洗涤,并用1:50稀释的小鼠抗HA抗体(艾博抗公司)染色1hr。洗涤细胞,并且然后用1:100稀释的alexa488标记的山羊抗小鼠第二抗体(生命科技公司)在冰上染色30分钟。洗涤细胞并在流式细胞术(Attune,生命科技公司)上分析。将转导效率评估为群体中alexa488阳性细胞的百分比。3.细胞内蛋白对于细胞内蛋白质,蛋白质表达的水平可以早在转染后8-12小时通过蛋白质印迹进行检测。如本文所述,培养类红细胞。通过如本文所述的吉普森装配构建编码在C末端具有HA标签的腺苷脱氨酶的转基因。如本文所述,通过慢病毒转导将转基因导入类红细胞。转染后两天,收集细胞,在PBS缓冲液中洗涤,并且在RIPA细胞裂解缓冲液(Pierce)中裂解。通过在100mMDTT中煮沸使细胞裂解物变性,然后荷载到NuPageSDS-PAGE预制凝胶上。电泳和转移到硝酸纤维素膜后,通过用1:5000稀释的小鼠抗HA抗体(艾博抗公司)染色,随后以1:5000稀释山羊抗小鼠HRP(Pierce),并且随后用HRP底物(SuperSignal,Pierce)处理,使蛋白带进行显影。使用Amersham成像仪(通用电气医疗集团(GEhealthcare))捕获图像。实例19:将细胞与化学修饰剂接触为了增加抗原呈递细胞吞噬作用并促进肝脏靶向,将本文所述的细胞组合物在37℃下用0.15微摩尔(microM)的钙离子载体A23187(西格玛奥德里奇,圣屈昂坦法拉维耶,法国)处理30min,或者在室温(RT)下用5mM的BS3(FischerBioblockScientific公司,伊尔基希,法国)进行处理。在处理后,将最终产物在2℃-8℃下储存在合适的缓冲液中。实例20:表达和活性的评估可以通过流式细胞术(如果蛋白质在表面上表达)或通过蛋白质印迹法(针对在细胞质中表达的蛋白质)定量评估培养细胞中及其上的外源蛋白的表达。1.定量流式细胞术抗小鼠Fc结合定量流式细胞术珠(简单细胞校准)购自Bangs实验室。针对相关细胞表面受体-血型糖蛋白A、Ckit和转铁蛋白受体-的荧光标记的小鼠抗体购自BioLegend公司。针对HA表位标签的荧光标记的小鼠抗体购自生命科技公司。如本文所述,培养类红细胞。通过如本文所述的吉普森装配构建编码在N末端具有HA标签的血型糖蛋白A的转基因。如本文所述,通过慢病毒转导将转基因导入类红细胞。转导后至少2天,收集2x10^5个细胞,在PBS缓冲液中洗涤,并用1:100稀释的以上列出的抗体之一染色1hr。洗涤细胞并在流式细胞术(Attune,生命科技公司)上分析。针对以上列出的四种抗体中的每一种重复该方案。根据制造商的说明书进行定量。简言之,将5种珠子样品中的每一种中的一滴与1:100稀释的以上列出的抗体一起孵育。将这些珠孵育1hr,在PBS中洗涤,并在流式细胞术(Attune,生命科技公司)上分析。针对以上列出的四种抗体中的每一种重复该方案。使用制造商提供的excel电子表格拟合校准曲线,从中导出基于细胞的信号的荧光强度的定量。2.定量蛋白质印迹法如本文所述,培养类红细胞。通过如本文所述的吉普森装配构建编码在C末端具有HA标签的腺苷脱氨酶的转基因。如本文所述,通过慢病毒转导将转基因导入类红细胞。转染后两天,收集细胞,在PBS缓冲液中洗涤,并且在RIPA细胞裂解缓冲液(Pierce)中裂解。通过使用lipofectamine2000(生命科技公司)的瞬时转染将转基因导入HEK293T细胞。将细胞培养一周,并且收获上清液。根据制造商的说明书,在HA亲和柱(Pierce)上纯化重组蛋白。通过在280nm处的吸光度评估蛋白质浓度。如本文所述进行蛋白质印迹法。除了细胞裂解物样品外,已知量的重组腺苷脱氨酶在同一凝胶上运行。在图像收集之后,使用重组条带的强度产生标准曲线以定量细胞样品中存在的蛋白质的量。高水平的转基因的稳健表达对最终细胞群的治疗能力具有重要影响。图2通过定量流式细胞术定量了指示分化的三个表面蛋白和一个外源转基因的表达,并且证实了转基因在高水平上稳健地表达。在四阶段体外分化过程中的七个时间点处收集培养物中的类红细胞。在第一时间点(“扩增D6”),细胞是有核造血前体。到最后的时间点(“Diff3D8”),细胞主要是去核类红细胞。GPA(实心三角形),类红细胞的典型标志物,在前体细胞中开始降低,并且迅速达到>1x10^6个拷贝/细胞。CKIT(虚线方形),干细胞因子的受体,开始升高,然后随着分化继续降低至<1x10^4个拷贝/细胞。TR(点状菱形),对于将铁转运到类红细胞中是必需的,初始增加,然后逐渐下降到<1x10^5个拷贝/细胞。在第二分化阶段(“Diff1”)结束时,引入转基因(空心圆),并且在整个分化过程中稳定地以约1x10^5个拷贝/细胞进行表达。上述数据证实转基因在培养细胞中稳健表达。可以如本文所述通过流式细胞术(如果蛋白质在表面上表达)或如本文所述的通过蛋白质印迹法(对于在细胞质中表达的蛋白质)来评估外源蛋白在培养细胞中及其上的表达。在外源基因在包含下游荧光报道蛋白的单转录本构建体中的情况下,报告蛋白的荧光可以用作用于表达上游基因的代表,并如本文所述的通过流式细胞术评估。图3A-S显示出去核培养的类红细胞上的表面和胞浆蛋白的外源性表达。上述数据最终证明多种蛋白质类别-包括细胞质的,表面的,完整的,与I型膜蛋白的融合体,与II型膜蛋白的融合体,与GPI连接的膜蛋白的融合体,细胞内的融合体,过表达的和重新表达的-可以在多种细胞类型上表达,这些细胞类型包括培养的去核类红细胞、培养的有核红细胞前体细胞、和K562红白血病细胞。图3B和3F证实在去核培养的细胞中两种外源蛋白的同时表达。在图3B中,如本文所述培养类红细胞。如本文所述的通过吉普森装配来装配编码血型糖蛋白A信号序列,HA表位标签,血型糖蛋白A编码序列,病毒T2A可切割序列和GFP的转基因构建体。如本文所述,通过慢病毒转导将转基因导入类红细胞。如本文所述,将细胞培养至终末分化。如本文所述,通过流式细胞术,使用荧光抗HA抗体和GFP荧光,分析细胞,来检测两种转基因的表达。在图3F中,如本文所述培养类红细胞。如本文所述的通过吉普森装配来装配编码血型糖蛋白A信号序列,针对乙型肝炎表面抗原具有特异性的抗体scFv,HA表位标签,血型糖蛋白A编码序列,病毒T2A可切割序列和GFP的转基因构建体。如本文所述,通过慢病毒转导将转基因导入类红细胞。如本文所述,将细胞培养至终末分化。如本文所述,通过流式细胞术,使用荧光抗HA抗体和GFP荧光,分析细胞,来检测两种转基因的表达。实例21:从血小板中的mRNA表达蛋白质通过流式细胞术测量从外源转染的mRNA翻译的外源蛋白在血小板中的表达。简言之,将富含血小板的血清以190g离心15分钟,以除去红细胞和白细胞。然后将上清液以2500g旋转5分钟以沉淀血小板。将血小板重新悬浮于5mL具有1uM前列腺素的Tyrode缓冲液中,洗涤,并重新悬浮于具有1uM前列腺素的750uL的Tyrode缓冲液中。将编码感兴趣基因的mRNA(在该实例中为GFP)与脂质体以1:1mg/mL比例混合。将混合物孵育5分钟,然后添加到洗涤的血小板群体中。将该组合在室温下在缓慢摇动的情况下孵育24小时。通过测量GFP荧光的流式细胞术测定转基因的血小板表达。表面蛋白也可以通过流式细胞术测定。细胞质或其他细胞内表达的蛋白质也可以通过蛋白质印迹法进行测定。存在将外源蛋白引入血小板中及其上的治疗相关性。由于血小板不具有核或RNA转录机制,因此DNA转染不是在血小板中诱导外源蛋白表达的可行手段。然而,mRNA转染和翻译是将外源蛋白引入细胞中的方式。据认为,血小板包含mRNA翻译机制,但到目前为止还不知道它们是否能够接受并且将外源mRNA翻译成蛋白质。图4是流式细胞术图的集合,其证明了编码转基因(在这种情况下是GFP)的外源mRNA通过血小板的翻译。用488nm激光激发后,在FL1通道中通过荧光检测GFP。(4A)未转染的血小板(1.7%GFP+)。(4B)用3ugGFPmRNA转染的血小板(8.6%GFP+)。(4C)用6.8ugGFPmRNA转染的血小板(3.3%GFP+)。数据最终首次证明外源mRNA通过血小板翻译为外源蛋白。实例22:酶的活性图5证实了针对类红细胞上包含的酶的活性。通过蛋白质印迹法评估细胞质酶的生物化学活性,用于在分化过程中保留蛋白质。通过体外酶活性测定评估细胞质酶的生物活性。图5显示出在培养的类红细胞中表达的两种不同细胞内酶的活性。1.腺苷脱氨酶。通过如本文所述的吉普森装配构建编码在C末端具有HA标签的腺苷脱氨酶的转基因。如本文所述,通过脂质体转染(生命科技公司),将转基因引入HEK-293T细胞中。使用衍生自海伦纽斯(Helenius)2012,生物化学与生物物理学报(BiochimBiophysActa)1823(10):1967的方案测定酶活性,其中酶的特定混合物将嘌呤转化成尿酸和H2O2,随后对产生的H2O2进行荧光检测。简言之,在转染后两天,收集细胞,吸出介质,并以2x10^5个细胞/mL向细胞中添加克雷布斯林格磷酸葡萄糖(KRPG;包括:145mMNaCl、5.7mM磷酸钠、4.86mMKCl、0.54mMCaCl2、1.22mMMgSO4、和5.5mM葡萄糖;pH7.35)。以50uM添加腺苷。反应6小时后,收集上清液,并且在60℃下热灭活5分钟。将上清液的等分试样转移到白色96孔微量培养板中的孔中,该微量培养板包含来自西格玛的0.25U/ml细菌嘌呤核苷磷酸化酶(PNP)和0.15U/ml微生物黄嘌呤氧化酶(XO)。在室温下孵育20分钟后,向微孔中添加30μl包含HRP(终浓度1U/ml,西格玛)和AmplexRed试剂(60μM,英杰公司,分子探针)的H2O2检测混合物,随后以545和590nm的发射和激发波长测量荧光强度(TecanInfiniteM200)。2.苯丙氨酸羟化酶如本文所述,培养类红细胞。通过如本文所述的吉普森装配构建编码在C末端具有HA标签的苯丙氨酸羟化酶的转基因。如本文所述,通过慢病毒转导将转基因导入类红细胞。转染后两天,收集细胞,在PBS缓冲液中洗涤,并且在RIPA细胞裂解缓冲液(Pierce)中裂解。将细胞裂解物(64ug总蛋白)添加到包含100mMTris-HCl(pH7.5)、4mMDTT、4mM苯丙氨酸、33μg过氧化氢酶、和0.4mMDMPH4(均来自西格玛)的1mL反应缓冲液中。反应在37℃下运行过夜。孵育后,通过在AmiconUltra-4离心过滤器10KD(密理博UFC801024)中以3700rpm旋转10min的离心过滤来将样品进行脱蛋白。收集样品,并且在540nm下通过吸光度测定酪氨酸浓度。这两个外源蛋白质的是通过终末分化的端部来保留的,一个非凡的创举因为在本领域中众所周知的是,类红细胞经历消除基本功能不必要的蛋白质的严格程序(刘(Liu)J等人,(2010)血液(Blood),115(10):2021-2027,罗迪西(Lodish)HF等人,(1975)发育生物学(DevelopmentalBiology)47(1):59)。在图5A中,在分化过程中的各种时间点,从有核前体细胞(“DiffID5”)到去核红细胞(“DiffIIID8”),通过抗HA蛋白质印迹法检测外源过表达的蛋白质腺苷脱氨酶。在图5C中,在分化过程中的各种时间点,从有核前体细胞(“DiffID5”)到去核红细胞(“DiffIIID8”),通过抗HA蛋白质印迹法检测外源表达的微生物蛋白苯丙氨酸羟化酶。此外,这两种酶都保持其将底物酶促转化成产物的能力。图5B显示出通过表达完整腺苷脱氨酶的293T细胞将腺嘌呤酶促转化为肌苷。图5D显示出培养的表达苯丙氨酸羟化酶的去核类红细胞的裂解物将苯丙氨酸酶促转化为酪氨酸。这些数据最终证明外源酶在整个培养过程中保留在类红细胞上,并且它们在类红细胞中具有酶活性,其具有深刻的治疗意义。实例23:CR1的活性图6显示出在培养的类红细胞表面上过表达的补体受体1(CR1)的生物化学和生物活性。通过流式细胞术评估CR1结合免疫复合物的生物化学活性。在共培养测定中通过将免疫复合物转移至巨噬细胞来评估CR1的生物活性。1.表达CR1的细胞的免疫复合物结合。如本文所述,培养类红细胞。如本文所述,通过吉普森装配构建编码补体受体1(CR1)的转基因构建体。如本文所述,通过慢病毒转导将转基因导入类红细胞。如本文所述,使用抗CR1抗体(艾博抗公司)通过流式细胞术评估转基因表达水平。如本文所述,将细胞培养至终末分化。在室温下,将Dylight650标记的牛血清白蛋白(BSA-650)与多克隆兔抗BSA(艾博抗公司)在过量的抗体中孵育30分钟。然后将复合物与人血清以1:1体积比在37℃下混合30分钟。对照复合物不与人血清混合或与热灭活的人血清混合。将复合物与表达CR1的细胞在37℃下孵育30分钟。洗涤细胞并针对捕获免疫复合物通过流式细胞术通过检测Dylight650荧光来分析。2.免疫复合物转移至巨噬细胞培养的U937单核细胞通过与100nM佛波醇乙酯(PMA)在37℃下孵育24小时来活化。用免疫复合物(见上文)包被的细胞与活化的U937巨噬细胞在37℃下孵育30分钟。通过流式细胞术分析共培养物。通过FSC/SSC门控鉴定巨噬细胞。通过检测巨噬细胞群中的Dylight650荧光来分析巨噬细胞上免疫复合物的存在。图6显示出在培养的类红细胞上外源性过表达的补体受体1(CR1)的生物化学和生物活性。图6A显示出CR1的生物化学活性,定义为体外免疫复合物的捕获。黑色直方图显示出在培养的类红细胞上过表达的CR1对基于BSA的免疫复合物的捕获。阴影直方图显示出与缺乏人补体的BSA和IgG的复合物结合的最小背景,证明结合事件是CR1介导的。图6B显示出CR1的生物活性,定义为捕获的免疫复合物从培养的类红细胞转移到巨噬细胞。这是本领域中的标准测定,参见,瑞皮克等人,2005,临床和实验免疫学,140:230;李等人,2010,传染免疫,78(7):3129。通过流式细胞术评估转移并且测量为巨噬细胞上标记的免疫复合物衍生的荧光的强度。在该测定中,与无免疫复合物孵育的巨噬细胞(黑色条)不变成荧光。独立于培养的过表达CR1的类红细胞的存在,与缺乏补体(并且因此不结合CR1)的BSA和IgG的复合物一起孵育的巨噬细胞仅摄取少量的免疫复合物(实心灰色条)。这种摄取可能是由于与IgG分子的Fc区相互作用的U937细胞上的Fc-γ受体。可以通过相同的Fc-γ介导的方法,在过表达CR1的细胞(散列条,左)的不存在下与免疫复合物(BSA+IgG+补体)一起孵育的巨噬细胞摄取与不补体存在下相同量的免疫复合物。然而,如通过荧光测量的,在过表达CR1的细胞(散列条,右)的存在下与免疫复合物一起孵育的巨噬细胞摄取近两倍数目的免疫复合物。这些数据最终证明在培养的类红细胞上CR1过度表达能够捕获所述类红细胞上的免疫复合物,促进免疫复合物从类红细胞转移到巨噬细胞,并且显着增加巨噬细胞摄取的免疫复合物的速率和数量。实例24:scFv的活性图7显示出作为与跨膜蛋白GPA融合的培养的类红细胞表面上外源表达的抗体scFv的生物化学和生物活性。如本文所述,培养类红细胞。如本文所述,通过吉普森装配装配编码血型糖蛋白A的前导序列、针对乙型肝炎表面抗原具有特异性的抗体scFv(scFv,描述于博斯2003,分子免疫学,40:617)、HA表位标签、[Gly-3-Ser]2柔性连接物、和血型糖蛋白A的主体的转基因构建体。如本文所述,通过慢病毒转导将转基因导入类红细胞。如本文所述,使用抗HA抗体(艾博抗公司)通过流式细胞术评估转基因表达。如本文所述,将细胞培养至终末分化。通过流式细胞术评估抗体scFv结合靶蛋白的生物化学活性,在这种情况下是乙型肝炎表面抗原(HBsAg)。用Dylight-650荧光团(Pierce)标记重组HBsAg蛋白(艾博抗公司)。将表达scFv的细胞与100nM标记的蛋白一起孵育,在PBS中洗涤,并如本文所述的通过流式细胞术针对Dylight650荧光进行分析。通过流式细胞术检测的HBsAg的体内捕获来评估抗体scFv的生物活性。用Dylight-650荧光团(Pierce)标记重组HBsAg蛋白(艾博抗公司)。用CFSE(西格玛)荧光标记表达scFv的细胞。将免疫受损的NSG小鼠(杰克逊实验室(Jacksonlabs))注射约400pmol标记的HBsAg至尾静脉中。几分钟后,向相同的小鼠注射2x10^7个表达scFv的细胞。定期通过下颌下穿刺在包含EDTA的试管中收集血液。如本文所述,收集的血细胞被洗涤并通过流式细胞术分析。人细胞被鉴定为CFSE阳性的细胞。HBsAg的捕获被检测为人细胞上的Dylight650荧光。图7A-7B显示出抗体scFv的生物化学活性,定义为其同源抗原,乙型肝炎表面抗原(HBsAg)的结合。在图7A中,将表达(黑色)或不表达(灰色阴影)抗体scFv的细胞与450nMHBsAg一起孵育,并用生物素化的抗HBsAg抗体和荧光链霉亲和素进行染色。表达抗体scFv(该培养物中约45%的细胞)的细胞结合抗原。在图7B中,将表达抗体scFv的细胞与各种浓度的HBsAg一起孵育并如上进行染色,显示出结合事件是剂量依赖性的,并且亲和力约10nM。图7C-7D显示出抗体scFv的生物活性,定义为在小鼠循环中同源抗原HBsAg的捕获。在该实验中,通过尾静脉向免疫受损的NSG小鼠注射约400pmol荧光标记的HBsAg。5分钟后,通过尾静脉内注射培养的去核类红细胞(7C)或表达外源抗体scFv的培养的去核类红细胞(7D)。在注射之前,将所有培养的细胞用CFSE荧光染料进行标记。6小时后收集血液,在流式细胞仪上进行分析,并且对CFSE+人细胞进行门控。裸培养的细胞不结合HBsAg(7C),而表达抗体scFv的细胞结合至HBsAg(7D)。与生物化学活性实验一致,该培养物中约45%的细胞表达抗体-scFv。这些数据证实,当在培养的类红细胞表面上表达时,抗体scFv是具有生物化学活性的,并且当在循环中时,类红细胞上的抗体scFv能够在体内结合其靶标。这对治疗方法具有深远的影响,其中需要循环中的物质的捕获,螯合和清除。实例25:活性-循环清除图8显示出用于靶的循环清除的表面分子捕获剂的生物化学和生物活性。通过流式细胞术评估捕获剂(在这种情况下为HA多肽和生物素)结合靶蛋白(在这种情况下为抗-HA抗体和抗生物素抗体)的生物化学活性。如通过流式细胞术和血浆蛋白质定量检测的,通过靶蛋白的体内捕获和清除来评估捕获剂的生物活性。1.通过化学修饰的细胞捕获抗生物素抗体购买来自正常人供体的红细胞(研究血液组分(ResearchBloodComponents))。根据制造商的说明书,将细胞用CFSE(西格玛)在37℃下标记20分钟。然后根据制造商的说明书,使用0.02体积的2mM储备生物素试剂在室温下用NHS-生物素(西格玛)将细胞生物素化30分钟。用Dylight650(Pierce)荧光标记抗生物素抗体(艾博抗公司)。通过使用标记的抗生物素抗体和CFSE荧光作为检测标志物的流式细胞术评估细胞的标记效率。将250ug标记的抗体通过尾静脉经静脉内注射到NSG小鼠(杰克逊实验室)中。4小时后,通过尾静脉经静脉内注射1x10^8个生物素化细胞。定期通过下颌下穿刺在包含EDTA的试管中收集血液。如本文所述,收集的血细胞被洗涤并通过流式细胞术分析。人细胞被鉴定为CFSE阳性的细胞。抗生物素抗体的捕获被检测为人细胞上的Dylight650荧光。根据制造商的说明书,使用生物素包被的微孔板(Pierce)通过ELISA分析来自所抽血液的血浆,以检测循环中的抗体水平。2.通过转基因培养细胞捕获抗HA抗体如本文所述,培养类红细胞。如本文所述的通过吉普森装配来装配编码血型糖蛋白A信号序列,HA表位标签,血型糖蛋白A编码序列,病毒T2A可切割序列和GFP的转基因构建体。如本文所述,通过慢病毒转导将转基因导入类红细胞。如本文所述,将细胞培养至终末分化。如本文所述,通过流式细胞术,使用用Dylight650(Pierce)和GFP荧光荧光标记的抗HA抗体(生命科技公司),分析细胞,来检测两种转基因的表达。将250ug标记的抗HA抗体通过尾静脉经静脉内注射到NSG小鼠(杰克逊实验室)中。4小时后,通过尾静脉经静脉内注射1x10^8个培养细胞。定期通过下颌下穿刺在包含EDTA的试管中收集血液。如本文所述,收集的血细胞被洗涤并通过流式细胞术分析。人细胞被鉴定为CFSE阳性的细胞。将抗HA抗体的捕获检测为人类细胞上的Dylight650荧光。根据制造商的说明书,使用HA肽包被的微孔板(Pierce)通过ELISA分析来自所抽血液的血浆,以检测循环中的抗体水平。图8显示出(8A-8B)作为与GPA融合体的培养的红细胞表面上表达的多肽HA和(8C-8D)与原代红细胞表面化学共轭的生物素的生物化学和生物活性。生物化学活性定义为体外靶蛋白的捕获。生物活性定义为提高的体外靶蛋白的清除。在图8A中,表达为与GPA的N末端的融合体的HA多肽捕获体内小鼠抗HA抗体。NSG小鼠注射荧光标记的小鼠抗HA抗体,然后注射培养的人类红细胞,这些类红细胞不表达(顶部)或(底部)表达在其表面上作为与GPA的融合体的HA表位标签。抽取血液并在流式细胞仪上分析细胞。该x轴数值为CFSE荧光。该y轴数值为抗HA抗体Dylight650荧光。在两个样品中都观察到CFSE阳性培养的人红细胞,但只有表达HA表位标签的细胞能够捕获循环的抗HA抗体。在图8B中,向小鼠注射抗HA抗体,然后任选地注射培养的人类红细胞,这些类红细胞不表达或不表达在其表面上作为与GPA的融合体的HA肽。在多个时间点收集血浆,并且使用HA肽包被的板作为底物通过ELISA评估血浆中抗HA抗体的水平。单独注射抗HA抗体的小鼠(空心圆,实线-120分钟后这只小鼠死于与治疗无关的原因)或注射抗HA抗体接着注射在其表面上不表达HA肽的细胞的小鼠(虚线)到注射细胞后24小时在循环中具有显着的抗体。相比之下,注射抗HA抗体随后注射在其表面上表达HA肽的细胞的小鼠在数分钟内耗尽靶抗体。该数据最终证明靶抗体通过在其表面上表达外源抗原多肽的培养的类红细胞快速且特异性地从循环中清除。在图8C中,通过胺官能化化学与类红细胞表面共轭的生物素分子捕获小鼠抗生物素抗体。在这两种情况下,通过流式细胞术评估捕获。当用荧光抗生物素抗体(下点图)染色时,CFSE标记和生物素化的细胞显示为双阳性,而未生物素化的CFSE标记的细胞仅显示为单阳性(上点图)。在图8D中,向小鼠注射抗生物素抗体,然后任选地注射培养人类红细胞,这些类红细胞不与在其表面上的生物素共轭或者与其共轭。在多个时间点收集血浆,并且使用生物素包被的板作为底物通过ELISA评估血浆中抗生物素抗体的水平。单独注射抗生物素抗体的小鼠(空心圆,实线)或注射抗生物素抗体接着注射不与在其表面上的生物素共轭的细胞的小鼠(虚线)到注射细胞后24小时在循环中具有显着的抗体。相比之下,注射抗生物素抗体随后注射与其表面上的生物素共轭的细胞的小鼠在数分钟内耗尽靶抗体。该数据最终证明靶抗体通过在其表面上包含外源抗原多肽的培养的类红细胞快速且特异性地从循环中清除。总之,数据最终证明表达外源抗原的EHC上的合适的外源抗原能够结合其体内靶分子,并且当在循环中时介导靶分子的快速循环清除,这具有深远的治疗影响。实例26:补体调节剂的活性通过本领域已知的标准CH50和AH50测定来评估表达外源抗原的EHC的补体调节活性(参见,例如,卡巴特(Kabat)等人,1961,实验室免疫化学(ExpImmunochem),第133-239页和普拉茨-米尔斯(Platts-Mills)等人,1974,免疫学杂志(JImmunol)113:348)。简言之,CH50测定利用绵羊红细胞(SRBC)作为靶细胞。简言之,在GVB(2+)缓冲液(具有Ca2+和Mg2+的明胶/维罗纳缓冲盐水)中制备包含1×10^9SRBC/ml的悬浮液,pH7.35。滴定溶血素(兔抗绵羊抗血清)以确定使SRBC敏化的最佳稀释度。将稀释的溶血素(1:800)与等体积的SRBC(1×109SRBC/ml)混合,并将整体在37℃下孵育15分钟。这导致5×10^8/ml抗体包被的红细胞(EA)。将EA(100μl)与100μl的5个连续两倍稀释度(1:20、1:40、1:80、1:160、和1:320)的正常人血清(NHS)的或者相似稀释度的NHS和表达外源抗原的EHC的混合物在37℃孵育1小时。将用GVB2+缓冲液孵育的NHS用作对照。通过将EA仅与缓冲液(不添加血清)一起孵育获得的背景对照,通过向EA中添加蒸馏水来确定总裂解(100%溶血)。使用1.2ml冰冷的0.15MNaCl终止反应,将混合物旋转以沉淀未裂解的细胞,并且经分光光度法(412nm)确定上清液的光密度。相对于100%裂解对照确定溶血百分比。通过确定裂解测定混合物中50%细胞所需的血清稀释度来定量补体活性。结果表示为在CH50单位/ml血清中的该稀释度的倒数。简言之,AH50测定取决于通过替代途径的活化由人血清对未敏化的兔红细胞(Erab)的裂解。通过向测定缓冲液中添加钙螯合剂乙二醇四乙酸(EGTA)来防止钙依赖性经典途径的活化,并将两种途径所必需的镁添加到缓冲液中。简言之,在GVB-Mg2+-EGTA缓冲液中制备兔RBC(2×10^8个细胞/ml)的细胞悬浮液。将1.5倍稀释度(1:4、1:6、1:9、1:13.5、和1:20.25)的正常人血清(NHS)或类似稀释度的NHS与表达外源抗原的EHC的混合物在GVB-Mg2+-EGTA缓冲液中制备,并且将100μl的每种血清稀释液添加到50μl的标准化Erab中。将用GVB-Mg2+-EGTA缓冲液孵育的NHS用作对照。然后将混合物在振荡水浴中在37℃下孵育60分钟,以保持细胞处于悬浮,并且使用1.2ml冰冷的NaCl(0.15M)停止反应。将管以1250g在4℃下旋转10分钟来沉淀细胞,并且经分光光度法(412nm)确定上清液的光密度。背景对照具有100μlGVB-Mg2+-EGTA缓冲液和50μlErab,并且不超过总裂解的10%在总裂解对照管中,向50μlErab悬浮液中添加100μl蒸馏水,并相对于100%裂解对照确定溶血百分比。计算测定结果,并且通过确定裂解测定混合物中50%细胞所需的血清稀释度来定量补体活性。结果表示为在AH50单位/ml血清中的该稀释度的倒数。实例27:负载有血小板的胸苷磷酸化酶的活性如本文所述,通过吉普森装配构建编码在C-末端具有HA标签的胸苷磷酸化酶的转基因。如本文所述,从前体细胞培养血小板。如本文所述,通过慢病毒转导将转基因导入培养的血小板前体细胞。如本文所述,通过使用抗HA检测抗体的蛋白质印迹法来评估培养的血小板中胸苷磷酸化酶的表达。通过定量胸苷向胸腺嘧啶的转化率,在血小板样品中,确定胸苷磷酸化酶活性。进行初步实验以确定关于时间和酶稀释的线性代谢物形成动力学;该方法显示为线性长达16分钟,超过4.0-719nmol/min/ml的胸苷磷酸化酶范围(对应于10-9088的样品稀释范围)。通过用125mMTris-HCl(pH7.4)以1:710稀释解冻样品制备透析前样品培养血小板和对照血小板样品的裂解物。然后将25ul的血小板裂解物添加到100μl磷酸钠缓冲液(100mM,pH6.5)和25ul胸苷标准品(10mM)中,混合并在37℃孵育10min。用25ul的40%TCA终止反应。在添加血小板裂解物之前,通过将TCA添加到磷酸钠缓冲液/胸腺嘧啶孵育混合物中来制备测定空白。将样品以13,400×g离心2min,并且在振荡器上在2分钟内将上清液用水饱和的乙醚洗涤2次,以提取TCA。为了避免醚干扰HPLC分离,通过将基质暴露于空气5分钟以允许醚的蒸发来实现有效去除。将10ul的样品体积注入HPLC中。使用反相色谱法,用无梯度洗脱,使用沃特斯AllianceHPLC2795系统,实现底物和产物的色谱分离。使用预填充的C18柱(SpherisorbODS125mm×4.6mmID,5um粒度,沃特斯)作为稳定阶段。使用具有离子配对试剂四丁基硫酸氢铵(5mM)的乙酸铵(40mM)流动相洗脱分析物,该离子配对试剂四丁基硫酸氢铵用HCl调节至pH2.70,以1.0ml/min的流速递送,运行时间8min。UV检测是在254nm和0.1吸光度单位满刻度下。通过将光谱与纯标准品进行比较来鉴定代谢物。实例28:展示的古德帕斯彻抗原的血小板的活性如本文所述的,通过吉普森装配构建编码与具有干扰HA标签的CD42b的N末端(GP1B,genbankAAH27955.1)融合的胶原α-3(IV)(COL4A3)NC1结构域抗原的转基因。如本文所述,从前体细胞培养血小板。如本文所述,通过慢病毒转导将转基因导入培养的血小板前体细胞。如本文所述,使用抗HA检测抗体,通过流式细胞术评估外源抗原在培养的血小板上的表达。从患有古德帕斯彻氏综合征的患者收集血清,并通过商业ELISA(MyBioSourceCOL4A3ELISA试剂盒)测试血清的抗COL4A3抗体。如本文所述,使用此抗COL4A3血清作为主要检测抗体和荧光抗人IgG作为第二检测抗体,通过流式细胞术评估表达抗原的血小板的结合能力。使用表达抗原的血小板在小鼠中模拟体内循环抗原的血小板促进清除。NSG小鼠注射100uL用Dylight650染料荧光标记的小鼠抗人COL4A3抗体(CreativeBioMart公司)。然后经尾静脉内注射表达外源抗原的CFSE标记的培养的血小板(每只小鼠10^8个)。在10min、30min、2h、12h、和24h处从下颌下位置抽取血液。将血液离心以收集富含血小板部分,然后如本文所述,将其染色并通过流式细胞术分析。通过跟踪CFSE-Dylight650双阳性群体确定血小板的抗体捕获。实例29:体内活性(小鼠)如本文所述,培养小鼠类红细胞。如本文所述的,使用慢病毒,用合适的外源抗原多肽转基因(例如编码补体受体1(CR1)的转基因)转导红细胞前体细胞。如本文所述,将细胞培养至终末分化。如本文所述,通过流式细胞术评估细胞中外源蛋白的存在。根据制造商的说明书,用荧光染料,例如CFSE(西格玛奥德里奇)标记细胞,以帮助它们的检测。将细胞注射到狼疮的NZBWF1/J小鼠模型或对应于合适的外源抗原多肽的其他合适的疾病或活性模型,经尾静脉内注射约1x10^8个细胞。通过下颌下穿刺在多个时间点收集血液。通过拉吉细胞测定检测血浆中的免疫复合物水平,参见,例如(Theofilopoulos)等人,976,临床调查杂志(JClinInvest)57(1):169。如本文所述,通过跟踪所抽取的血液样品中CFSE荧光细胞的百分比,通过流式细胞术评估培养细胞的药代动力学。通过肉眼尸检评估小鼠总体健康,包括肾组织的组织学,来跟踪免疫复合物沉积和炎症介导的损伤的减少。实例30:快速筛选与培养的类红细胞(天-周)相比,细胞系,例如293T和K562,具有更短的表达和培养周期(约1天)。通过编码合适的外源抗原多肽的基因文库,这些细胞系可用于快速迭代,以鉴定具有最高表达或活性的外源抗原多肽。如本文所述,通过聚合酶链反应和吉普森装配来构建合适的外源抗原多肽转基因文库,例如补体受体1(CR1)的全长和更短的变体。如本文所述的,使用脂质体,在微量滴定板中以平行方式将转基因文库转染到HEK293T细胞中,并使如本文所述的,用慢病毒转导入K562细胞中。在24-48小时后,如本文所述,通过流式细胞术来评估外源抗原的表达。如本文所述,通过用流式细胞术检测的荧光免疫复合物的捕获,以及如本文所述,通过用流式细胞术检测的荧光免疫复合物转移到培养的单核细胞,来评估文库中每种外源抗原的活性。然后将来自最具功能性的-例如最高表达,捕获大多数免疫复合物或最佳转移免疫复合物至单核细胞-文库的外源抗原使用本文所述的慢病毒单独转导到如本文所述的平行类红细胞培养物中。如本文所述,通过流式细胞术评估在培养的类红细胞上的每种外源抗原的表达。如本文所述,通过用流式细胞术检测的荧光免疫复合物的捕获,以及如本文所述,通过用流式细胞术检测的荧光免疫复合物转移到培养的单核细胞,来评估培养的类红细胞上的每种外源抗原的活性。实例31:评估体内RBC的清除率在免疫受损的小鼠模型中体内评估类红细胞的清除率。在第-1天用100uL氯膦酸脂质体(Clodrosomes.com)溶液处理NSG小鼠以选择性地耗尽巨噬细胞。用荧光标签CFSE标记细胞,并且经尾静脉将约1x10^8个细胞注射到每只小鼠中。定期通过下颌下穿刺收集血液,并且收集血液细胞。将细胞用抗人GPA抗体共染色并且通过流式细胞术进行分析。通过CFSE信号和人GPA信号将人类红细胞与小鼠类红细胞区分开。针对治疗应用,重要的是培养的类红细胞和细胞内或在表面上包含外源蛋白质的培养的类红细胞在通常在体内循环。这在使用标准的免疫受损的小鼠模型的图9中显示。在图9A中,在流式细胞仪上分析从注射的小鼠收集的血液。在图的右上象限中鉴定培养的人类红细胞,针对CFSE和人-GPA是双阳性的。在图9B中,向小鼠注射人血红细胞(实心圆),培养的去核类红细胞(虚线菱形),表达细胞内外源蛋白的、培养的去核类红细胞(点线正方形)和表达表面外源蛋白的、培养的去核类红细胞(空心三角形)。人细胞的清除率被测量为随时间剩余的CFSE+细胞百分比,缩放至初始时间点(注射后20分钟)。四个样品之间的清除率没有显着差异。这些数据明确地证实培养的去核类红细胞具有与正常人血红细胞基本相似的循环。此外,在胞内空间或细胞表面上表达的外源蛋白质基本上不影响这些细胞的循环行为。这针对该技术的治疗性翻译是一个重要结果。实例32:不良循环事件的评估通过检测血液中的纤维蛋白原分解产物和在注射培养的类红细胞的动物中的组织学来评估由循环中培养的类红细胞引起的不良事件的发生率。纤维蛋白原分解产物的检测。如本文所述,向小鼠注射培养的类红细胞。通过在包含EDTA的管中下颌下穿刺从小鼠收集血液。通过离心分离细胞并且收集血浆。遵循制造商的说明书,通过ELISA(MyBiosource)在小鼠血浆中测量纤维蛋白原分解产物纤维蛋白肽A和纤维蛋白肽B的水平。组织学。尸检后收集来自相同小鼠的组织样品。修剪组织,包埋在石蜡中,并切片。通过H&E染色和三色染色来染色组织切片。在10x和20x放大下拍摄显微镜图像。针对治疗应用,重要的是,培养的类红细胞和包含外源蛋白质(细胞内或在表面上)的培养的类红细胞不诱导不良事件,如凝血级联的激活和组织血栓形成。图10A和10B显示针对注射以下各项的小鼠的小鼠血浆中纤维蛋白肽A和B的水平:(1)人血红细胞、(2)培养的去核类红细胞、(3)表达细胞内外源蛋白的、培养的去核类红细胞、(4)表达表面外源蛋白的、培养的去核类红细胞,和(5)单独的重组蛋白。针对所有样品,纤维蛋白肽A和B(纤维蛋白原分解和凝血级联激活的标志物)的水平基本相似。图10C和10D显示出注射有培养的去核类红细胞(10C)和重组蛋白(10D)的小鼠的脾的组织学染色切片。组织之间没有实质性差异,并且在任何样品之间没有观察到脾、肝、肺、脑、心脏、和肾中的可识别的组织损伤。这些数据最终证明,在有或没有外源蛋白质的情况下,培养的类红细胞当在循环中时在小鼠中不诱导任何不良事件。实例33:循环中外源蛋白保留的评估通过流式细胞术和蛋白质印迹法评估在培养的去核类红细胞中及其上的外源蛋白的保留。1.通过流式细胞术评估的外源蛋白的保留如本文所述,培养类红细胞。如本文所述,通过吉普森装配来装配编码血型糖蛋白A信号序列、针对乙型肝炎表面抗原具有特异性的抗体scFv,HA表位标签和血型糖蛋白A编码序列的转基因构建体。如本文所述,通过慢病毒转导将转基因导入类红细胞。如本文所述,将细胞培养至终末分化。用CFSE对细胞进行荧光标记,并且经尾静脉内注射到免疫受损的NSG小鼠(杰克逊实验室)中(每只小鼠1x10^8个细胞)。定期通过下颌下穿刺收集血液。收集的细胞用荧光抗HA抗体(艾博抗公司)进行染色,并通过流式细胞术进行分析。人细胞被鉴定为CFSE+细胞,并且通过同样针对表位标签染色为阳性的CFSE+细胞的部分来评估外源蛋白质保留。2.通过蛋白质印迹法评估的外源蛋白的保留如本文所述,培养类红细胞。如本文所述,通过吉普森装配装配编码腺苷脱氨酶和HA表位标签的转基因构建体。如本文所述,通过慢病毒转导将转基因导入类红细胞。如本文所述,将细胞培养至终末分化。用CFSE对细胞进行荧光标记,并且经尾静脉内注射到免疫受损的NSG小鼠(杰克逊实验室)中(每只小鼠1x10^8个细胞)。定期通过下颌下穿刺收集血液。将收集的细胞洗涤,裂解,并且如本文所述通过蛋白质印迹法用针对HA表位标签的检测抗体进行分析。针对治疗应用,重要的是当在循环中时细胞内或表面上包含外源蛋白的培养的类红细胞保留这些转基因。这个创举是非凡的,因为在该领域中广泛假设,当它们成熟时,当在循环中时,类红细胞经历严格的成熟程序和基本功能不必要的蛋白质的消除(刘(Liu)J等人,(2010),血液(Blood)115(10):2021-2027,罗迪西(Lodish)HF等人,(1975),发育生物学(DevelopmentalBiology)47(1):59)。图11显示出在培养的去核类红细胞中及其上表达的外源蛋白质保留在循环中。在图11A中,向小鼠注射在其表面上表达抗体scFv的培养的去核类红细胞。在整个多天循环研究期间,抗体scFv阳性细胞的百分比开始并稳定地保持在约50%。在图11B中,向小鼠注射表达具有HA标签的细胞质酶的培养的去核类红细胞,或注射具有HA标签的重组酶。当通过蛋白质印迹法分析时,显然在实验期间酶保留在培养的细胞内。条带强度的降低归因于在实验期间细胞的清除,而不是来自所述细胞中外源酶的去除。数据明确地证实,在培养的去核类红细胞中及其上表达的外源蛋白保留在循环中的细胞中及其上,这对治疗相关性具有巨大的和前所未有的影响。实例34:体内半衰期延长的评估如本文所述,培养类红细胞。如本文所述,通过吉普森装配装配编码腺苷脱氨酶和HA表位标签的转基因构建体。如本文所述,通过慢病毒转导将转基因导入类红细胞。如本文所述,将细胞培养至终末分化。用CFSE对细胞进行荧光标记,并且经尾静脉内注射到免疫受损的NSG小鼠(杰克逊实验室)中(每只小鼠1x10^8个细胞)。定期通过下颌下穿刺收集血液。将收集的细胞洗涤,裂解,并且如本文所述通过蛋白质印迹法用针对HA表位标签的检测抗体进行分析。通过如本文所述的吉普森装配构建编码在C末端具有HA标签的腺苷脱氨酶的转基因。如本文所述,通过脂质体转染(生命科技公司),将转基因引入HEK-293T细胞中。根据制造商的说明书,使用HA亲和树脂(Pierce),在7天后从细胞培养上清液中纯化蛋白质。蛋白质浓度通过280nm处的吸光度来评估。将蛋白质(40ug)经尾静脉内注射到免疫受损的NSG小鼠(杰克逊实验室)中。定期通过下颌下穿刺收集血液。如本文所述,通过蛋白质印迹法用针对HA表位标签的检测抗体分析血浆。在图11B中,向小鼠注射表达具有HA标签的细胞质酶的培养的去核类红细胞,或注射具有HA标签的重组酶。当通过蛋白质印迹法分析时,清楚的是当在循环细胞中表达时,与当以可溶形式注射时相比,酶循环半衰期显著延长。实例35:体内清除率的评估-血小板使用本文详细说明的程序培养表达外源胸苷磷酸化酶的血小板群,并用CFSE标记,并且经尾静脉内注射到NSG小鼠中。类似地用CFSE标记天然人来源的血小板群并且注射到另一只小鼠中。在10min、1h、4h、8h、24h、和48h处,从两只小鼠采集样品,并且使用流式细胞术来定量血小板循环水平。比较天然与培养的血小板的半衰期。实例36:不良循环事件的评估-血小板针对治疗应用,重要的是,培养的血小板和包含外源蛋白质(细胞内或在表面上)的培养的血小板不诱导不良事件,如凝血级联的激活和组织血栓形成。当经尾静脉将培养的血小板注射到NSG小鼠中时,遵循制造商的方案(MyBiosource),通过ELISA在小鼠血浆中检测纤维蛋白原分解产物纤维蛋白肽A和纤维蛋白肽B。尸检后收集来自NSG小鼠的组织样品。修剪组织,包埋在石蜡中,并切片。通过H&E染色和三色染色来染色组织切片。在10倍和20倍放大下拍摄显微镜图像,并且由受过训练的病理学家针对任何致病特征进行评估。实例37:循环中外源蛋白保留的评估-血小板通过流式细胞术和蛋白质印迹法评估外源蛋白在培养的血小板中及其上的保留。将包含细胞内外源蛋白的CFSE标记的血小板经尾静脉内注射到小鼠中。定期通过下颌下穿刺收集血液。离心血液以分离富含血小板的血浆,然后将其裂解,并且通过蛋白质印迹法,对存在于外源蛋白上的表位标签进行染色来分析。实例38:获得用于生产的供体细胞获得知情同意后,健康的CD34+干细胞供体接受rhG-CSF(格拉诺赛特或优保津)、10ug/kg/天s.c.,持续5天用于外周血干细胞动员,并且然后连续2天经历血液成分单采术,收集动员的CD34+HSC。通过菲可密度梯度离心从动员的外周血中分离单核细胞(MNC),并且分成两部分。相对于美天旎方案,通过使用抗CD34包被的磁珠(美天旎生物技术公司,德国),将一部分用于来纯化CD34+细胞。控制CD34+部分的纯度。然后将富含CD34+的HSC立即用于两步培养方法中或冷冻直到在一步培养方法中使用。使用的完全培养基(CM)是补充有2mML-谷氨酰胺和100IU/ml青霉素-链霉素(Gibco公司,格兰德岛(GrandIsland),纽约州,美国)和10%热灭活的FBS(Gibco公司)的RPMI1640(Eurobio,法国)。将补充有10%热灭活的FBS的IMDM(Gibco公司)用于扩增。从R&D系统(明尼阿波里斯市,明尼苏达州,美国)购买重组人干细胞因子(rhSCF)、血小板生成素(TPO),胎肝酪氨酸激酶3配体(Flt-3L),GM-CSF和TNF-α。实例39:扩大生产类红细胞按体积逐渐放大,在静态培养中维持细胞在1x10^5和2x10^6个细胞/mL之间的密度。扩增阶段以10^5/ml进行接种,并且包括3-7个渐进体积转移;100ml、500ml、1L、10L、50L、100L、100L。在生产过程中,细胞培介质括IMDM、FBS、BSA、全转铁蛋白、胰岛素、谷氨酰胺、地塞米松、β雌二醇、IL-3、SCF和促红细胞生成素的组合。当细胞达到适于接种生产生物反应器的体积时,将其转移到生产生物反应器中用于最终的放大和分化。实例40:在生物反应器(波型)中培养细胞根据操作手册设置波型生物反应器2/10系统。简言之,Cellbag组装在摇摆单元上,该摇摆单元放置在灌注模块上。用空气充气该袋后,将重量设置为零。随后,用适当量的培养基填充袋并孵育至少两小时,允许培养基达到37℃。将介质和细胞通过转移瓶,一种具有两个端口的特殊设计的DURAN玻璃瓶,转移到袋中。在烧瓶的上部,将过滤器连接到端口。在另一个端口中,通过烧瓶的底部装配管。转移瓶上的管子与Cellbag上的进料接头连接。将转移烧瓶保持在LAF罩中,以降低污染的风险。在灌注开始之前,用于收获和进料的管和容器连接到Cellbag上。如下制备管:将分别具有3.2和6.4mm内外直径的50或70cm长的SaniflexASTP-ELP硅胶管(Gore/SaniflexAB)在两端装备阳卢尔锁连接。将硅胶管通过阴卢尔锁连接到C-Flex管的一端。在C-Flex管的另一端,装配阳卢尔锁,并且然后对管进行高压灭菌。将卢尔锁用所有管上的拉链固定。在灌注之前,将硅氧烷部分连接到Cellbag和C-Flex部分,至5L容器(HycloneLabtainer)用于进料和收货。所有连接都在层流柜中进行。环境和代谢因素的控制可以改变培养中的类红细胞的转录因子和基因调节蛋白的表达或活性,参见,例如,塞萨尔(Csaszar)等人,2009,生物技术与生物工程(BiotechnolBioeng)103(2):402;(Csaszar)等人,2012,细胞-干细胞(CellStemCell)10(2):218。为了提供对反应器中的输入和输出的控制,创建了微量体积递送系统,其中的关键组分是60-80cm长的熔融石英毛细管(#TSP100375,聚微技术公司(PolymicroTechnologies)),内径为100μm。在输入端,用通过PEEK卢尔连接到MicroTight适配器(#P-662,UpchurchScientific公司)的luer-lok尖端原料注射器(#309585BD)给毛细管进料。将原料注射器荷载在冰箱中保持在4℃的模型33双注射泵(#553333,哈佛仪器公司(HarvardApparatus))上。在输出端,毛细管进入生物反应器:两端口FEP细胞培养袋(#2PF-0002,VueLife)置于37℃下,5%CO2的细胞培养箱中的定轨摇床上。该毛细管通过自密封橡胶隔膜(#B-IIS,InterLink)用针进入生物反应器的中点进行进料。该生物反应器上的相对连接器用另外的自密封橡胶隔膜替换。在与具有10%胎牛血清的PBS溶液一起使用之前,将原料注射器和递送毛细管封闭过夜,以防止蛋白质粘附到注射器和毛细管壁上。使用美国国家仪器公司(NationalInstruments)LabVIEW7.1创建一个程序来控制注射泵的注射。程序的基本给予策略是初始注射至浓度L1,随后是等待时间t1并且随后注射,每次至浓度L2,并且随后等待时间t2,重复n次。用户输入流速,原料浓度,初始培养体积,注射后所希望的浓度,注射之间的时间和总注射次数。实例41:免疫原性和耐受性诱导的评估1.小鼠中的耐受性诱导在小鼠中,可以通过用包含抗原蛋白(在该实施例中为卵清蛋白(OVA))的本发明的细胞组合物的3次连续静脉内注射来诱导耐受性。在第-7,-3和-1天用游离OVA或在本发明的细胞组合物(细胞-OVA)内表达的OVA(细胞OVA)注射原始小鼠。然后通过两次注射与多聚I:C佐剂(英杰公司,圣迭哥,加利福尼亚州)混合的抗原来诱导强免疫应答,使小鼠对OVA免疫。2.抗体滴度的评估通过标准ELISA评估小鼠血清中的IgG水平。简言之,在不同时间点,从已经注射了包含抗原蛋白(例如,卵清蛋白(OVA))的本发明的细胞组合物的小鼠的血液样品,和从已经注射游离或重组OVA的小鼠获得血清。使用标准ELISA测定,其中OVA作为抗原(1μg/ml,在50mM碳酸盐缓冲液中,pH9.7)吸附到测定板上。将血清样品在1:50-1:200的范围内连续稀释用于预处理或无处理血清,并且1:400-1:500,000用于后处理血清,并一式两份进行测试。使用与辣根过氧化物酶共轭的第二抗小鼠免疫球蛋白,随后用显色底物处理,比色检测血清中抗OVA抗体与吸附的重组OVA的结合。3.T细胞应答的分析如本文所述,诱导针对抗原蛋白OVA的耐受性。在OVA的免疫相注射的第二次给予后7天对小鼠实施安乐死,并收集它们的脾脏。通过使器官通过70微米的细胞粗滤器过滤并且在用0.8%氯化铵溶液(干细胞技术公司(Stem-CellTechnologies),格勒诺布尔,法国)的RBC裂解后,获得脾细胞悬浮液。在与用于分析的抗体孵育之前,将所有样品与抗-Fc受体抗体(纯化的抗CD16/32,Ozyme,圣迭哥,加利福尼亚州)一起孵育以防止非特异性结合。以下单克隆抗体(Abs)用于脾细胞染色:PC5-抗-CD62L(MEL14)和PC7-抗-CD8,购自Biolegend公司。OVA肽-MHC四聚体(PE-Kb-SIINFEKL四聚体)购自贝克曼库尔特商贸公司(BeckmannCoulter)。OVA特异性T细胞通过流式细胞术鉴定为通过用抗CD8和OVA肽-MHC四聚体染色而为双阳性的细胞。在该细胞群中,被激活的OVA特异性CD8T细胞的百分比由通过染色抗CD62L抗体为阳性的细胞的部分来确定。4.体内T细胞裂解测定用10微克/ml的SIINFEKL肽(Genscript公司,皮斯卡特维,新泽西州)在37℃下将原始脾细胞脉冲1小时,并且然后用0.4微摩尔CFSE(CFSE低)进行标记。用4微摩尔CFSE(CFSE高)标记未处理的脾细胞的对照群。CFSE低和CFSE高细胞以1:1的比例组合,并且通过i.v.途径注射1E7个细胞/小鼠到先前已经耐受本文所述的OVA抗原的小鼠或到已经用本文所述的OVA抗原免疫的小鼠。十六小时后,如本文所述制备脾单细胞悬浮液,并使用流式细胞术分析以确定CFSE低/CFSE高细胞比率。实例42:评估培养的类红细胞的扩增和分化重要的是评估体外分化细胞的扩增,分化和去核,以确保转基因的引入不会负面影响培养中细胞的质量。通过细胞计数评估扩增。通过流式细胞术,蛋白质印迹法和RT-PCR评估分化。通过流式细胞术评估去核。通过细胞计数评估扩增率。如本文所述培养类红细胞。在各个时间点,收集细胞,用PBS洗涤,并使用伯爵夫人自动细胞计数仪器(CountessAutomaticCellCounterinstrument)(生命技术公司)进行计数。通过细胞数随时间的增长来确定细胞的扩增速率。通过流式细胞术评估分化。如本文所述培养类红细胞。在各个时间点,收集细胞,用PBS洗涤,并用购自生命科技公司的针对细胞表面标志物GPA(CD235a)、CKIT(CD117)和TR(CD71)的荧光抗体的1:100稀释物进行染色。如本文所述通过流式细胞术分析标记的细胞。通过蛋白质印迹法评估分化。如本文所述培养类红细胞。在各个时间点,收集细胞,用PBS洗涤,用RIPA缓冲液裂解,并使用针对分化标志物GATA1、GATA2、Band3、CD44和肌动蛋白(艾博抗公司)的抗体如本文所述通过蛋白质印迹法进行分析。通过流式细胞术评估去核。如本文所述培养类红细胞。在各个时间点,收集细胞,用PBS洗涤,并以制造商推荐的稀释度用针对血型糖蛋白A(生命技术公司)的荧光抗体和核酸着色剂DRAQ5(Pierce)进行染色,并如在此描述的,在Attune流式细胞仪上进行分析。通过显微镜检术评估去核(联苯胺-吉姆萨)。如本文所述,培养类红细胞。在各个时间点,收集细胞,用PBS洗涤,并使用Cytospin(赛墨科技公司)在载玻片上旋转。细胞是在用-20℃甲醇在室温下细胞离心2分钟后的固定细胞,用水漂洗并风干。用10mLPBS溶解联苯胺片剂(西格玛#D5905),向其中添加10μLofH2O2。将溶液用0.22um注射器过滤器进行过滤。载玻片上的细胞斑点用300-500uL联苯胺溶液覆盖,在室温下孵育1小时,然后用水洗涤。用水以1:20稀释吉姆萨着色剂(西格玛#GS500)。载玻片上的细胞斑点用300-500uL的吉姆萨溶液覆盖,在室温下孵育40分钟,用水洗涤并风干。然后在显微镜上成像之前,将载玻片安装并密封。图12A显示针对包含转基因的细胞(短划线和点线)和不包含转基因的细胞(实线),在扩增和分化的七天窗口期间,培养物中的类红细胞的扩增速率。值得注意的是,包含转基因的培养细胞的扩增速率与不含转基因的细胞的扩增速率是不可区分的。图12B是用针对细胞表面分化标志物GPA和CKIT的抗体染色的细胞的流式细胞术图的集合。在这个特定的分化阶段,当细胞接近末端成熟时,培养物失去其CKIT表达并增加其GPA表达。值得注意的是,通过这种分化度量,包含转基因的培养细胞与不含转基因的培养细胞无法区分。图12C是用针对表面标志物GPA的抗体和荧光DNA着色剂染色的细胞的流式细胞术图的集合。三个细胞群是明显的:(1)GPA高和DNA低的细胞,包括去核类红细胞;(2)GPA高和DNA高的细胞,包括仍包含遗传物质的类红细胞;和(3)GPA低和DNA高的细胞,其包括红细胞或来自去核细胞的膜包裹的排出的核。值得注意的是,通过这种去核度量,包含转基因的培养细胞与不含转基因的培养细胞无法区分。将转基因引入细胞培养物不显着影响培养物中细胞的扩增速率,分化或去核速率。实例43:评估血红蛋白含量1.总血红蛋白根据制造商的说明书,通过Drabkin试剂(西格玛-奥德里奇,产品D5941)确定红细胞血红蛋白含量。简言之,将血细胞与试剂在缓冲水溶液中合并,充分混合,并且使用标准分光光度计测量540nm波长下的吸光度。可溶性血红蛋白标准曲线用于定量细胞中的血红蛋白含量。2.通过RT-PCR进行的血红蛋白分型裂解细胞并且收集总RNA。根据制造商的方案,使用用于RT-PCR的SuperScript第一链合成系统(生命技术公司)进行反转录。简言之,将总RNA(5ug)与150ng随机六聚体引物和10nmoldNTP混合物在10uLH2O中在65℃下孵育5分钟,然后在冰上孵育1分钟。用2uL10xRT缓冲液,4uL的25mMMgCl2、2uL的0.1MDTT、和1uL的RNAseOUT制备反应主混合物。将反应混合物添加到RNA/引物混合物中,简单混合,然后在室温下放置2min。向每个管中添加1uL(50单位)SuperScriptIIRt,混合,并在25℃下孵育10min。将反应在42℃下孵育50min,在70℃下热灭活15min,然后保存在冰上。添加1uLRNaseH并且在37℃下孵育20min。然后将该反应产物,第一链cDNA储存在-20℃下,直到需要进行RT-PCR反应。从IDT-DNA购买扩增不同血红蛋白基因和对照基因的引物。引物如下:hHBB_F-tcctgaggagaagtctgccgt(Seq.IDNo.9);hHBB_R-ggagtggacagatccccaaag(Seq.IDNo.10);hHBA_F1-tctcctgccgacaagaccaa(Seq.IDNo.11);hHBA_R1-gcagtggcttagcttgaagttg(Seq.IDNo.12);hHBA_F2-caacttcaagctaagccactgc(Seq.IDNo.13);hHBA_R2-cggtgctcacagaagccag(Seq.IDNo.14);hHBD_F-gactgctgtcaatgccctgt(Seq.IDNo.15);hHBD_R-aaaggcacctagcaccttctt(Seq.IDNo.16);hHBG2_F-cactggagctacagacaagaaggtg(Seq.IDNo.17);hHBG2_R-tctcccaccatagaagataccagg(Seq.IDNo.18);hHBE_F-aagagcctcaggatccagcac(Seq.IDNo.19);hHBE_R-tcagcagtgatggatggacac(Seq.IDNo.20);h18S-RNA-F-cgcagctaggaataatggaatagg(Seq.IDNo.21);h18S-RNA-R-catggcctcagttccgaaa(Seq.IDNo.22)。在总体积50uL的H2O中,用25uLSYBRGreenMix(2倍)(应用生物系统公司(AppliedBiosystems))、0.5uL第一链cDNA、2uL正向/反向引物对混合物(每种引物为5pmol/uL)制备RTPCR反应混合物。使用以下扩增循环在ABIPrismSDS7000仪器(应用生物系统公司)中进行反应:50℃2min,1个循环;95℃10min,1个循环;95℃15s->60℃30s->72℃30s,40个循环;72℃10min,1个循环。用SDS7000仪器进行解离曲线分析和RT-PCR结果。实例44:评估培养的血小板的分化-FACS可以通过流式细胞术评估培养物中血小板的分化状态。巨核细胞(MK)代表在末端血小板分化之前的独特的细胞形态。为了确定MK的成熟程度,洗涤1×10^6个培养细胞(LAMA-84和CD34+细胞),并且然后用(a)抗CD41-FITC(GpIIb/IIIa;BD生物科技公司,圣何塞,加利福尼亚州,美国)或抗CD71-FITC或(b)抗CD33-FITC、抗CD41-PE、抗CD45-PerCp和CD34-APC(贝克曼库尔特商贸公司,富勒顿,加利福尼亚州,美国)进行标记,并且针对产生的CD41细胞的百分比进行分析。为了测定倍性的数量,将分化的LAMA-84细胞在75%乙醇中在4℃下固定过夜并用碘化丙啶(PI,50μg/ml)标记并使用FACScalibur(BD公司)进行分析,而在通过定量每细胞核的数量和用该着色剂的MK的特定形态的May-Grunwald/吉姆萨染色后在显微镜下定量地分析第14天分化的CD34+细胞。只分析具有MK形态的细胞。在细胞离心涂片制剂中多核细胞的存在指示多倍体MK的存在。通过形态学针对多核成熟MK的存在评估分化的CD34+细胞。实例45:评估培养的血小板的分化-qPCR可以通过定量PCR评估培养物中血小板的分化状态。提取血小板RNA以进一步表征培养的细胞。使用TRIzol试剂(英杰公司)提取总RNA。通过血小板(GPIIIa)和白细胞(CD45)标记物的PCR分析评估每种血小板制剂的纯度。在进一步分析之前,使用生物分析器2100(安捷伦)评估血小板RNA的完整性。从细胞裂解物中收集总RNA,并使用商业合成试剂盒(Clontech公司)产生cDNA文库。用Quant-iTPicoGreendsDNA试剂盒(英杰公司)对标记的cDNA进行定量,并稀释至3pM以加载到单个泳道中并在Illumina1G基因组分析仪(Solexa公司)上进行测序。通过连续质量控制标准过滤原始序列。首先,验证至少6nt的3’Solexa衔接子的存在。不符合该标准的序列读数被丢弃,而修剪其他序列以去除在3’末端包含的衔接子序列。关于其长度(>10nt),拷贝数(>4次读取)和可读性(<9个未鉴定的核苷酸,注释N),将剩余标签进一步过滤。随后将符合所有这些标准的读取定义为可用读取。所有可用的读取与从miRBase数据库提取的前微小RNA进行比对。与不止一个前体完全匹配的序列标签在它们之间平均分布。为了解释Drosha和Dicer不完全切割,与成熟微小RNA区域中的前微小RNA完全匹配的任何序列标签(与参照成熟微小RNA位置相比允许高达4nt移位)被认为是成熟微小RNA。考虑到小RNA的总数是不变的,微小RNA表达水平定义为映射标准化为可用次数的每个成熟微小RNA的读取次数。每个微小RNA的相对丰度定义为与映射成熟微小RNA的总读取数相比,映射每个微小RNA的读取次数。实例46:通过离心纯化可以纯化培养的细胞部分,并且通过离心从核和污染的交替密度细胞类型中分离。将细胞以200g离心15分钟以分离红细胞和富含网织红细胞的部分。移液管吸掉上清液,并且然后将所需的细胞部分在改良的Tyrode缓冲液(包含138mMNaCl、5.5mM葡萄糖、12mMNaHCO3、0.8mMCaCl2、0.4mMMgCl2、2.9mMKCl2、0.36mMNa2HPO4和20mMHepes,pH7.4)中,在1μM前列腺素I2存在下进行洗涤,并重新悬浮于相同的缓冲液中。实例47:通过化学去核纯化培养细胞的去核可以通过培养物的化学添加剂来刺激,这可以帮助在纯化之前增加细胞的去核部分。如本文所述培养类红细胞。在收集前48小时,将细胞与210mMMe2SO一起孵育。然后通过在室温下以350Xg离心5min收集细胞,以3X105个细胞/ml的水平重新悬浮于包含210mMMe2SO和5ug/mL的细胞松弛素B(或其他肌动蛋白或核操纵分子,即p38MAPK,补骨脂素)的新鲜介质中,并在37℃下孵育。如本文所述,通过流式细胞术,使用DRAQ5作为核酸着色剂和抗血型糖蛋白A的抗体作为分化的红细胞表面标志物来评估无核细胞的比例。实例48:通过声泳进行纯化可以使用若干种机械分离系统来获得均匀的细胞群。自由流动声泳代表一种机械分离方法(彼得森(Petersson)2007,美国化学学会(AmericanChemicalSociety))。当悬浮在具有营养添加剂(包括CsCl(0.22g/mL))的盐溶液(0.9mg/mL)中时,添加到盐溶液中。使用具有两个主动出口(流率为0.10mL/min/出口)的声泳芯片(Cell-Care)处理包含培养的类红细胞的样品悬浮液。注射泵(WPISP260P,世界精密仪器公司(WorldPrecisionInstrumentsInc.),萨拉索塔,佛罗里达州)用于控制芯片中的流速。所有出口通过注入器使用特氟龙管单独连接到高精度玻璃注射器(1005TLL和1010TLL,哈美顿博纳图斯股份公司(HamiltonBonaduzAG),博纳杜茨,瑞士),允许独立控制出口流速。清洁流体入口连接到注射泵,并且用浸没在烧杯中的其另一端将细胞悬浮液入口连接到特氟隆的50mm长(0.3-mmi.d.)片,样品悬浮液以一定速率从该烧杯中吸出,该速率由净出口流量和净流体入口流量之间的差定义。用于在分离通道的壁之间诱导驻波的超声是使用附接到芯片背面的20×20mm压电陶瓷(Pz26,FerropermPiezoceramics公司,Kvistgard,丹麦)产生的。超声凝胶(AquasonicClear,帕克实验室公司(ParkerLaboratoriesInc.),费尔菲尔德,新泽西州)确保两者之间的良好的声耦合。压电陶瓷通过连接到函数发生器(HP3325A,惠普公司(Hewlett-PackardInc.),帕洛阿尔托,加利福尼亚州)的功率放大器(型号75A250,放大器研究,索德顿,宾夕法尼亚州)致动。即使声波从背面进入芯片,但由于沿着晶体结构的三个轴的机械振动的耦合,在分离通道的侧壁之间诱发驻波。使用标准显微镜和瓦特计(43ThrulineWattmeter,BirdElectronic公司,克里夫兰,俄亥俄州)监测分离过程。随后可以通过调节信号频率,致动功率和流速来控制该过程。使用库尔特计数器(Multisizer3,贝克曼库尔特公司(BeckmanCoulterInc.),富勒顿,加利福尼亚州)分析样品中的细胞大小分布。将每个样品与电解质(IsotonII,贝克曼库尔特公司)混合,并使用100-um孔径进行分析。使用光度计(等离子/低HB光度计,HemoCueAB,Angelholm,瑞典)测量溶血水平,即来自损伤的红细胞的游离血红蛋白的浓度。实例49:通过离体成熟纯化不完全成熟的类红细胞可以在模拟自然体内成熟触发物的系统中通过离体孵育来驱动。1.与基质细胞共培养在培养的最后阶段,在没有细胞因子的新鲜介质中,在粘附基质层上培养类红细胞。在37℃下在5%CO2的空气中保持培养物。粘附细胞层,在补充有10%胎牛血清的RPMI(英杰公司)中,由MS-5基质细胞系或从整个正常成人骨髓建立的间充质基质细胞(MSC)组成(参见,珀克普(Prockop),DJ(1997),科学(Science)276:71)。在用于共培养之前,通过至少两个连续传代扩增和纯化粘附的MSC。2.在纤连蛋白包被的板中培养在培养的最后阶段,在吸附有人纤连蛋白的平板中培养类红细胞。为了产生这些板,用1mL无菌H2O/mg蛋白质重建纤连蛋白(西格玛奥德里奇),并允许其在37℃下溶解至少30分钟。可能残留少量未溶解的材料。这不会影响产品性能。纤连蛋白溶液在无菌平衡盐溶液中稀释100倍,并以最小体积添加到培养表面。允许培养表面在室温下风干至少45分钟。通过抽吸除去过量的纤连蛋白。实例50:通过磁导入进行纯化通过磁导入分离、富集和/或纯化类红细胞的策略是本领域已知的,参见,例如,(Zborowski)等人,2003,生物物理学杂志(Biophys)J84(4)2638和金与查尔斯(Jin&Chalmers),2012,公共科学图书馆期刊(PLOSOne)20127(8):e39491。使用商业磁分离系统(QuadroMACSTM分离器,组合四个MidiMACSTM分离单元和LD柱,美天旎生物技术公司,奥本,加利福尼亚州)用于从HSC衍生的红细胞培养物的磁性红细胞富集。在填充有氮气(MedipureTM氮气,浓度>99%,普莱克斯公司(Praxair,Inc.),丹伯里,康涅狄格州)的Glove-BagTM可充气手套室(科尔帕默公司(ColeParmer),弗农希尔斯,伊利诺伊州)中对细胞脱氧。在脱氧之前,将包括分离系统、脱气的无菌缓冲液(PBS+2mMEDTA+0.5%BSA)和无菌收集管的所有材料和设备置于手套袋中,然后将其密封。将脱氧培养物直接加载到LD柱中,将该柱置于在填充有N2气体的可充气手套室内保持在缺氧条件下的QuadroMACSTM分离器中。通过包含在磁体内的柱的细胞被标记为阴性部分,并且预期它们在最终成熟之前是“非磁性”的,包括HSC和类红细胞。保留在分离柱中的细胞被标记为阳性部分,其是“磁性的”并且由几乎富有功能性血红蛋白的成熟的RBC样细胞组成。它们在从磁体移除之后从LD柱中洗脱。一旦分离完成,通过将收集的细胞暴露于空气来可逆地回收含氧细胞。实例51:通过FACS纯化使用BD公司AriaIiu细胞分选仪分选红细胞培养的细胞群。在分选之前,收集细胞,用PBS洗涤,并以制造商推荐的稀释度用针对血型糖蛋白A(生命技术公司)的荧光抗体和核酸着色剂DRAQ5(Pierce)进行染色。以28,000滴/秒的液滴驱动频率使用100μm喷嘴。样本阈值速率约为4000事件/秒。温度控制选项用于在整个分选过程中将样品和收集管保持在4℃。此外,在200rpm下使用样品搅拌特征,以防止样品在整个分选中沉降。将样品在从注射器分配的约750μl的等分试样中进行分选。同时,在这些暂停期间,收集管保持在4℃下,避光,并且在恢复分选之前轻轻混合。将分选的样品收集到包含补充有10%FCS的250μlDMEM的12×75mm硼硅酸盐玻璃收集管中。实例51:通过酶促处理细胞进行纯化同种异体红细胞源可以从A和B抗原去除中获益以产生普遍兼容的产物。这可以通过能够选择性切割半乳糖基团的一组酶来促进,使得类红细胞在神经学上更有利。使用标准克隆方法在大肠杆菌BL-21中产生两种类型的内切-β-半乳糖苷酶的重组蛋白,这些重组蛋白最初从产气荚膜梭菌中鉴定。制备用于释放A/BAg和内切-β-半乳糖苷酶C(EndoGalC)以释放Galα1-3Galβ1-4GlcNAc(GalAg)的ABase,其已知在异种移植中是具有高度免疫原性的,并且具有类似于A/BAg的碳水化合物结构。ABase在血型A[GalNAcα1-3(Fucα1-2)Galβ1-4GlcNAc]和血型B[Galα1-3(Fucα1-2)Galβ1-4GlcNAc]中切割Galβ1-4GlcNAc键。简言之,在克隆ABase后,在没有信号肽的pET-15b载体eabC中构建具有C末端His标签的表达质粒。将该外源基因转化到大肠杆菌BL-21细胞中。在镍-氨三乙酸柱(凯杰有限公司(QIAGENGmbH),希尔登,德国)上纯化作为可溶性蛋白部分的细胞中产生的酶。最后,获得5mL的纯化的重组ABase,其浓度为3.6mg/mL,比活性为1500U/mg。一个单位的酶活性定义为水解1μmol底物/min所需的酶量。检查ABase处理对Ag存在,Ab结合和补体激活的影响。将人A/BRBC用ABase进行消化,并在与交叉反应性(包含抗A或抗B或抗A和B;分别为B型,A型或O型)人血清一起孵育后进行流式细胞术分析。使用平均荧光强度(MFI)定量血型A、B和GalAg的表达水平。消化水平表示为在不存在ABase的情况下孵育后在RBC上表达的血型A或BAg的百分比。从三个健康人志愿者收集新鲜血型O血清,并在-80℃下冷冻以保持内源补体活性直到使用。将热灭活(在56℃下30min)血清用于Ab结合的分析。具有和不具有酶(ABase)消化的RBC与在37℃下用包含0.2%牛血清白蛋白(PBS/BSA)的磷酸盐缓冲盐水稀释的50%血液O型血清(100μL)孵育30分钟。洗涤后,RBC与FITC标记的抗人IgG/IgM(达科公司(DAKO),格洛斯楚普,丹麦)(×30,100μL)在4℃下反应30分钟,并且然后进行流式细胞术分析。酶处理对补体激活的抑制作用也通过C3d沉积的变化来评估。在补体活性存在下,将RBC与50%人血清在37℃下孵育15min后,在4℃下将RBC与FITC标记的兔抗人C3dAb(达科公司,格洛斯楚普,丹麦)(×100,100μL)反应30min,并且然后应用于流式细胞术分析。基于MFI计算对照水平(在酶不存在下)的百分比,以评估酶处理对Ab结合和C3d沉积的抑制作用。实例52:通过离心纯化血小板可以通过离心从混合的细胞悬浮液中纯化血小板。将约40ml全血分布在血液收集管中,用3.2%的柠檬酸钠作为抗凝血剂。将管以400×g离心10min。在该阶段后,三个层被清楚地分界:血浆、血红细胞和中间区。该血浆在顶部具有血小板,血红细胞因为它们较重的密度处于底部;并且细的发白的中间区由较大的血小板和白细胞组成,并且称为血沉棕黄层。使用Jelco18G针,抽出具有血小板的血浆的上部,并将血沉棕黄层置于其他两个管中,这次没有添加剂:一个管产生血浆(P管),另一个产生凝血酶(T管)。仅使用1.5ml血浆来产生凝血酶,向其中添加10%的0.5ml的葡萄糖酸钙,在37℃下的双锅炉中15min。然后将两个管再次离心,这次以800×g离心相同的时间长度(T=10min)。在最终离心后,T管包含富含凝血酶的液体,而P管包含血小板沉淀和一些血红细胞(红细胞-血小板结块)。在该阶段通过去除总血浆体积的三分之二来减小体积。去除的部分是血小板缺乏的,而具有沉降的血小板(容易通过搅拌分散)的剩余部分是富含血小板的。实例53:胸苷掺入可以使用本领域已知的胸苷掺入测定来评估细胞群的自复制潜力,参见,例如,哈科年(Harkonen)等人,1991,实验细胞研究(ExpCellRes),186L288,和田中(Tanaka)等人,1992PNAS89:8928。简言之,从马泰克生物科学公司(MartekBiosciences)(哥伦比亚,马里兰州)获得均匀的富含13C-和15N-的胸苷[U-13C,15N-TdR],并且从ICN放射化学(欧文(Irvine),加利福尼亚州)购买3H-TdR(80Ci/mmol)。介质和缓冲液获自飞世尔科技公司(匹兹堡,宾夕法尼亚州)。除磷酸二酯酶外的所有酶都来自宝灵曼公司(BoehringerMannheim)(印第安纳波利斯,印第安纳州)。磷酸二酯酶II获得自沃辛顿生化有限公司(WorthingtonBiochemicalCorporation)(莱克伍德,新泽西州)。高效液相色谱(HPLC)溶剂来自EM科学公司(EMScience)(吉布斯敦,新泽西州),并且包含<0.1ppm蒸发残余物。如本文所述培养类红细胞。培养后,收集细胞用于胸苷掺入测定。将细胞用1.6μg/ml的[U-13C,15N]-TdR标记18小时,添加未富集的胸苷以达到1μM的最终胸苷浓度。在用磷酸盐缓冲盐水洗涤细胞后,在指定浓度(0.1-10μCi/ml)下添加3H-TdR再培养18小时前,在补加的DMEM中将细胞培养6小时以上。向样品中添加未标记的胸苷,使最终的胸苷浓度达到0.13μM,这相当于接受10μCi放射性标记/ml的样品中3H-TdR的浓度。除去3H-TdR后,在分离DNA前,将细胞在补充的DMEM中孵育另外6-54h。使用修饰的纯基因DNA分离试剂盒(GentraSystems公司,明尼阿波里斯,明尼苏达州)提取DNA。基于样品中的细胞数量,使用放大/缩小程序来确定添加的试剂体积。例如,当使用1×10^7个细胞时,将包含328μg蛋白酶K的21μl添加到3ml细胞裂解溶液中。混合后,将样品在室温下放置过夜。第二天,添加10μgRNA酶并将样品混合并且在37℃下孵育2h。添加蛋白质沉淀溶液(1ml),将样品在冰上孵育5min。以2000g离心10min后,将包含DNA的上清液与3ml100%2-丙醇混合,并且轻轻倒置50次,或直到可以看到白色的DNA线。然后将样品以2000g离心5min。在3ml的70%乙醇中洗涤之前,将所得DNA沉淀干燥5min,并以2000g再离心5min。将最终沉淀物进行空气干燥,并且然后在去离子H2O中再水化,并且在260nm下通过吸水性进行定量。相同的程序应用于CD34+干细胞作为复制能力的对照。任何DNA通过煮沸3分钟来变性,然后在冰上快速冷却。酶水解程序以0.5mg/ml的DNA浓度进行。以下方案描述每毫升DNA溶液中添加的试剂体积。在45℃下,在10μl包含200mMMgCl2、100mMZnCl2、和1MTris(pH7.2)的缓冲液中,用10μl核酸酶P1(0.5U/μl)和5μlDNA酶I(4U/μl)水解DNA持续2h,随后添加20μl磷酸二酯酶(4mU/μl)并在37℃下进一步孵育2h。最后,添加5μl的10M乙酸铵(pH9.0)和10μl碱性磷酸酶(1U/μl),并且将样品在37℃下孵育另一个2h。消化的DNA样品用0.22-μm尼龙滤器进行过滤。使用4.6×250mmSupelcosilLC-18-SHPLC柱(色谱科(Supelco),贝尔丰特,宾夕法尼亚州),用HPLC/CRI/IRMS系统分析该样品。以1ml/min使用相同的溶剂系统,并且在15分钟内线性梯度为5%至25%B。通过HPLC分离后,使用化学反应界面质谱(CRIMS)分析脱氧核苷。在该过程中,脱氧核苷流入由氦气流驱动的雾化和去溶剂化系统,其中它们作为干燥粒子束出现。使用Finnigan/MATδS同位素比质谱仪(ThermoFinnigan公司,圣何塞,加利福尼亚州)及其伴随的Isodat数据系统确定来自该在线产生的CO2的13CO2/12CO2丰度。5-氟脱氧尿苷(西格玛)用作内部同位素比率标准。从每个样品:T,dA和dG获得三个核苷酸的同位素比率(以下方程中的IR)。从每个DNA衍生的脱氧核苷放出的CO2的富集通过等式(13)CO2(每毫升)=1000×(IR实验-IR标准)/IR标准来计算。为了保持最高水平的内部一致性并避免任何实验间漂移,在所有实验中,从T的同位素比率中减去dG的同位素比率。评估从稳定同位素标记期的结束(第0天)至清除结束(第3天)的数据。实例54:核材料的定量使用DNA着色剂DRAQ5(Pierce)通过流式细胞术评估包含DNA的混合群体中的细胞数量。根据制造商的说明书,将细胞与着色剂一起孵育,并且在流式细胞仪(Attune细胞计数器)(生命科技公司)上进行分析。量化高于核材料含量的预定阈值的细胞的百分比。实例55:体外致肿瘤性测定为了评估细胞的复制潜力,可以进行软琼脂集落形成测定。简言之,通过制备0.5%Agar+1xRPMI+10%FCS溶液来制备基本琼脂层,所有组分加热至40℃,并将1.5mL溶液添加至35mm培养皿中。在使用前,允许琼脂在室温下固化30min。通过在微波中熔化0.7%琼脂糖并冷却至40℃制备顶层琼脂糖层。将2xRPMI+20%FCS溶液加热至40℃。计数细胞并且制备用于以5000个细胞/板,以200,000个细胞/mL的密度接种。将0.1mL细胞悬浮液添加到10mL管中,随后添加3mL温热的0.7%琼脂糖和3mL温热的RPMI/FCS溶液。通过涡旋轻轻混合该溶液,并且向三个或四个重复的基本琼脂板中的每一个添加(1.5mL)。将板在37℃下在潮湿的培养箱中孵育10-30天。每周用细胞培养基(0.75mL/板)给细胞进料1-2次。为了评估集落形成,将板用0.5mL的0.005%结晶紫染色>1hr。使用解剖显微镜计数菌落。实例56:体内致肿瘤性测定将终末分化的培养的类红细胞植入各种动物模型中以评估致瘤性的可能性。从各种模型收集若干种组织,并且用组织学,免疫化学和荧光测定法分析以定量致肿瘤性。动物在移植前两天开始每天接受腹膜内注射CsA(10mg/kg,山地明,诺华制药(NovartisPharma),纽伦堡)。对于NK细胞的耗尽,除了CsA腹膜内注射单克隆抗体(mAb),一些大鼠接受了抗NKR-P1A(克隆10/78,小鼠IgG1,BD生物科技公司,海德堡,德国)或各自的同型对照(克隆PPV-06,小鼠IgG1,Exbio,布拉格、捷克共和国)。抗-NKR-P1AmAb(克隆10/78)针对与mAb相同的表位(克隆3.2.3)。在注射类红细胞前一天给予1mg各自的抗体,随后在细胞移植后第4天给予0.5mg。在开始这些实验之前,在类红细胞移植后第0天和第4天以及在尸体解剖时(第92天)采集血液样品,以便通过流式细胞术确定血液中NK细胞的比例。对于皮下肿瘤生长的分析,将在100μl磷酸盐缓冲盐水(PBS)中的类红细胞注入至动物的侧腹。通过触诊每隔一天监测肿瘤生长,并使用线性测径器记录大小。在第100天之前,当肿瘤体积达到小鼠中1cm3和大鼠中5cm3的时,当发生大于10%的重量损失时,或者当可观察到任何疼痛或痛苦的行为迹象时,处死动物。对所有动物进行尸体解剖。在注射部位附近的小鼠组织立即在液氮中冷冻或置于磷酸盐缓冲的4%福尔马林中16h,并且然后包埋在石蜡中。取出脾脏和淋巴结用于随后的免疫分析。进行类红细胞移植到单侧6-OHDA损伤的大鼠的纹状体中。在移植后6周处死这些动物。通过流式细胞术分析动物组织。添加针对已建立的CD133、CD3、CD、CD16、CD19、CD20、CD56、CD44、CD24、和CD133的癌细胞生物标志物的合适的荧光和PE-共轭的抗体到切下的组织样品中,并且进行分析以定量致瘤潜力。实例57:通过EKTA的变形性通过细胞计数术,针对变形性特征,相对于天然红细胞样品评估如本文所述培养的类红细胞。血细胞计数器由与氦氖激光组合的Couette型粘度计组成,该激光用于产生悬浮在两个圆柱体之间的粘性流体中的红细胞的衍射图像。当粘度计旋转时,正常红细胞在剪切场中伸长,导致衍射图像变为椭圆形。通过对沿衍射图案的主(A)轴和次(B)轴的光强度进行定量,并将其表达为比率(A-B)/(A+B),变形性指数(DI)或伸长指数(EI),测量图像的椭圆率。选择介质的粘度大于最密类红细胞的内部粘度。31g/升聚乙烯吡咯烷酮(PVP)溶液,mw=360,000,在由K2HP04和KH2P04组成的0.04M的磷酸盐缓冲液中,在蒸馏水中,在25℃下产生0.20泊的粘度并且在37℃下产生12泊的粘度。用NaCl将渗透压调节至所希望的水平,并在Roebling冰点渗透压计中测量。通过使用少量添加1-M的NaOH和HCl溶液来改变最终pH,并在TechniconBGI1血气分析仪中进行测量。将叠氮化钠作为防腐剂添加至储备溶液中以获得0.4g/l。该血细胞计数器从红细胞样品收集三个主要指标,并且将其与原生红细胞进行比较;渗透压最小值(Omin),变形性指数(Dimax)和DI达到其最大值(Ohyp)的一半时的渗透压。Omin与细胞的表面积对体积比有关,并且已经发现其等于经典渗透脆性测试中的50%溶血点。Dimax是变形性指数的最大值,通常达到290mosmol(生理渗透压值)。这表明细胞在剪应力下的最大变形性,并且与许多因素有关,如表面积、体积、内部粘度和细胞膜的机械性能。Ohyp是DI达到其最大值的一半时的渗透压。这给出了曲线的高渗部分的位置的指示,其与细胞的内部粘度以及膜的机械性质相关,例如其将如何在力(刚度)下弯曲。将针对培养的类红细胞获得的参数与针对原代红细胞的相同值进行比较。实例58:通过LORCA的变形性通过激光衍射技术(LORCA,激光辅助旋光细胞分析仪,R&RMechatronics)测量纯化的cRBC群的变形性。简言之,将高度稀释的细胞悬浮液在Couette系统中剪切,在2个圆柱之间具有0.3mm的间隙,其中一个能够旋转以诱导剪切应力。使激光束通过悬浮液,并在37℃下测量衍射图案。在低剪切应力下,细胞是圆盘,而在高剪切应力下,细胞变成椭圆形。细胞变形性用伸长指数(EI)表示,伸长指数(EI)取决于变形细胞的椭圆率。将包含12.5uL沉淀的RBC颗粒的等分试样在5mL聚乙烯吡咯烷酮溶液(分子量360000)中稀释。选择在30Pa(称为EImax)和3Pa下的EI值作为变形性的代表值,以便在各种剪切应力下容易地在样品之间进行比较。实例59:血管闭塞的评估-离体大鼠脉管系统可以使用本领域已知的方法,用分离的人工灌注的大鼠血管系统评估类红细胞的血管闭塞的可能性,参见,例如,考尔(Kaul)等人,1983,临床研究杂志(JClinInvest)72:22。简言之,在Wistar系的麻醉的(戊巴比妥钠30mg/kg)大鼠(120-150g)中,用肝素化(100uL/mL)硅橡胶管在距离回结肠连接的3cm处插管右回肠动脉和静脉。在使用包含1%牛血清白蛋白的林格氏液的稳态灌注下,在结扎之间切割升结肠和末端回肠(每个3cm)。在实现所有血管连接的止血结之后,隔离组织。将分离的中阑尾系膜轻轻地铺展在显微镜载物台上的光学透明的Lucite块上。除了插管和显微镜物镜的出口,整个制备物用塑料偏氯伦包装膜包裹。对照动脉灌注压力(Ppa)和静脉出口压力(Pv)分别保持恒定在80和3.8mmHg,并通过Statham-GouldP-50压力传感器(Stathan仪器公司,奥克斯纳德,加利福尼亚州)监测。使用光电滴计数器监测静脉流出(Fv)速率,并以mL/min来表示。允许10-12min的间隔用于组织平衡和Fv的稳定化。仅使用显示阑尾系膜微脉管系统不含宿主血细胞并具有4.6+/-0.5(平均值+/-SD)的稳定Fv的制剂。实验在37℃下进行。如本文所述分离类红细胞。在Ppa和Fv的对照测量后,通过在动脉插管部位远端15cm处的注射端口轻轻地递送红细胞(0.2mL,Hct30%),并且Ppa和Fv的变化记录在草多道记录仪(Grass仪器有限公司,昆西,马萨诸塞州)的带状记录纸上。在用林格氏溶液输注样品之前,灌注组织制备物10-15min,以允许组织的稳定化并且清除宿主动物的剩余血细胞的脉管系统。通过在高压(100mmHg)下用完全氧合的林格氏溶液短暂(2-3分钟)灌注脉管系统,可以清除输注细胞后产生的阻塞,并且恢复血流。在每个实验结束时,称重整个组织制备物(不含插管和腔内容物)。计算外周阻力单位(PRU),并表示为PRU=ΔP/Q=mmHg/mL/(min-g),其中ΔP(mmHg)是动静脉压力差,并且Q(mL/min-g)是是每克组织的静脉流出速率。在每个实验中,在样品的推注输注之后确定压力-流动恢复时间(Tpf)。Tpf定义为Ppa和Fv在递送给定样品后返回到其基线水平所需的时间(秒),并且其表示贯穿阑尾系膜脉管系统的总传输时间。将针对培养的类红细胞获得的参数值与针对原代类红细胞获得的值进行比较。实例60:血管闭塞的评估-体外流动小室使用体外渐变高度流动室评估类红细胞血管闭塞的方法是本领域已知的,参见,例如,泽娜迪等人,2004,血液,104(12):3774。简言之,使用渐变高度流动室来定量类红细胞与内皮细胞(EC)的粘附。将用涂覆有EC的载玻片用预先加热至37℃的具有1.26mMCa2+,0.9mMMg2+(Gibco公司,格兰德岛,纽约州)的Hanks平衡盐溶液(HBSS)洗涤,并且然后装入可变高度流动室中。流动室安装在连接到设置为37℃的热板(东海希多公司(TokaiHit),富士宫市,日本)的反相相差显微镜(Diaphot;尼康公司(Nikon),梅尔维尔,纽约州)上。使用附接到显微镜并连接到MacintoshG4计算机(苹果公司(Apple),库比蒂诺,加利福尼亚州)的摄像机(RSPhotometrics公司,图森,亚利桑那州)观察细胞。如本文所述培养类红细胞,并遵循制造商的说明书,用荧光染料PKH26红色荧光细胞连接子试剂盒(西格玛)进行标记。将悬浮于具有Ca2+、Mg2+的0.2%(体积/体积)的HBSS中的细胞(3mL)注入流动室中,并允许其在没有流动的情况下粘附于载玻片15分钟。在暴露于流动之前,针对荧光细胞的总数,检查沿着垂直于未来流动定向的线的7个不同位置中的每一个的最少3个视野。然后使用校准的注射泵开始流体流动(具有Ca2+、Mg2+的HBSS)。暴露于流动后,再次检查视野并计数粘附细胞的数量。粘附细胞的部分如下:暴露于流体后附着的细胞数量/流动前每个视野存在的细胞。壁面切应力计算如下:τw=(6μQ)/(wH[x]2),其中τw表示壁面切应力(达因/cm2);Q,体积流率(cm3/s);μ,介质粘度;w,流动通道的宽度;和H(x),作为沿着显微镜载片的位置的函数的流动室的高度。实例61:评估血管闭塞-活体镜检法使用活体镜检法评估类红细胞血管闭塞的方法是本领域已知的,参见,例如,泽娜迪等人,2007,血液110(7):2708。简言之,通过腹膜内注射100mg/kg氯胺酮(雅培实验室,芝加哥,伊利诺州)和10mg/kg甲苯噻嗪(拜耳司(Bayer),尼米逊,堪萨斯州)实现测试动物的全身麻醉。在无菌条件下使用层流罩,将双侧钛框架窗室经手术植入背部皮肤褶皱中。手术涉及小心地去除背部皮肤褶皱一侧的表皮和真皮层,暴露与相对的皮肤褶皱的横纹肌相邻的皮下组织的血管,并且然后使用不锈钢螺钉和缝线将室的两侧固定到皮肤上。将玻璃窗放置在室中以覆盖暴露的组织并用扣环固定。随后,将动物保持在32℃至34℃下,直到在手术后3天进行体内研究。将具有窗室的麻醉动物放置在Axoplan显微镜(卡尔蔡司公司(CarlZeiss),索恩伍德,纽约州)的台上;使用恒温控制的加热垫将温度保持在37℃。所有输注都通过背尾静脉。如本文所述培养类红细胞。然后根据制造商的说明书,用Dil或DiO(分子探针公司(MolecularProbes),尤金,俄勒冈州)染料标记细胞。注入标记的细胞(300μL;在具有Ca2+和Mg2+的PBS中的血细胞比容[Hct]0.50[50%]),并且使用LDAchroplan20×/0.40Korr和Fluar5×/0.25物镜在皮下血管中观察RBC粘附和血流动力学至少30分钟。使用连接到数字摄像机C2400(松光子学公司(HamamatsuPhotonicsKk),滨松市,日本)的Trinitron彩色视频监视器(PVM-1353MD;索尼(Sony),东京,日本)和JVC盒式磁带录像器(BR-S3784;VCRKing公司,达拉谟,北卡罗来纳州),同时记录微循环事件和细胞粘附。对于每组条件检查三十节的小静脉。基于以下各项,心房不同于小静脉:(1)观察到发散流而不是收敛流;(2)使用透照法的血管壁的双折射外观,其是动脉血管平滑肌的特征;和(3)没有曲折证据的相对直的血管轨迹。通过检查使用×20放大率产生的录像带来进行红细胞通量和粘附的测量。通过考虑附着于血管壁上并静止1分钟的细胞来定量细胞粘附。由SSRBC占据的直径高达25μm或大于25μm的血管的长度的百分比定量如下:SSRBC占据的小静脉长度%=(具有粘附细胞的血管壁的长度/所分析的血管段的总长度)×100。RBC通量的变化计算如下:通量=穿过在每分钟直径小于50μm的血管上标记的单个点的循环荧光人RBC的数目。实例62:血管闭塞的评估-血小板使用人血管内皮细胞(HUVEC)评估血小板的血管闭塞的方法可以从用于针对红细胞的类似方法改变。简言之,将2-mL体积的0.05%血细胞比容悬浮液添加到组织培养皿上的汇合的HUVEC中。将锥板装置在添加血小板后1分钟内进行组装,并置于尼康Diaphot-TMD反相相差显微镜(南方微仪器公司(SouthernMicroInstruments),亚特兰大,乔治亚州)上。启动电机以转动锥体,并在0.1或1达因/cm2剪切应力下连续监测粘附性30min。通过吹在粘附装置上的气幕培养箱(尼科尔森精密仪器公司(NicholsonPrecisionInstruments,Inc.),贝塞斯达,马里兰州)将温度保持恒定在37℃。通过针对每个时间点,聚焦在8个不同视野20sec/视野,每5min将血小板粘附进行可视化和记录。通过CCD-72系列相机(Dage-MTI公司,密歇根市,印第安纳州)在400×总放大率下观察整个实验,并用SVO2000盒式磁带录像器(索尼电子公司(SonyElectronics),圣何塞,加利福尼亚州)记录在录像带上。在每个实验结束时通过在手动回放记录的视频图像期间对单个粘附细胞计数来离线定量粘附。将每个时间点的8个视野中的细胞计数取平均并且归一化为每平方毫米内皮的粘附红细胞。实例63:用共振器评估质量/体积/密度基于布赖恩(Bryan)等人,芯片实验室(LabChip),2014,使用双悬浮微通道共振器(SMR)系统表征终末分化的类红细胞群的质量、体积和密度。在细胞密度测量开始时,系统首先用作为高密度流体的过滤的珀可介质冲洗。接下来,用稀释的细胞样品填充样品旁路,并且调节样品入口和出口处的管形瓶高度以引导流体流入第一SMR。高密度流体入口处的压力用于设定流体2的密度,并且废物出口处的压力控制装置中的总流速。为了最小化由于较重的细胞沉降在样品瓶或管的底部而引起的尺寸偏差的可能性,通过冲洗样品旁路通道以定期引入新鲜样品。通过LabVIEW获取数据并且使用MATLAB进行处理。使用Coulter计数器监测细胞浓度。在生长至5×10^5-1×10^6个细胞/ml的培养物上进行细胞测量。在第二SMR中引入用于测量的高密度流体被配制为50%(v/v)珀可(西格玛),1.38%(w/v)粉末的L15介质(西格玛),0.4%(w/v)葡萄糖,100IU青霉素和100μgmL-1链霉素。将培养基pH调节至7.2。将该珀可介质在4℃下储存,并且在用于双重SMR之前立即过滤。实例64:通过膜联蛋白V评估磷脂酰丝氨酸含量如本文所述培养类红细胞。将50μL细胞悬浮液在包含5mMCaCl2的林格氏溶液中洗涤,然后在37℃下在避光条件下用膜联蛋白-V-FITC(1:200稀释;免疫工具(ImmunoTools),弗里索伊特(Friesoythe),德国)在该溶液中染色20min。如本文所述,洗涤细胞并通过进行流式细胞术染色,并且使用488nm的激发波长和530nm的发射波长测量膜联蛋白-V荧光强度。从膜联蛋白-V荧光评估相对磷脂酰丝氨酸暴露。实例65:通过色谱法评估脂质含量在抗氧化剂BHT(西格玛奥德里奇)存在下,通过在室温下用甲醇-氯仿1:1进行三次提取,从洗涤的表达外源抗原的EHC中提取脂质。在福尔奇(Folch)、利斯(Lees)和斯隆斯坦利(SloaneStanley)1957,生物化学杂志(JBiolChem)226:497的方法中用0.05MKCl洗涤合并的提取物。简言之,针对第一次提取,在离心管中将包含0.05mg/mLBHT的15mL甲醇添加到洗涤的复合物中,并且允许静置30min,偶尔搅拌以破碎沉淀物。然后添加15mL氯仿,并且允许混合物静置30min,偶尔搅拌以打碎团块。将管在1500g下离心5分钟,并且将上清液部分倒入配有特氟龙活塞的分离漏斗中。类似地,除了提取物仅静置10分钟,每次添加后偶尔搅拌以外,向残余物中添加15mL甲醇-BHT,然后添加15mL氯仿,进行第二次和第三次提取。离心后,将上清液部分汇集在分液漏斗中,然后添加并且混合48mL的氯仿和28mL的0.05MKCl。允许混合物在4℃的黑暗中静置过夜以进行相分离。在升温至室温后,收集两个澄清相中的较低相,并且在40℃下在旋转真空蒸发器中真空蒸发至干。将脂质用氯仿定量转移至10mL容量瓶中并储存在-22℃下。如下确定脂质提取物中游离胆固醇的浓度。将脂质提取物在在己烷-二乙醚-冰醋酸80:20:1中的0.5mm硅胶HR(Brinkmann仪器公司,韦斯特伯里,纽约州)层上进行层析,通过用2,7-二氯荧光素溶液喷雾对薄层色谱板进行染色(见下文),将游离胆固醇点刮到锥形离心管中,并且用2.0ml氯仿提取一次,并且用1.0ml氯仿提取三次,提取物在40℃下在旋转真空蒸发器中蒸发至干,并且通过没有皂化的曼(Mann),1961,临床化学(ClinChem),7:275的氯化铁方法估算胆固醇。将通过二溴化物衍生物分离从商业认证的试剂级材料制备的游离胆固醇标准品(参见,例如,费塞尔(Fieser),美国化学会志(JAmerChemSoc)195375:5421)通过层析分析程序得到,并且用每组确定来估算。在每次针对标准回收率的确定中校正游离胆固醇的值,平均值为95%。TLC对于除去BHT是必要的,否则其通过产生在560nm下吸收的棕色产物而干扰氯化铁方法。磷脂分布通过在4℃下在硅胶HR(0.5mm厚)的氯仿甲醇-冰乙酸-水(25:15:4:2)中的总脂质提取物的等分试样的TLC中一式三份来确定,向其中添加50mg/100ml浓度的BHT,以防止色层分析过程中的自动氧化;用水(“中性”板)制备TLC板。使用“楔尖技术”在板的原点处应用脂质样品(参见,例如,斯特尔(Stahl),1965,薄层层析(Thin-LayerChromatography),学术出版社有限公司(AcademicPressInc.))导致单个磷脂的优异分离。特别地,该方法提供了磷脂酰乙醇胺(PE)、磷脂酰丝氨酸(PS)、卵磷脂和鞘磷脂之间的完全分离;离散斑点在PS和被鉴定为磷脂酰肌醇(PI)的卵磷脂之间迁移。通过喷射2,7-二氯荧光素(33.3mg/100ml的2mMNaOH水溶液)的溶液并且然后直接刮到Kramer-Gittleman管中,使斑点在UV光下可见,其中磷脂在190℃下用1.0ml的70%高氯酸消化60min。除了在显色后之外,如上所述进行剩余的程序,通过以3000g离心5min除去硅胶并且在透明上清液溶液上测定吸光度。对空白泳道的相应区域的吸光度进行校正。气相-液相色谱法使用Barber-Colman仪器(型号5000)在己烷溶解的样品上进行,该Barber-Colman仪器装备有EGSS-X(与硅酮结合的丁二酸乙二醇酯聚酯)8%在Gas-ChromP(100-120目)(应用科学实验室公司(AppliedScienceLaboratoriesInc.))上的配对的8-ft柱,和双火焰离子化检测器。氮气流速在入口处为50ml/min。注射样品后,柱温度保持在165℃持续10min,然后以2℃/min升至200℃。实例66:膜粘度的评估细胞群的膜粘度可以通过荧光光漂白测定法来评估。收集0.5ml类红细胞样品,并在HEPES缓冲盐水(132mMNaCl、4.7mMKCl、2.0mMCaCl2、1.2mMMgSO4、20mMHEPES,调节至pH7.4)中洗涤一次。然后将填充的细胞在145mMNaCl-10mMNaHCO3(pH9.5)中洗涤一次,并且重悬于具有1mg/mlDTAF(获自研究有机物公司(ResearchOrganics),克利夫兰(Cleveland),俄亥俄州)的相同缓冲液中。将细胞在冰上孵育1h,然后在50mM甘氨酸-95mMNaCl-10mMNaHCO3(pH9.5)中洗涤两次,以除去任何不与蛋白质共价结合的染料。最后,将细胞洗涤两次,并且在具有1mg/ml牛血清白蛋白的HEPES缓冲盐水中重悬浮至-2%血细胞比容。应用相同的处理以控制天然红细胞。流动室安装在装备用于入射荧光显微镜的LeitzDiavert(罗克利,新泽西州)倒置显微镜的台上。分色镜和激发/发射滤光片是与荧光素染料(Leitz名称12)一起使用的标准组合,激发波长在450-490nm范围内。目标是具有100x放大率和1.25数值孔径的油浸式。具有适当电源和外壳(Oriel,斯坦福德,康涅狄格州)的100瓦高压水银弧光灯(欧司朗,慕尼黑)用作荧光激发源。计算机控制的电子快门(VincentAssociates公司,罗契斯特市,纽约州)限制曝光持续时间,并与用于测量荧光强度的光子计数电子系统同步。入射光照明器的视场光阑用于将激发限制到直径为20-40um的圆形区域。定期地,来自计算机的输出脉冲使快门打开持续典型的20ms的持续时间。来自短暂荧光图像的光被一系列棱镜分开,使得一半光被引导到低光水平SIT摄像机(型号66-SIT,Dage-MTI公司,密歇根市,印第安纳州),一半光到光电倍增管(型号8850,美国无线电公司(RCA),哈里森,纽约州)封闭在环境温度外壳中。在电子快门打开的时间期间,触发视频图像处理器(型号794,休斯飞机公司(HughesAircraft),卡尔斯巴德(Carlsbad),加利福尼亚州)以获取荧光图像,提供可以被监视以确保对象保持在焦点以及没有外物侵入视野的视频快照。用视频卡尺测量视频屏幕上的距离,并通过与台式千分尺的视频图像比较来校准。同样在快门打开的时间期间,将光电倍增管信号用光子计数技术进行处理。放大器/鉴别器(型号AD6,太平洋仪器公司(PacificInstruments),康科德,加利福尼亚州)针对高于给定幅度的每个信号脉冲产生数字逻辑脉冲,并且这些数字脉冲在100-MHz门控计数器(型号770,EG&GOrtec公司,橡树岭,田纳西州)上进行计数。微型计算机控制光子计数的门控,复位和记录。典型的实验由在激发光的短暂(20ms)脉冲期间进行的多个初步荧光测量,随后是样品细胞被漂白期间延长的辐射周期(通常为30s),随后是另一系列的短暂曝光,每15-30s,直到荧光似乎已经完成其恢复。将针对培养的类红细胞获得的恢复时间及其他参数值与针对原代类红细胞获得的相同值进行比较。实例67:用Advia血液学分析仪评估平均红细胞容积在Advia120血液分析仪(西门子医疗公司(SiemensHealthcare))中使用电阻抗测量培养的类红细胞的平均红细胞体积(MCV)。将结果与天然人红细胞的结果进行比较。实例68:培养的类红细胞的病原体测试使用RT-PCR来定量培养的类红细胞群中存在的外来病毒并且证实无污染(测定号003000.BSV,BioReliance公司)。通过将红细胞群直接接种到分别支持有氧和厌氧细菌生长的2种不同类型的介质中,进行未处理和最终大量,最终小瓶,预分枝细胞,和细胞和病毒库的无菌性测试。将样品孵育14天,然后按照BioReliance无菌测试方案USP71测试微生物污染物。实例69:渗透脆性的评估评估渗透脆性以测量当暴露于低渗溶液时类红细胞对裂解的抗性。NaCl在水中的溶液以跨越0%至1%的浓度下制备。将细胞在每种盐溶液中孵育15分钟。将样品离心以沉淀完整的细胞。通过使用分光光度计在540nm下吸收光,针对血红蛋白含量,来测定上清液。计算发生50%溶血的点,并且与原发性红细胞获得的值进行比较。实例70:玫瑰花结/免疫原性的评估直接抗球蛋白测试(也称为库姆斯测试)评估由多克隆抗体从血清与细胞上的表面抗原的结合引起的类红细胞凝集或复位。其可以用合并的人血清进行用于一般同种异体免疫原性评估,或用来自预期接受者的血清进行用于特异性免疫原性预测。简言之,将1-2滴储存在EDTA管中的细胞添加到反应管中。用等渗盐水洗涤该管三次。第三次洗涤后,从洗涤的细胞制备3%的悬浮液。标记2个管A和B。向每个管中添加一滴经洗涤的3%悬浮液。再次洗涤这些管。当倾析时,放置试管,这样使得细胞按钮在顶部。这将防止太多的细胞在洗涤过程中丢失。排干好,用生物擦拭干燥印迹。立即向两个管中添加一滴人类测试血清,并且摇动混合。允许B管在室温下孵育5分钟。离心A管持续针对库姆斯旋入式血清离心机校准的时间。立即轻轻再悬浮并且使用点亮的凝集观察器(BD公司)检测凝集。如果A管是阳性的,则不必读取B管,也不必在显微镜下检查A管。如果A管由点亮的凝集观察器为阴性,则在显微镜下检查凝集。如果通过显微镜读数A管为阴性,在其孵育期后离心B管,并且用B管样品重复步骤2-4。如果B管也是阴性,添加一滴IgG包被的库姆斯对照细胞(检查细胞)到管和离心机。针对凝集进行检查。此步骤中应存在凝集,或该测试无效。如果在添加检查细胞(ccc)之前的任何步骤中没有凝集,则该测试被解释为阴性。如果在添加检查细胞之前的任何步骤中观察到凝集,则该测试被解释为阳性。实例71:氧结合能力的评估在与1cm路径长度比色皿连接的血压计中确定37℃下的平衡氧结合曲线。使用分光光度计(Cary50;Variant公司)进行光谱测量,并且用帕尔贴模块控制温度。分析在包含140mMNaCl和2mM葡萄糖的50mMbis-Tris缓冲液(pH7.2)中进行。在氮气下彻底脱氧后,通过用哈美顿注射器通过橡胶帽将已知体积的纯氧注入血压计,使红细胞悬浮液在不同的氧分压下平衡。通过模拟可见光和Soret区域中的吸收光谱作为RBC悬浮液的完全脱氧和氧合光谱的线性组合通过最小二乘回归来估计部分饱和度。实例72:细胞代谢状态的评估可使用多种不同的基于酶的测定法经代谢活性来验证类红细胞群,以量化重要的代谢终产物。活性糖酵解是评估的关键代谢途径,并且可以用以下测定(基于糖酵解细胞的测定试剂盒,曼化学品公司(CaymanChemical),项目600450)来测量。将450ul测定缓冲液等分到试管中,然后添加50uL的L-乳酸标准品并充分混合。使用乳酸浓度标准物构建滴定曲线,从1mM稀释度开始。将细胞添加96孔板中并以1000RPM离心5分钟。将100uL标准品转移到单独的96孔板中。然后向每个孔中添加90uL测定缓冲液。然后将每个细胞壁中的10ul上清液转移到相应的新孔中。使用重复移液器向每个孔中添加100ul反应溶液。然后将板在定轨振荡器上在室温下孵育30分钟。用读板器在490nm下读取吸光度。将结果与天然细胞进行比较以鉴定任何代谢差异。实例73:血小板聚集的评估可以监测培养的或原始来源血小板的聚集倾向。通过在光源前摇动它们将血小板进行涡旋分析,结果表示为双折射的存在或不存在。将体积为50-70mL的产生的血小板浓缩物的单元静置1小时,并在22℃±2℃(71.6°F±3.6°F)的控制温度为下以70rpm放置在线性摇床中。血小板浓缩物的测试(血小板计数,血小板聚集和pH)在处理后第1、3和5天进行;仅在第1天进行白细胞计数,并且仅在储存的第5天进行微生物控制。为了从血小板浓缩物的样品获得等分试样,使用无菌连接其确保环境的完整性。在血液收集的四小时内使用双通道Chronolog(Crono-Log)使用比浊聚集技术实现血小板聚集。为此,首先通过以1000rpm轻离心5分钟获得细胞,并且然后以3000rpm离心15分钟将样品在自动计数器(Human)中进行血小板计数。在调整血小板浓度后,使用不同浓度的诱导激动剂来评估聚集:胶原2.0μg/mL和ADP7.0μg/mL(Crono-Log)。对于每个测试,每个在不同的比色杯中等待自发聚集后,使用400μL的PRP和400μL的PPP。在通过诱导激动剂刺激5分钟后观察聚集曲线,并且紧接着测量凝集并且根据在测试期间形成的曲线以百分比表示。测试的结果通常表示为通过透射过测试溶液的光的量的聚集百分比;聚集分为正常,低或高。实例74:自体培养过程使用自体来源的祖细胞CD34+细胞进行类红细胞培养以优化患者的细胞免疫相容性。如本文所述,使用GM-CSF将来自骨髓的CD34+细胞动员到患者的外周。收集介于10^6-10^8个之间的CD34+细胞并使用上述22天方案使用确定的介质进行培养。在第4天期间,用包含编码表达治疗剂的基因的慢病毒载体进行转染细胞。完成培养方案时,纯化细胞,并且跨多个质量控制度量进行评估,这些质量控制度量包括与循环生存力,免疫原性,复制潜力,纯度和治疗剂量相关的物理性质。然后将细胞储存在合适的稳定溶液中并配制在注射器或合适的递送载体中。然后将细胞输注到捐赠初始CD34+细胞的相同患者中。实例75:自体荷载过程为了制备荷载有合适的外源抗原的治疗性类红细胞,自体来源的红细胞可用于优化患者的细胞免疫相容性。从患者抽取血液,并且以5000g离心20分钟。去除血沉棕黄层,并且将剩余的红细胞以10^8个细胞/ml的密度重新悬浮于抗凝血缓冲液中,得到总共10^10个细胞。通过上述方法之一使细胞荷载感兴趣的治疗性外源抗原。完成荷载方案时,纯化细胞,并且跨多个质量控制度量进行评估,这些质量控制度量包括与循环生存力,免疫原性,复制潜力,纯度和治疗剂量相关的物理性质。然后将细胞储存在合适的稳定溶液中并配制在注射器或合适的递送载体中。将细胞输注到捐赠初始红细胞的相同患者中。实例76:同种异体培养过程为了产生可扩展的通用治疗剂,可以从同种异体来源培养类红细胞。使用同种异体来源的祖细胞CD34+细胞进行类红细胞培养以简化过程并培养一定体积的能够在规模上治疗患者的治疗剂。供体是主要血液抗原的血型,包括A、B、Rh,以鉴定通用供体(例如,ORh-或孟买Rh-)。如本文所述,使用GM-CSF将来自骨髓的CD34+细胞动员到适合供体的外周。收集介于10^6-10^8个之间的CD34+细胞并使用上述22天方案使用确定的介质进行培养。在第4天期间,用包含编码表达治疗剂的基因的慢病毒载体进行转染细胞。完成培养方案时,纯化细胞,并且跨多个质量控制度量进行评估,这些质量控制度量包括与循环生存力,免疫原性,复制潜力,纯度和治疗剂量相关的物理性质。然后将细胞储存在合适的稳定溶液中并配制在注射器或合适的递送载体中。然后不管患者的主要血型,将细胞输注到患者中。实例77:同种异体荷载过程使用同种异体来源的祖细胞CD34+细胞进行类红细胞培养以简化过程从而制备能够大规模治疗患者的更大量的治疗细胞。供体是主要血液抗原的血型,包括A、B、Rh,以鉴定通用供体(例如,ORh-或孟买Rh-)。通过上述方法之一使细胞荷载感兴趣的治疗性外源抗原。完成荷载方案时,纯化细胞,并且跨多个质量控制度量进行评估,这些质量控制度量包括与循环生存力,免疫原性,复制潜力,纯度和治疗剂量相关的物理性质。然后将细胞储存在合适的稳定溶液中并配制在注射器或合适的递送载体中。然后不管患者的主要血型,将细胞输注到患者中。实例78:存储1.冷藏的缓冲液中存储用于储存血红细胞的标准方案是本领域已知的,参见,例如,梅里曼(Meryman)和霍恩布洛尔(Hornblower)1986,输注(Transfusion)26(6):500。用于储存血红细胞(长达42天)的标准方案是将血液收集到抗凝血剂溶液(柠檬酸盐-右旋糖-磷酸盐)中。如本文所述培养类红细胞。通过离心除去血浆制备红细胞浓缩物。将细胞在4℃±2℃下在轻微高渗添加剂溶液SAGM(钠、腺嘌呤、葡萄糖、甘露醇,376mOsm/L)中储存。2.冷冻的缓冲液中存储用于类红细胞的甘油化,冷冻和解冻的方法是本领域已知的,参见,例如,梅里曼和霍恩布洛尔1977,输注,17(5):4348。将柠檬酸盐-磷酸盐-右旋糖中的人血液在收集4天内脱甘油并冷冻。为了制备脱甘油化RBC,首先将约10mL的全血以1,400g离心10-15min,并且除去血浆。然后使用具有以下组合物的甘油水溶液在两步中将所得填充细胞甘油化:57.1g甘油、0.03g氯化钾、0.085g氯化镁六水合物、0.08g磷酸二钠和1.6g乳酸钠,总体积为100mL,调节至pH6.8.42。在第一步中,在3min的时间内用温和搅拌将1.5mL该甘油溶液逐滴添加到包装的细胞中。然后允许混合物未扰动平衡至少5min。在第二甘油化步骤中,逐滴添加5mL甘油溶液,同时在3分钟时间内温和搅拌混合物,产生约40%w/v的最终甘油组合物。整个甘油化过程在室温下进行。然后将脱甘油化的RBC在低温小瓶中分成0.6-1.1mL的等分试样,放置在NalgeneCryo“弗罗斯蒂先生(Mr.Frosty)”冷冻容器(赛墨科技公司,北卡罗来纳州),并且在-80℃冷冻机中储存至少12h以及长达10年。通过将低温小瓶置于37℃水浴中1分钟来解冻冷冻的RBC。所有的脱甘油化血液样品在解冻的2小时内用于脱甘油实验。3.呈注射器状的配制品细胞群可以通过注射器经静脉内给予。使用标准盐水缓冲液在37℃下,将治疗细胞稀释至10^7个细胞/ml的密度,这样使得递送100ml体积或10^9个细胞。将细胞溶液荷载到150cc注射器,20计量注射针中,并以5cc/min通过贵要静脉内注射到患者体内。在注射期间,监测患者的生命体是否有任何免疫原性或凝血反应。4.呈袋状的配制品细胞群可以通过连接到袋和滴室的注射器经静脉内给予(即,IV滴注)。使用标准盐水缓冲液在37℃下,将治疗细胞稀释至10^7个细胞/ml的密度,这样使得递送100ml体积或10^9个细胞。将细胞溶液荷载到1L塑料袋中,连接到导管,并允许通过重力经由贵要静脉排出到患者体内。在输注期间,监测患者的生命体是否有任何免疫原性或凝血反应。实例79:疾病的治疗1.血友病诊断患有血友病A的患者。如本文所述制备表达外源FVIII的去核造血细胞的组合物。将10^9个细胞经静脉内给予患者。凝血速率用本领域已知的标准体外凝血时间测定来评估。如本文所述在血清中检测针对FVIII的循环抗体。评估循环抗体的水平以跟踪免疫耐受诱导的有效性。如果凝血级联活性不足以确保健康凝血,则重组或分离的FVIII同时经静脉内给予以减少血友病A的症状。2.非典型溶血性尿毒症综合征诊断患有非典型溶血性尿毒症综合征(aHUS)的患者。如本文所述制备表达外源CFH的去核造血细胞的组合物。将10^9个细胞经静脉内给予患者。用本领域已知的标准尿液溶血测定来评估症状性溶血率。如本文所述在血清中检测针对CFH的循环抗体。评估循环抗体的水平以跟踪免疫耐受诱导的有效性。给患者给予治疗,直到使用本文所述的测定观察到疾病的症状得到改善。3.多发性硬化症具有多发性硬化症(MS)的个体接受1x10^9个如本文所述产生和配制的表达抗原的去核造血细胞的单次输注,这些细胞表达抗原性多肽髓磷脂碱性蛋白(MBP)。在研究药物给予的当天,在1期住院单位中监测患者24小时。主要结果的测量在第3个月进行,并且进行额外的安全性随访直到第6个月,伴随连续进行临床检查、MRI检查和一般身体检查以及临床和实验室分析,以评估不良事件并监测MS疾病活动。重复该过程,直到诱导出耐受性,使得个体中MS症状得到改善。参见,例如,安德里亚斯卢特罗蒂(AndreasLutterotti)等人,科学转化医学期刊(SciTranslMed)5,188ra75(2013)。通过具有以下抗体组的流式细胞术在全血(EDTA试管)中分析不同细胞亚群的频率:对于免疫细胞亚群(粒细胞,嗜酸性粒细胞,单核细胞,和B、T、NK和NKT细胞)-抗CD45(PE-Cy7,eBioscience公司),抗CD16,(APC-Cy7,BioLegend公司),抗CD19[异硫氰酸荧光素(FITC),BD公司],抗CD14(V450,BD公司),抗CD3[荧光素叶绿素蛋白(PerCP),BD公司]和抗CD56[藻红蛋白(PE),eBioscience公司];对于T细胞亚群包括CD4+,FoxP3+Treg,调节性CD8+CD57+ILT2+,和促炎性CD8+CD161高T细胞-抗CD3(PE-Cy7,eBioscience公司),抗CD4(APC,eBioscience公司),抗CD8[太平洋蓝(PB),Dako-Biozol公司],抗FoxP3(PE,美天旎公司),抗CD25(APC,eBioscience公司),抗CD57(FITC,BD公司),抗ILT2(PE,贝克曼公司(Beckman))和抗CD161(APC,美天旎公司)。相应的同型对照包括在所有染色中。用LSR-II流式细胞仪(BD公司)和FACSDiva软件(BD公司)分析细胞。通过菲可密度梯度离心(PAA)分离外周血单核细胞(PBMC),并通过细胞内细胞因子染色如下评估T细胞的功能表型:将5×10^5个新鲜分离的PBMC在200ml的X-VIVO15(龙沙公司(Lonza))在无菌FACS管中孵育过夜。第二天,在布雷菲德菌素A(10mg/ml,eBioscience公司)存在下,用佛波醇12-豆蔻酸酯13-乙酸酯(50ng/ml,西格玛)和离子霉素(1mg/ml,西格玛)刺激细胞5小时。用磷酸盐缓冲盐水洗涤后,将细胞用LiveDead试剂盒(AmCyan,英杰公司)染色,固定,透化,并用不同抗体:抗IL-17(AlexaFluor647;eBioscience公司),抗IL-4(PE-Cy7,BioLegend公司),抗IFN-g(FITC,BioLegend),抗IL-10(PE;BioLegend公司),抗CD3(PE,DakoCytomation公司),抗-CD4(PB,DakoCytomation公司),和抗-CD8(PB,BioLegend公司)或相应的同型对照进行染色。在耐受程序之前和3个月后,在新鲜分离的PBMC中测量针对研究中使用的髓鞘肽的抗原特异性T细胞应答。通过用胸苷掺入的增殖测定分析抗原特异性T细胞应答。简言之,将分离的PBMC以1.5×10^5个PBMC/孔接种在具有1mM肽的X-VIVO15介质(龙沙公司)中的96孔板中。每个抗原接种48个孔,并且每个平板中只有6个孔用介质作为阴性对照。TTx(5mg/ml)(NovartisBehring公司)用作阳性对照。在第7天,将板与1mCi的[3H]胸苷(HartmannAnalytic公司)孵育15小时。将[3H]胸苷脉冲板用闪烁b计数器(Wallac1450,珀金埃尔默公司(PerkinElmer))进行分析。测量每个孔的闪烁计数(CPM)。显示高于未刺激孔的平均+3SD的CPM的孔被认为是阳性的。本发明所属领域的技术人员可从前文描述和相关附图中获益进而想出这些发明的许多改进形式和其他实施例。因此,应当理解本发明不限于公开的具体实施例,改进和其他实施例意欲包括于附加的权利要求范围内。尽管其中使用特殊术语,它们仅以通用和描述性意义而非限制目的使用。说明书中提到的所有出版物和专利申请指示了本发明所属领域技术人员的水平。将所有出版物和专利申请引入本文作为参考,其程度就像明确且个别指出引入每一篇个别出版物或专利申请作为参考一样。表A-J表A:循环细胞表A1:类红细胞表B:循环细胞相关蛋白CD1CD23CD46CD72CD120CD195CD2CD24CD47CD73CD122CD197CD3CD25CD48CD74CD127CD199CD4CD26CD49aCD80CD132CD209CD5CD27CD49bCD81CD133CD202aCD6CD28CD49cCD82CD134CD220CD7CD29CD49dCD83CD135CD221CD8CD30CD49eCD86CD138CD235aCD9CD31CD49fCD87CD141CD271CD10CD32CD53CD88CD142CD279CD11aCD33CD54CD89CD143CD303CD11bCD34CD55CD90CD144CD304CD11cCD35CD56CD91CD147CD309CD12wCD36CD57CD95CD151CD326CD13CD37CD58CD96CD152TLR1CD14CD38CD59CD100CD154TLR2CD15CD39CD61CD103CD156TLR4CD16CD40CD62ECD105CD158TLR5CD17CD41CD62LCD106CD163TLR6CD18CD42CD62PCD107CD165CD19CD43CD63CD107aCD166CD20CD44CD68CD107bCD168CD21CD45CD69CD109CD184CD22CD71CD117CD186表C:红细胞相关蛋白表C1:红细胞跨膜蛋白表C2:红细胞GPI-连接的蛋白表C3:红细胞细胞内蛋白表D:共轭方法表D1:酶促共轭方法酶促反应SpyCatcher/SpyTagSpy0128衍生物转肽酶异肽酶分选酶DD-转肽酶肽基转移酶G-谷氨酰转肽酶D-谷氨酰转肽酶法尼基转移酶异戊烯基转移酶二甲丙烯基转移酶焦磷酸香叶酯合酶脱氢多萜醇二磷酸合酶表E:反应性基团的化学表F:自身免疫性疾病和抗原表G:炎性疾病和抗原疾病抗原克罗恩病鞭毛蛋白,微生物抗原溃疡性结肠炎嗜中性粒细胞胞浆抗原,微生物抗原乳糜泻谷蛋白炎性肠病微生物抗原表H:过敏性疾病触发物表1:治疗疾病的治疗性蛋白表J:治疗疾病的治疗性蛋白类别表1-8表3.补体受体1的序列4A.CR1亚型S前体,智人NCBI参照序列号NP_000642.34B.CR1亚型F前体,智人NCBI参照序列号NP_000564.24C.预测的CR1亚型X1,智人NCBI参照序列号XP_005273121.1权利要求书:当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1