利用噻吩并三唑并二氮杂*化合物治疗非小细胞肺癌和/或小细胞肺癌的方法与流程
本发明描述了一种利用噻吩并三唑并二氮杂化合物治疗小细胞肺癌的方法,所述化合物具有改善的溶解度和生物利用度,其可以以固体分散体的形式提供。
背景技术:
晚期小细胞肺癌(“SCLC”)的标准化疗在过去二十年中没有实质上的变化。鉴定新型SCLC疗法时的挑战在于肿瘤抑制基因(TP53、PTEN、RBI)中失活突变的普遍性,并且仅在致癌基因(例如PIK3CA或MYC家族成员)中罕见的获得失效突变。溴结构域蛋白和外部(BET)-溴结构域(BRD)蛋白的抑制剂可以通过抑制BRD与组蛋白蛋白上的乙酰化赖氨酸结合,防止转录协同剂装配在靶基因启动子上,从而调节致癌基因(如MYC家族)的转录。
下文所述的式(1)化合物已经显示出可抑制乙酰化的组蛋白H4与包含串联的溴结构域(BRD)的转录调节子家族(称为BET(溴结构域和额外末端)蛋白,包括BRD2、BRD3和BRD4)的结合。参见美国专利申请公开No.2010/0286127A1,其全文以引用的方式并入本文。Denis,G.V.在文献“Bromodomain coactivators in cancer,obesity,type 2 diabetes,and inflammation,”Discov Med 2010;10:489-499中报道了如下内容:BET蛋白作为增殖和分化的主要表观遗传调节子出现,并且还与倾向于血脂异常或脂肪生成不当调节、心血管病和2型糖尿病的升高的炎症性质和风险、以及自体免疫疾病(例如类风湿性关节炎和系统性红斑狼疮)的易感性增加有关,该文献的全部内容以引用方式并入本文。因此,式(1)化合物可以用于治疗多种癌症、心血管疾病、2型糖尿病和自体免疫失调(例如类风湿性关节炎和系统性红斑狼疮)。
技术实现要素:
在一些实施方案中,本说明书提供了利用本文所述的组合物治疗小细胞肺癌的方法。在一些实施方案中,本说明书提供了利用本文所述的组合物治疗非小细胞肺癌的方法。
在一些实施方案中,本说明书提供了治疗哺乳动物中非小细胞肺癌和/或小细胞肺癌的方法,该方法包括:向需要的患者施用药学可接受量的组合物,该组合物包含根据本文第III、IV、V和VI部分所描述的任意组合物。
在一些实施方案中,本说明书提供了治疗哺乳动物中非小细胞肺癌和/或小细胞肺癌的方法,该方法包括:向需要的患者施用药学可接受量的组合物,该组合物包含根据本文第III、IV、V和VI部分所描述的任意组合物的固体分散体。
在一些实施方案中,本说明书提供了用于治疗非小细胞肺癌和/或小细胞肺癌的式(1)化合物,特别是式(1A)化合物。
在一些实施方案中,本说明书提供了根据本文第III、IV、V和VI部分所描述的任意组合物的固体分散体,其用于治疗非小细胞肺癌和/或小细胞肺癌。
在一些实施方案中,本说明书提供了利用式(1)的噻吩并三唑并二氮杂化合物、或其可药用的盐或水合物或溶剂合物治疗非小细胞肺癌和/或小细胞肺癌的方法,
其中:
R1为具有1至4个碳的烷基;
R2为氢原子、卤素原子、或任选地被卤素原子或羟基取代的具有1至4个碳的烷基;
R3为卤素原子;任选地被卤素原子、具有1至4个碳的烷基、具有1至4个碳的烷氧基、或氰基取代的苯基;-NR5-(CH2)m-R6,其中R5为氢原子、或具有1至4个碳的烷基,m为0至4的整数,并且R6为任选地被卤素原子取代的苯基或吡啶基;或-NR7-CO-(CH2)n-R8,其中R7为氢原子或具有1至4个碳的烷基,n为0至2的整数,并且R8为任选地被卤素原子取代的苯基或吡啶基;以及
R4为-(CH2)a-CO-NH-R9,其中a为1至4的整数,并且R9为具有1至4个碳的烷基;具有1至4个碳的羟烷基;具有1至4个碳的烷氧基;或任选地被具有1至4个碳的烷基、具有1至4个碳的烷氧基、氨基或羟基取代的苯基或吡啶基;或-(CH2)b-COOR10,其中b为1至4的整数,并且R10为具有1至4个碳的烷基。
在一些实施方案中,式(I)选自式(IA)、其可药用的盐或溶剂化物:
其中,X为卤素,R1为C1-C4烷基,R2为C1-C4烷基,a为1至4的整数,R3为C1-C4烷基、C1-C4羟烷基、C1-C4烷氧基、任选地具有如式(1)中R9所限定的取代基的苯基、或任选地具有如式(1)中R9所限定的取代基的杂芳基。
在一个这样的实施方案中,将噻吩并三唑并二氮杂化合物配制成含有无定形噻吩并三唑并二氮杂化合物和可药用的聚合物的固体分散体。
在一个实施方案中,式(1)选自由以下化合物构成的组:(i)(S)-2-[4-(4-氯代苯基)-2,3,9-三甲基-6H-噻吩并[3,2-f][1,2,4]三唑并-[4,3-a][1,4]二氮杂-6-基]-N-(4-羟苯基)乙酰胺或其二水合物;(ii)(S)-{4-(3'-氰基联苯-4-基)-2,3,9-三甲基-6H-噻吩并[3,2-f][1,2,4]三唑并[4,3-a][1,4]二氮杂-6-基}乙酸甲酯;(iii)(S)-{2,3,9-三甲基-4-(4-苯基氨基苯基)-6H-噻吩并[3,2-f][1,2,4]三唑并[4,3-a][1,4]二氮杂-6-基}乙酸甲酯;以及(iv)(S)-{2,3,9-三甲基-4-[4-(3-苯基丙酰基氨基)苯基]-6H-噻吩并[3,2-f-][1,2,4]三唑并[4,3-a][1,4]二氮杂-6-基}乙酸甲酯。
在一个实施方案中,式(1)所示的噻吩并三唑并二氮杂化合物为(S)-2-[4-(4-氯代苯基)-2,3,9-三甲基-6H-噻吩并[3,2-f][1,2,4]三唑并[4,3-a][1,4]二氮杂-6-基]-N-(4-羟苯基)乙酰胺二水合物。
在一个实施方案中,式(1)所示的噻吩并三唑并二氮杂化合物为(S)-2-[4-(4-氯代苯基)-2,3,9-三甲基-6H-噻吩并[3,2-f][1,2,4]三唑并[4,3-a][1,4]二氮杂-6-基]-N-(4-羟苯基)乙酰胺。
在一些实施方案中,本说明书提供了使用式(1)的噻吩并三唑并二氮杂化合物与一种或多种化疗药物联合治疗非小细胞肺癌和/或小细胞肺癌的方法,所述化疗药物选自由m-TOR抑制剂和ALK抑制剂构成的组。
在一些实施方案中,本说明书提供了使用式(1A)的噻吩并三唑并二氮杂化合物与一种或多种化疗药物联合治疗非小细胞肺癌和/或小细胞肺癌的方法,所述化疗药物选自由m-TOR抑制剂和ALK抑制剂构成的组。
在一些实施方案中,该方法进一步包括向患者使用mTOR抑制剂。在一个此类的实施方案中,mTOR抑制剂选自由雷帕霉素、替西罗莫司、地磷莫司和依维莫司所构成的组。在一个此类的实施方案中,mTOR抑制剂是依维莫司。噻吩并三唑并二氮杂化合物和mTOR抑制剂可以同时或按顺序施用。在一些实施方案中,噻吩并三唑并二氮杂化合物和mTOR抑制剂的如此组合产生协同效应。
在一些实施方案中,所述方法进一步包括向患者施用ALK抑制剂。在一个此类的实施方案中,ALK抑制剂选自由色瑞替尼和克唑替尼所构成的组。在一个此类的实施方案中,ALK抑制剂是克唑替尼。噻吩并三唑并二氮杂化合物和ALK抑制剂可以同时或按顺序施用。在一些实施方案中,噻吩并三氮卓二氮化合物和ALK抑制剂的如此组合产生协同效应。
在一个实施方案中,非小细胞肺癌是EML4-ALk阳性的。在一个此类的实施方案中,非小细胞肺癌在治疗后表现出N-MYC mRNA水平的下调。在另一个此类的实施方案中,非小细胞肺癌表达BRD4/3/2、c-MYC、BCL-2、p21和细胞周期蛋白D1。在另一个实施方案中,所述治疗诱导STAT3的瞬时上调,随后在24小时和最多72小时暴露后下调。
在另一个实施方案中,非小细胞肺癌是EML4-ALk阴性的。在一个这样的实施方案中,非小细胞肺癌表达BRD4/3/2、c-MYC、BCL-2、p21和细胞周期蛋白D1。在另一个这样的实施方案中,治疗诱导STAT3的瞬时上调,随后在24小时和最多72小时暴露后下调。
在一个实施方案中,NMYC被下调。在一个实施方案中,HEXIM被上调。
在治疗非小细胞肺癌和/或小细胞肺癌的方法的一些实施方案中,EFR3B基因被上调。在治疗非小细胞肺癌和/或小细胞肺癌的方法的一些实施方案中,FOS基因被上调。在治疗非小细胞肺癌和/或小细胞肺癌的方法的一些实施方案中,IL7R基因被下调。在治疗非小细胞肺癌和/或小细胞肺癌的方法的一些实施方案中,IL6相关基因被下调。在一些实施方案中,EFR3B和FOS基因被上调,IL7R和IL6相关基因被下调。
在一个实施方案中,使式(1)的噻吩并三唑并二氮化合物形成固体分散体。在一个实施方案中,固体分散体包含无定形的式(1)的噻吩并三唑并二氮杂化合物或其可药用的盐或水合物;以及可药用的聚合物。在一个实施方案中,在固体分散体的X射线粉末衍射图中,基本上不存在与式(1)的结晶型噻吩并三唑并二氮杂化合物有关的衍射线。在一个实施方案中,可药用的聚合物为醋酸羟丙基甲基纤维素琥珀酸酯,其中噻吩并三唑并二氮化合物与醋酸羟丙基甲基纤维素琥珀酸酯(HPMCAS)的重量比为1:3至1:1。在一个实施方案中,固体分散体表现出约130℃至约140℃的单一的玻璃化转变温度(Tg)拐点。
在一个实施方案中,固体分散体包含无定形噻吩并三唑并二氮杂化合物(S)-2-[4-(4-氯代苯基)-2,3,9-三甲基-6H-噻吩并[3,2-f][1,2,4]三唑并[4,3-a][1,4]二氮杂-6-基]-N-(4-羟苯基)乙酰胺二水合物、或其可药用的盐或水合物;以及可药用的聚合物。在一个实施方案中,在固体分散体的X射线粉末衍射图中,基本上不存在与结晶型噻吩并三唑并二氮杂化合物(S)-2-[4-(4-氯代苯基)-2,3,9-三甲基-6H-噻吩并[3,2-f][1,2,4]三唑并[4,3-a][1,4]二氮杂-6-基]-N-(4-羟苯基)乙酰胺二水合物有关的衍射线。在一个实施方案中,可药用的聚合物为醋酸羟丙基甲基纤维素琥珀酸酯,其中噻吩并三唑并二氮化合物与醋酸羟丙基甲基纤维素琥珀酸酯(HPMCAS)的重量比为1:3至1:1。在一个实施方案中,固体分散体表现出约130℃至约140℃的单一的玻璃化转变温度(Tg)拐点。
应当理解,除非另有说明,本文所述的根据式(1)的化合物的任何实施方案可用于本文所述的药物组合物的任何实施方案中。此外,除非另有说明,本文作为本发明实施方案描述的任何化合物或药物组合物可用作药物,特别是用于治疗如本文实施方案中所述的非小细胞肺癌和/或小细胞肺癌。
附图说明
参照示例性实施方案的附图进行阅读,将会更好的理解前面的发明概述以及下面的对本发明的包含噻吩并三唑并二氮杂制剂的药物组合物和方法的实施方案的详述。但是,应该理解的是本发明不限于所示的精确设置和手段。
在附图中:
图1A示出了包含固体分散体的对比制剂的溶解情况,其中所述固体分散体包含25%的化合物(1-1)和Eudragit L100-55;
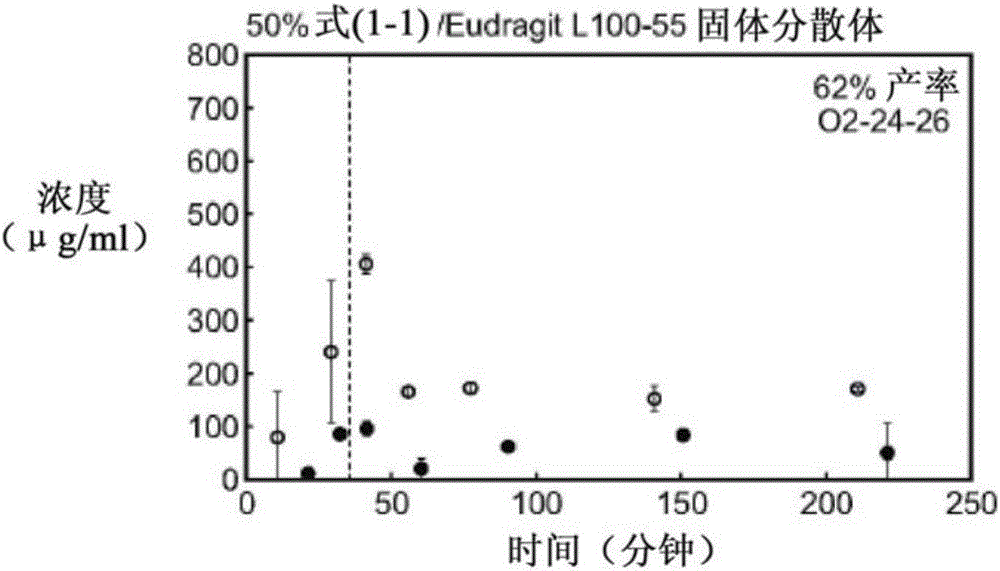
图1B示出了包含固体分散体的对比制剂的溶解情况,其中所述固体分散体包含50%的化合物(1-1)和Eudragit L100-55;
图1C示出了包含固体分散体的示例性制剂的溶解情况,其中所述固体分散体包含25%的化合物(1-1)和聚乙烯吡咯烷酮(PVP);
图1D示出了包含固体分散体的示例性制剂的溶解情况,其中所述固体分散体包含50%的化合物(1-1)和PVP;
图1E示出了包含固体分散体的示例性制剂的溶解情况,其中所述固体分散体包含25%的化合物(1-1)和PVP-乙酸乙烯酯(PVP-VA);
图1F示出了包含固体分散体的示例性制剂的溶解情况,其中所述固体分散体包含50%的化合物(1-1)和和PVP-VA;
图1G示出了包含固体分散体的示例性制剂的溶解情况,其中所述固体分散体包含25%的化合物(1-1)和醋酸羟丙基甲基纤维素琥珀酸酯(HPMCAS-M);
图1H示出了包含固体分散体的示例性制剂的溶解情况,其中所述固体分散体包含50%的化合物(1-1)和HPMCAS-M;
图1I示出了包含固体分散体的示例性制剂的溶解情况,其中所述固体分散体包含25%的化合物(1-1)和羟丙基甲基纤维素邻苯二甲酸酯(HPMCP-HP55);
图1J示出了包含固体分散体的示例性制剂的溶解情况,其中所述固体分散体包含50%的化合物(1-1)和HPMCP-HP55;
图2A示出了示例性制剂的体内筛选结果,该示例性制剂包含25%化合物(1-1)和PVP的固体分散体;
图2B示出了示例性制剂的体内筛选结果,该示例性制剂包含25%化合物(1-1)和HMPCAS-M的固体分散体;
图2C示出了示例性制剂的体内筛选结果,该示例性制剂包含50%化合物(1-1)和HMPCAS-M的固体分散体;
图3示出了化合物(1-1)的固体分散体的X射线粉末衍射图谱。
图4A示出了在环境条件下平衡的25%化合物(1-1)和PVP的固体分散体的修正的差示扫描量热轨迹;
图4B示出了在环境条件下平衡的25%化合物(1-1)和HMPCAS-M的固体分散体的修正的差示扫描量热轨迹;
图4C示出了在环境条件下平衡的50%化合物(1-1)和HMPCAS-M的固体分散体的修正的差示扫描量热轨迹;
图5示出了25%化合物(1-1)和PVP或HPMCAS-M的固体分散体、以及50%化合物(1-1)和HPMCAS-MG的固体分散体的玻璃化转变温度(Tg)对相对湿度(RH)的图;
图6示出了在75%相对湿度下平衡的25%化合物(1-1)和PVP的固体分散体的修正的差示扫描量热轨迹;
图7示出了1mg/kg静脉定量给药后的化合物(1-1)(实心矩形)、3mg/kg口服定量给药的25%化合物(1-1):PVP(空心圆圈)、3mg/kg口服定量给药的25%化合物(1-1):HPMCAS-MG(空心三角形)和3mg/kg口服定量给药的50%化合物(1-1):HPMCAS-MG(空心倒三角)的血浆浓度对时间的曲线。插图描绘了在半对数标度上绘制的相同的数据;
图8示出了化合物(1-1)作为25%化合物(1-1):PVP(空心圆圈)、25%化合物(1-1):HPMCAS-MG(空心三角形)和50%化合物(1-1):HPMCAS-MG(空心倒三角)在3mg/kg口服定量给药后的血浆浓度对时间的曲线。插图描绘了在半对数标度上绘制的相同的数据;
图9示出了在稳定性测试时间为0时,化合物(1-1)在HPMCAS-MG中的固体分散体的X射线粉末衍射图谱;
图10示出了在40℃和75%相对湿度的环境下暴露1个月后,化合物(1-1)在HPMCAS-MG中的固体分散体的X射线粉末衍射图谱;
图11示出了在40℃和75%相对湿度的环境下暴露2个月后,化合物(1-1)在HPMCAS-MG中的固体分散体的X射线粉末衍射图谱;
图12示出了在40℃和75%相对湿度的环境下暴露3个月后,化合物(1-1)在HPMCAS-MG中的固体分散体的X射线粉末衍射图谱;
图13示出了72小时后的化合物(1-1)抗增殖作用和在NSCLC细胞中发现的常见突变的表征;
图14示出了暴露于化合物(1-1)24小时后S期细胞分数的减少;
图15A示出了NSCLC细胞中以化合物(1-1)敏感性(GI50值)的顺序分类的BRDs、C-MYC、N-MYC、BCL2和P21mRNA水平;
图15B示出了NSCLC细胞中BRD2、BRD3、C-MYC、N-MYC、P21、BCL2和细胞周期蛋白D1的基础蛋白表达;
图16A示出了暴露于500nM化合物(1-1)后,HOP62、HOP92、H2228、H3122和A549细胞中的C-MYC mRNA水平;
图16B示出了暴露于500nM化合物(1-1)后,HOP62、HOP92、H2228、H3122和A549细胞中的N-MYC mRNA水平;
图16C示出了暴露于500nM化合物(1-1)后,HOP62、HOP92、H2228、H3122和A549细胞中C-MYC蛋白和N-MYC蛋白的调节;
图17示出了在NSCLC细胞系中化合物(1-1)与依维莫司或critzotinib联合使用的效果;
图18A示出了相比于载体对照处理的小鼠,在H3122肿瘤小鼠中,化合物(1-1)减缓了肿瘤的发展;
图18B示出了在化合物(I-1)治疗的3周期间没有观察到重量减轻;
图19示出了化合物(1-1)(OTX015)在NCI-H69、NCI-H82、DMS79和DMS114细胞系中72小时后的抗增殖作用和在这样的细胞系中发现的常见突变的表征;
图20A示出了当用化合物(1-1)处理NCI-H69、NCI-H82、DMS79和DMS114细胞系4和24小时时所观察到的与C-MYC相关的荧光强度;
图20B示出了当用化合物(1-1)处理NCI-H69、NCI-H82、DMS79和DMS114细胞系4和24小时时所观察到的与N-MYC相关的荧光强度;
图20C示出了当用化合物(1-1)处理DMS114细胞系4和24小时时所观察到的与HEXM1、HIST2H2BE和HIST1H2BK相关的荧光强度;
图20D示出了当在NCI-H69、NCI-H82细胞系中用化合物(1-1)处理0、4、24、48和72小时时,C-MYC和β-肌动蛋白水平的蛋白质印迹结果;
图20E示出了当在DMS79和DMS114细胞系中用化合物(1-1)处理0、4、24、48和72小时时,C-MYC和β-肌动蛋白水平的蛋白质印迹结果;
图21A示出了用化合物(I-1)处理72小时的NCI-H69、NCI-H82、DMS79和DMS114细胞系中BRD2水平的荧光强度;
图21B显示了用化合物(1-1)处理72小时的NCI-H69、NCI-H82、DMS79和DMS114细胞系中BRD3水平的荧光强度;
图21C示出了用化合物(1-1)处理72小时的NCI-H69、NCI-H82、DMS79和DMS114细胞系中的BRD4水平的荧光强度;
图21D示出了用化合物(1-1)处理72小时的NCI-H69、NCI-H82、DMS79和DMS114细胞系中C-MYC水平的荧光强度;
图21E示出了在用化合物(1-1)处理72小时的NCI-H69、NCI-H82、DMS79和DMS114细胞系中N-MYC水平的荧光强度;
图21F示出了用化合物(1-1)处理72小时的NCI-H69、NCI-H82、DMS79和DMS114细胞系中Bc12水平的荧光强度;
图21G示出了用化合物(1-1)处理72小时的NCI-H69、NCI-H82、DMS79和DMS114细胞系中p21水平的荧光强度;
图21H示出了用化合物(1-1)处理4小时和24小时的NCI-H69、NCI-H82、DMS79和DMS114细胞系中,BRD2、BRD3、C-MYC、N-MYC、Bc12、p21和β-肌动蛋白水平的蛋白质印迹结果;
图22A示出了用化合物(I-1)和对照处理72小时的NCI-H69、NCI-H82、DMS79和DMS114细胞系中G1期的细胞数目;
图22B示出了用化合物(I-1)和对照处理72小时的NCI-H69、NCI-H82、DMS79和DMS114细胞系中的p27和β-肌动蛋白水平的蛋白质印迹。
发明详述
现在,下文将参照附图和实施例来更全面地描述本发明的主题,所述附图和实施例示出了代表性的实施方案。但是,本发明的主题可以以不同的形式体现,并且不应该解释成局限于本发明列出的实施方案。提供这些实施方案旨在描述并使本领域的技术人员能够实施。除非另有说明,否则本发明所用的所有技术和科学术语都具有所述主题所属技术领域的普通技术人员通常所理解的相同的含义。本发明中所提及的所有公开、专利申请、专利和其他参考文献均以引用的方式全文并入本文中。
I.定义
本文所用的术语“烷基”是指饱和的直链或支链烃。
术语“取代的烷基”是指具有一个或多个取代基的烷基部分,其中所述的取代基替代了烃链上的氢或者一个或多个碳。
无论术语“烯基”是单独使用还是作为取代基的一部分(例如“C1-4烯基(芳基)”)使用,都是指具有至少一个碳-碳双键部分的部分不饱和支链或直链一价烃基团,其中所述的双键是通过由母体烷基分子的两个相邻碳原子的每个碳原子上除去一个氢原子而得到的,并且所述基团是通过由一个碳原子上除去一个氢原子而得到的。原子可以在双键周围以顺式(Z)或反式(E)构象来排布。典型的烯基基团包括但不限于乙烯基、丙烯基、烯丙基(2-丙烯基)、丁烯基等。例子包括C2-8烯基或C2-4烯基。
术语“C(j-k)”(其中j和k为整数,表示碳原子的指定数量)是指烷基以包含j至k个碳原子(包含端值)的前置代号的脚注来显示的烷基、烯基、炔基、烷氧基或环烷基基团,或者指基团中的烷基部分。例如C(1-4)表示包含1、2、3或4个碳原子的基团。
如本文所用,术语“卤代”或“卤素”是指F、Cl、Br或I。
术语“可药用的盐”是本领域公认的,并且是指化合物的相对无毒性的无机酸和有机酸加成盐,或者有机碱或无机碱加成盐,包括(例如)在本发明的组合物中所包含的那些。
如本文所用,术语“固体分散体”是指一组固体产物,其由至少两种不同的组分组成,通常为亲水性载体和疏水性药品(活性成分)。
术语“手性的”是本领域公认的,并且是指具有镜像对应部分的非重叠性性质的分子,而术语“非手性”是指可重叠在它们镜像对应部分上的分子。“前手性分子”是指在特定的过程中具有转化成手性分子的潜力的分子。
符号用于表示键,其可以是单键、双键或三键。
术语“对映异构体”和用于描述对映异构体的结构式意指包含“纯的”对映异构体,其不包含光学异构体以及对映异构体及其光学异构体的混合物,其中对映异构体以对映异构体过量而存在,例如至少10%、25%、50%、75%、90%、95%、98%或99%对映异构体过量。
当在本文中使用术语“立体异构体”时,由所有的几何异构体、对映异构体或非对映异构体组成。本发明涵盖了这些化合物和它们的混合物的多种立体异构体。此外,所公开的化合物的构象异构体和旋转异构体也被包括在内。
如本文所用,术语“立体选择合成”是指这样的化学或酶反应,其中单一的反应物在建立新的立构中心的过程中或者在转化现有的立构中心的过程中形成了立体异构体的不均匀的混合物,并且这些反应是本领域公知的。立体选择合成涵盖了对映选择性和非对映选择性的转化。例如参见Carreira,E.M.和Kvaerno,L.,Classics in Stereoselective Synthesis,Wiley-VCH:Weinheim,2009。
术语“喷雾干燥”是指其中涉及将供料悬液或溶液雾化成小滴并在处理室(其中具有用于蒸发的强驱动力,例如热干气、部分真空或它们的组合)中快速除去混合物中的溶剂的工艺。
如本文所用,术语“治疗有效量”是指这样的任何量,与未接受此量的噻吩并三唑并二氮杂或者任何其他药物活性剂的相应患者相比,该量的本发明的噻吩并三唑并二氮杂或者任何其他药物活性剂改善性地治疗、治愈、预防或减轻疾病、病症或副作用,或者降低疾病或病症的进展的速度。
术语“约”是指+/-10%。在一个实施方案中,术语“约”是指+/-5%。
在本申请及随附的权利要求书的全文中,除非上下文中必须另外说明,否则词语“包括”或其变体(例如“包含”或“包含有”)都应该解释为包括所述的整数或步骤、或者一组整数或步骤,但不排除任何其他的整数或步骤、或一组整数或步骤。此外,术语“包含”应当被理解为隐含着“由…组成”。
目前已经发现,下文所述的式(1)的噻吩并三唑并二氮杂化合物可以与可药用的聚合物一同配制为固体分散体,从而提供可以使药物组分由胃肠道高度地吸收至血液循环中的口服制剂。在一个实施方案中,可药用的聚合物为醋酸羟丙甲纤维素琥珀酸酯(也称为醋酸羟丙基甲基纤维素琥珀酸酯或HPMCAS)。在一个实施方案中,可药用的聚合物为聚乙烯吡咯烷酮(PVP)。
在一些实施方案中,醋酸羟丙基甲基纤维素琥珀酸酯(HPMCAS)可包括:具有9%乙酰基/11%琥珀酰基的M级(例如,平均粒径为5μm的HPMCAS(即,HPMCAS-MF,细粉级)或平均粒径为1mm的HPMCAS(即,HPMCAS-MG,颗粒级));具有12%乙酰基/6%琥珀酰基的H级(例如,平均粒径为5μm的HPMCAS(即,HPMCAS-HF,细粉级)或平均粒径为1mm的HPMCAS(即,HPMCAS-HG,颗粒级));以及具有8%乙酰基/15%琥珀酰基的L级(例如,平均粒径为5μm的HPMCAS(即,HPMCAS-LF,细粉级)或平均粒径为1mm的HPMCAS(即,HPMCAS-LG,颗粒级))。
在一些实施方案中,聚乙烯吡咯烷酮的分子量可为约2,500(12PF,重均分子量在2,000至3,000之间)、约9,000(17PF,重均分子量在7,000至11,000之间)、约25,000(25,重均分子量在28,000至34,000之间)、约50,000(30,重均分子量在44,000至54,000之间)或约1,250,000(90或90F,重均分子量在1,000,000至1,500,000之间)。
II.治疗方法
在一些实施方案中,本说明书提供了使用本文所述的组合物治疗小细胞肺癌(SCLC)的方法。在一些实施方案中,本说明书提供了使用本文所述的组合物治疗非小细胞肺癌(NSCLC)的方法。
在一些实施方案中,本说明书提供了使用本文所述的组合物治疗非小细胞肺癌和/或小细胞肺癌的方法。关于本发明的方法,术语“和/或”是指该方法治疗这样的病症,其中1)非小细胞肺癌和小细胞肺癌都存在于患者体内,或2)非小细胞肺癌或小细胞肺癌,但不是两者都存在于患者体内。
在一些实施方案中,本说明书提供了在哺乳动物中治疗非小细胞肺癌和/或小细胞肺癌的方法,其包括:向需要的患者施用药学上可接受量的组合物,所述组合物包含根据任何的本文所述的III、IV、V和VI部分所述的任意组合物。
在一些实施方案中,本说明书提供了治疗哺乳动物中非小细胞肺癌和/或小细胞肺癌的方法,该方法包括:向需要的患者施用药学可接受量的组合物,该组合物包含根据本文第III、IV、V和VI部分所描述的任意组合物的药物制剂。
在一些实施方案中,本说明书提供了用于治疗非小细胞肺癌和/或小细胞肺癌的式(1)化合物,特别是式(1A)化合物。
在一些实施方案中,本说明书提供根据本文第III、IV、V和VI部分所描述的任意组合物的固体分散体,其用于治疗非小细胞肺癌和/或小细胞肺癌。
在一些实施方案中,本说明书提供了利用式(1)的噻吩并三唑并二氮杂化合物,包括其任意的盐、异构体、对映异构体、消旋物、水合物、溶剂化物、代谢物和多型变体,治疗非小细胞肺癌和/或小细胞肺癌的方法,
其中:
R1为具有1至4个碳的烷基;
R2为氢原子;卤素原子;或任选地被卤素原子或羟基取代的具有1至4个碳的烷基;
R3为卤素原子;任选地被卤素原子、具有1至4个碳的烷基、具有1至4个碳的烷氧基、或氰基取代的苯基;-R5-(CH2)m-R6,其中R5为氢原子、或具有1至4个碳的烷基,m为0至4的整数,并且R6为任选地被卤素原子取代的苯基或吡啶基;或-NR7-CO-(CH2)n-R8,其中R7为氢原子或具有1至4个碳的烷基,n为0至2的整数,并且R8为任选地被卤素原子取代的苯基或吡啶基;并且
R4为-(CH2)a-CO-NH-R9,其中a为1至4的整数,并且R9为具有1至4个碳的烷基;具有1至4个碳的羟烷基;具有1至4个碳的烷氧基;或任选地被具有1至4个碳的烷基、具有1至4个碳的烷氧基、氨基或羟基取代的苯基或吡啶基;或-(CH2)b-COOR10,其中b为1至4的整数,并且R10为具有1至4个碳的烷基。
在一些施方案中,式(I)选自式(IA)、其可药用的盐或溶剂化物:
其中,X为卤素,R1为C1-C4烷基,R2为C1-C4烷基,a为1至4的整数,R3为C1-C4烷基、C1-C4羟烷基、C1-C4烷氧基、任选地具有如式(1)中R9所限定的取代基的苯基、或任选地具有如式(1)中R9所限定的取代基的杂芳基。
在一个此类的实施方案中,将噻吩并三唑并二氮杂化合物配制成含有无定形噻吩并三唑并二氮杂化合物和可药用的聚合物的固体分散体。
在一些实施方案中,小细胞肺癌是复合型小细胞癌。复合型小细胞癌可以指小细胞癌加一种或多种分化形式的肺癌,例如鳞状细胞癌或腺癌。在其他实施方案中,小细胞肺癌是小细胞癌(也称为燕麦细胞癌)。
在本发明方法的一些实施方案中,小细胞肺癌具有纯合突变C637C>T。在本发明方法的一些实施方案中,治疗诱导小细胞肺癌中HEXIM的上调。
在一些实施方案中,非小细胞肺癌是EML4-ALk阳性的。在一个实施方案中,非小细胞肺癌在治疗后展示出N-MYC mRNA水平的下调。在另一个实施方案中,非小细胞肺癌表达BRD4/3/2、c-MYC、BCL-2、p21和细胞周期蛋白D1或它们的组合。在另一个实施方案中,治疗诱导STAT3的瞬时上调,随后在暴露后约24小时至约72小时之间下调。
在另一个实施方案中,所述非小细胞肺癌是EML4-ALk阴性的。在一个实施方案中,非小细胞肺癌表达BRD4/3/2、c-MYC、BCL-2、p21和细胞周期蛋白D1或它们的组合。在另一个实施方案中,治疗诱导STAT3的瞬时上调,随后在24小时和最多72小时暴露后下调。
在一些实施方案中,非小细胞肺癌在KRAS基因中具有突变。在一些实施方案中,非小细胞肺癌在LKB1基因中具有突变。在一些实施方案中,非小细胞肺癌在KRAS和LKB1基因中都具有突变。
在一些实施方案中,治疗诱导N-MYC的下调(例如在一些实施方案中,非小细胞肺癌在治疗期间或之后展示出N-MYC mRNA水平的下调)。在一些实施方案中,治疗诱导IL7R的下调。在一些实施方案中,治疗诱导IL6相关基因的下调。在一些实施方案中,治疗诱导HEXIM的上调。在一些实施方案中,治疗诱导FOS的上调。在一些实施方案中,治疗诱导EFR3B的上调。
在一些实施方案中,本说明书提供了使用式(1)的噻吩并三唑并二氮杂化合物化疗药物联合治疗非小细胞肺癌和/或小细胞肺癌的方法,所述化疗药物选自由m-TOR抑制剂和ALK抑制剂构成的组。
在一些实施方案中,本说明书提供了使用式(1A)的噻吩并三唑并二氮杂化合物与化疗药物联合治疗非小细胞肺癌和/或小细胞肺癌的方法,所述化疗药物选自由m-TOR抑制剂和ALK抑制剂构成的组。
在一些实施方案中,该方法进一步包括向患者使用mTOR抑制剂。在一个此类的实施方案中,mTOR抑制剂选自由雷帕霉素、替西罗莫司、地磷莫司和依维莫司所构成的组。在一个此类的实施方案中,mTOR抑制剂是依维莫司。噻吩并三唑并二氮杂化合物和mTOR抑制剂可以同时或按顺序施用。在一些实施方案中,噻吩并三唑并二氮杂化合物和mTOR抑制剂的此类组合产生协同效应。
在本发明的方法中,与式(1)的噻吩并三唑并二氮杂联合使用的针对哺乳动物的雷帕霉素(mTOR)抑制剂的例子包括但不限于下表A中所列出的mTOR抑制剂。
表A:可以与式(1)的噻吩并三唑并二氮杂联合使用的示例性的mTOR抑制剂化合物:
在一些实施方案中,所述方法还包括向患者施用ALK抑制剂。在一个此类的实施方案中,ALK抑制剂选自由ceritinib和克唑替尼所构成的组。在一个此类的实施方案中,ALK抑制剂是克唑替尼。噻吩并三唑并二氮杂化合物和ALK抑制剂可以同时或按顺序施用。在一些实施方案中,噻吩并三氮杂二氮化合物和ALK抑制剂的此类组合产生协同效应。
在本发明的方法中,与式(1)的噻吩并三唑并二氮杂联合使用的ALK抑制剂的例子包括但不限于下表B中所列出的ALK抑制剂。
表B:可以与式(1)的噻吩并三唑并二氮杂联合使用的示例性的ALK抑制剂化合物:
在一个实施方案中,使式(1)的噻吩并三唑并二氮化合物形成固体分散体。在一个实施方案中,固体分散体包含无定形的所述化合物。
在本发明中,“治疗”是指向由医生诊断为患有小细胞肺癌或具有发展成小细胞肺癌的风险的人(患者)施用本发明的活性成分的行为或行动,其旨在(例如)减轻小细胞肺癌或症状、预防小细胞肺癌或症状的发作、或恢复小细胞肺癌发病前的状态。
III.噻吩并三唑并二氮杂化合物:
在一个实施方案中,在本发明的制剂中使用的噻吩并三唑并二氮杂化合物如式(1)所示,包括其任意的盐、异构体、对映异构体、消旋物、水合物、溶剂化物、代谢物和多型变体:
其中R1为具有1至4个碳的烷基;
R2为氢原子、卤素原子、或任选地被卤素原子或羟基取代的具有1至4个碳的烷基;
R3为:卤素原子;任选地被卤素原子、具有1至4个碳的烷基、具有1至4个碳的烷氧基、或氰基取代的苯基;-NR5-(CH2)m-R6,其中R5为氢原子、或具有1至4个碳的烷基,m为0至4的整数,并且R6为任选地被卤素原子取代的苯基或吡啶基;或-NR7-CO-(CH2)n-R8,其中R7为氢原子或具有1至4个碳的烷基,n为0至2的整数,并且R8为任选地被卤素原子取代的苯基或吡啶基;并且
R4为:-(CH2)a-CO-NH-R9,其中a为1至4的整数,并且R9为具有1至4个碳的烷基;具有1至4个碳的羟烷基;具有1至4个碳的烷氧基;或任选地被具有1至4个碳的烷基、具有1至4个碳的烷氧基、氨基或羟基取代的苯基或吡啶基;或-(CH2)b-COOR10,其中b为1至4的整数,并且R10为具有1至4个碳的烷基。
在一个实施方案中,合适的烷基包括含有1个碳原子至4个碳原子的直链或支链烷基基团。在一个实施方案中,合适的烷基包括含有1个碳原子至3个碳原子的直链或支链烷基基团。在一个实施方案中,合适的烷基包括含有1个碳原子至2个碳原子的直链或支链烷基基团。在一个实施方案中,示例性的烷基基团包括但不限于甲基、乙基、丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基。在一个实施方案中,示例性的烷基基团包括但不限于甲基、乙基、丙基、异丙基、2-甲基-1-丙基和2-甲基-2-丙基。
在一些实施方案中,本发明提供了本文所述的噻吩并三唑并二氮杂化合物的可药用的盐、溶剂化物(包括水合物)和同位素标记的形式。在一个实施方案中,噻吩并三唑并二氮杂化合物的可药用的盐包括与无机酸形成的酸加成盐。在一个实施方案中,噻吩并三唑并二氮杂的可药用的无机酸加成盐包括盐酸、氢溴酸、氢碘酸、磷酸、偏磷酸、硝酸和硫酸的盐。在一个实施方案中,噻吩并三唑并二氮杂化合物的可药用的盐包括与有机酸形成的酸加成盐。在一个实施方案中,噻吩并三唑并二氮杂化的可药用的有机酸加成盐包括以下酸的盐:酒石酸、乙酸、三氟乙酸、柠檬酸、苹果酸、乳酸、富马酸、苯甲酸、甲酸、丙酸、乙醇酸、葡萄糖酸、马来酸、琥珀酸、樟脑硫酸、异硫羰酸(isothionic acid)、粘酸、龙胆酸、异烟酸、糖二酸、葡萄糖醛酸、糠酸、谷氨酸、抗坏血酸、邻氨基苯甲酸、水杨酸、苯乙酸、扁桃酸、扑酸(帕莫酸)、甲烷磺酸、乙烷磺酸、泛酸、硬脂酸、磺胺酸、褐藻酸、半乳糖醛酸和芳基磺酸(例如苯磺酸和4-甲基苯磺酸)。
本发明提供了本发明所述的噻吩并三唑并二氮杂化合物的药学可接受的同位素标记形式,其中一个或多个原子被原子数相同、但是原子量或质量数与天然常见的原子量或质量数不同的原子所替代。适于包含在噻吩并三唑并二氮杂化合物中的同位素的实例包括以下元素的同位素:氢,例如2H和3H;碳,例如11C、13C和14C;氯,例如36Cl;氟,例s如18F;碘,例如123I和125I;氮,例如13N和15N;氧,例如15O、17O和18O;以及硫,例如35S。通常可以通过本领域那些技术人员已知的常规技术来制备噻吩并三唑并二氮杂化合物的同位素标记形式。
式(1)所示化合物的某些同位素标记形式(例如引入了放射性同位素的那些)可用于药品和/或底物的组织分布研究中。由于放射性同位素氚(3H)和碳-14(14C)易于引入和即时的检测手段,所以它们特别适用于上述目的。使用较重的同位素(例如氘(2H))进行取代可以提供由于更广的代谢稳定性而产生的某些治疗益处,例如体内半衰期增长或者剂量需求降低,因此,在一些情况下可以是优选的。使用发射正电子的同位素(例如11C、18F、15O和13N)进行取代可以用于正电子发射断层成像术研究(PET)中,以用于检测底物受体的占用。
在一些实施方案中,本发明公开的噻吩并三唑并二氮杂化合物可以与药学可接受的溶剂以溶剂化和非溶剂化的形式存在。本领域技术人员应该理解的是,溶剂化物为溶质(在这种情况下为本发明所述的噻吩并三唑并二氮杂化合物)和溶剂形成的化学计量可变的复合物。优选的是此类溶剂不会干扰溶质(噻吩并三唑并二氮杂化合物)的生物活性。用于形成溶剂化物的合适的溶剂的实例包括但不限于水、甲醇、二甲基亚砜、乙醇和乙酸。所用的合适的溶剂为药学可接受的溶剂。合适的是,所用的溶剂为水。在一个实施方案中,本发明所述的噻吩并三唑并二氮杂化合物的药学可接受的溶剂化物包括乙醇溶剂化物、异丙醇溶剂化物、二氧戊烷溶剂化物、四氢呋喃溶剂化物、二甲基亚砜溶剂化物、叔丁醇溶剂化物、2-丁醇溶剂化物、二氧戊烷溶剂化物、1,3-二甲基-3,4,5,6-四氢-2(1H)-嘧啶酮(“DMPU”)溶剂化物、1,3-二甲基咪唑啉酮(“DMI”)溶剂化物和1,3-二甲基咪唑啉酮(“DMP”)溶剂化物、或它们的混合物。
在一些实施方案中,本发明所述的噻吩并三唑并二氮杂化合物可以包含一个或多个手性中心和/或双键,因此可以以几何异构体、对映异构体或非对映异构体的形式存在。可以根据Cahn–Ingold–Prelog规则来命名噻吩并三唑并二氮杂化合物的对映异构体和非对映异构体,所述的规则将“R”或“S”描述符指定给每个立构中心(通常也称为手性中心),并将E或Z描述符指定给每个碳-碳双键(以命名几何异构体),这样可以通过在系统名中包含描述符而独特地描述整个分子的构造。
在一些实施方案中,本发明所述的噻吩并三唑并二氮杂化合物可以以外消旋混合物或外消旋物的形式存在,其可以包含手性分子的等量的左旋和右旋对映异构体。可以通过前缀(±)-或dl-来表示此类外消旋混合物,表明右旋和左旋异构体的相等(1:1)的混合物。此外,前缀rac-(或racem-)或符号RS和SR也可以用于命名外消旋混合物。
此外,由取代基围绕碳-碳双键的排列或者取代基围绕环烷基或杂环的排列而产生的几何异构体也可以存在于本发明的化合物中。在一些实施方案中,符号可用于表示可以为单键、双键或三键的键。围绕碳-碳双键的取代基可以命名为呈“Z”或“E”构型,其中根据IUPAC标准使用术语“Z”和“E”。除非另有说明,否则描述双键的结构涵盖了“Z”和“E”异构体。围绕碳-碳双键的取代基也可以称为“顺式”或“反式(”,其中“顺式”表示取代基在双键的同侧上,而“反式”表示取代基在双键的对侧上。取代基围绕碳环的排列也可以命名为“顺式”或“反式”。术语“顺式”表示取代基在环的平面的同侧上,而术语“反式”表示取代基在环的平面的对侧上。其中取代基置于环的平面的同侧和对侧上的化合物所形成的混合物命名为“顺式/反式”或“Z/E”。
在一些实施方案中,本发明所公开的噻吩并三唑并二氮杂化合物可以以单晶或多晶形式或多型物存在。在一个实施方案中,本发明所公开的噻吩并三唑并二氮杂化合物包含其无定形形式。在一个实施方案中,本发明所公开的噻吩并三唑并二氮杂化合物包含其单一多晶型物。在另一个实施方案中,本发明所公开的噻吩并三唑并二氮杂化合物包含其多晶型物的混合物。在另一个实施方案中,所述化合物为结晶形式。
在一些实施方案中,本发明所公开的噻吩并三唑并二氮杂化合物可以以单一对映异构体或者对映异构体的富集形式存在。在一个实施方案中,本发明所公开的噻吩并三唑并二氮杂化合物以对映异构体过量百分比(entiomeric excess)超过80%的形式存在。在一个实施方案中,本发明所公开的噻吩并三唑并二氮杂化合物以对映异构体过量百分比超过90%的形式存在。在一个实施方案中,本发明所公开的噻吩并三唑并二氮杂化合物以对映异构体过量百分比超过98%的形式存在。在一个实施方案中,本发明所公开的噻吩并三唑并二氮杂化合物以对映异构体过量百分比超过99%的形式存在。在一个实施方案中,本发明所公开的噻吩并三唑并二氮杂化合物以选自由至少10%、至少25%、至少50%、至少75%、至少90%、至少95%、至少98%和至少99%所构成的组中的对映异构体过量百分比的形式存在。
对于一对对映异构体对而言,可以使用以下等式(等式(1))来计算对映异构体E1相对于对映异构体E2的对映异构体过量百分比:
可以通过手性高效液相色谱(HPLC)、核磁共振(NMR)或任何其他合适的方法来测定E1和E2的相对量。在一些实施方案中,对映异构体化合物的纯度可以指对映异构体E1和E2相对于其他材料的量,这些其他物质特别包括副产物和/或未反应的反应物或试剂。
在一些实施方案中,式(1)的噻吩并三唑并二氮杂化合物包括但不限于噻吩并三唑并二氮杂化合物(1-1)至(1-18),其列于下表C中。
本文将表C的化合物(1-1)也称为OTX-015、OTX015或Y803。
表C:本发明的制剂中使用的示例性化合物:
表C(续)
表C(续)
在一些实施方案中,式(1)所示的噻吩并三唑并二氮杂化合物包括:(i)(S)-2-[4-(4-氯代苯基)-2,3,9-三甲基-6H-噻吩并[3,2-f][1,2,4]三唑并-[4,3-a][1,4]二氮杂-6-基]-N-(4-羟苯基)乙酰胺或其二水合物;(ii)(S)-{4-(3'-氰基联苯-4-基)-2,3,9-三甲基-6H-噻吩并[3,2-f][1,2,4]三唑并[4,3-a][1,4]二氮杂-6-基}乙酸甲酯;(iii)(S)-{2,3,9-三甲基-4-(4-苯基氨基苯基)-6H-噻吩并[3,2-f][1,2,4]三唑并[4,3-a][1,4]二氮杂-6-基}乙酸甲酯;以及(iv)(S)-{2,3,9-三甲基-4-[4-(3-苯基丙酰基氨基)苯基]-6H-噻吩并[3,2-f-][1,2,4]三唑并[4,3-a][1,4]二氮杂-6-基}乙酸甲酯。
在一些实施方案中,式(1)所示的噻吩并三唑并二氮杂化合物包括(S)-2-[4-(4-氯代苯基)-2,3,9-三甲基-6H-噻吩并[3,2-f][1,2,-4]三唑并[4,3-a][1,4]二氮杂-6-基]-N-(4-羟苯基)乙酰胺二水合物。
IV.制剂:
式(1)所示的化合物在盖伦组合物的常规给药和制备方面表现出非常特有的困难,特别包括药物生物利用度以及患者间和患者内的剂量应答的可变性的特定问题,由此需要针对几乎水不溶性的所述化合物开发非常规的剂型。
之前,已经发现可以利用载体丙烯酸乙酯-甲基丙烯酸甲酯-三甲基铵乙基甲基丙烯酸酯氯化物共聚物(Eudragit RS,由Rohm公司制造)将式(1)所示的化合物配置成固体分散体,从而提供在下肠道中优先释放药物组分以用于治疗炎性肠病(例如溃疡性结肠炎和克罗恩病)的口服制剂(2009年1月8日公开的美国专利申请20090012064A1)。通过多个实验(包括动物试验),发现就炎性肠病而言,药物在病变部位的释放及其对炎性病变的直接作用比其由胃肠道吸收至循环中更为重要。
现在,意外地发现根据式(1)所示的噻吩并三唑并二氮杂化合物、其可药用的盐、溶剂化物(包含水合物)、消旋物、对映异构体以及同位素标记的形式可以使用可药用的聚合物配制成固体分散体,从而提供口服制剂,该口服制剂能够使药物组分由胃肠道高度吸收至循环中,从而治疗炎性肠病以及其他疾病。在狗和人类中的研究证明,与之前研发的用于治疗炎性肠病的Eudragit固体分散体制剂相比,所述这些固体分散体具有高的口服生物利用度。
固体分散体为改善水溶性差的药物的口服生物利用度的一种手段。
如本文所用,术语“固体分散体”是指一组固体产物,其包含至少2种不同的成分,通常为亲水性载体和疏水性药物(根据式(1)的噻吩并三唑并二氮杂化合物)。基于药物在分散体内的分子排布,可以区分为6种不同类型的固体分散体。通常,固体分散体分为:简单低共熔混合物、固溶体、玻璃溶液和悬浮液、以及结晶载体中的无定形沉淀。此外,可存在一些组合,例如在同一样品中,一些分子可以成簇存在,而一些分子以分子形式分散。
在一个实施方案中,根据式(1)的噻吩并三唑并二氮杂化合物可以以分子形式分散于无定形颗粒(簇)中。在另一个实施方案中,根据式(1)的噻吩并三唑并二氮杂化合物可以以结晶颗粒形式分散。在一个实施方案中,载体可以是结晶的。在另一个实施方案中,载体可以是无定形的。
在一个实施方案中,本发明提供了包含固体分散体的药物组合物,其中所述固体分散体由以下形成:根据式(1)的噻吩并三唑并二氮杂化合物或其可药用的盐、溶剂化物(包括水合物)、消旋物、对映异构体、异构体或其同位素标记形式;以及可药用的聚合物。在一个实施方案中,可药用的聚合物为醋酸羟丙甲纤维素琥珀酸酯(也称为醋酸羟丙基甲基纤维素琥珀酸酯或HPMCAS)。在一个实施方案中,分散体中噻吩并三唑并二氮杂化合物与醋酸羟丙基甲基纤维素琥珀酸酯(HPMCAS)的重量比为1:3至1:1。在一个实施方案中,至少一部分噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在另一个实施方案中,噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在一些实施方案中,固体分散体表现出单一拐点的玻璃化转变温度(Tg)。在一些实施方案中,单一的Tg在130℃至140℃之间出现。在其他这样的实施方案中,单一的Tg在约135℃出现。在一些这样的实施方案中,固体分散体在40℃、相对湿度为75%的环境下暴露至少1个月。在一些实施方案中,在固体分散体的X射线粉末衍射图案中,基本上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。就本申请的目的而言,“基本上不存在”是指在约21°2θ下,在无定形晕(amorphous halo)上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。
在一个实施方案中,本发明提供了一种药物组合物,其包含式(1)所示的噻吩并三唑并二氮杂化合物或其可药用的盐、溶剂化物(包括水合物)、消旋物、对映异构体、异构体或同位素标记形式在可药用的聚合物中的固体分散体。在一个实施方案中,可药用的聚合物为聚乙烯吡咯烷酮(也称为聚维酮或PVP)。在一个实施方案中,分散体中噻吩并三唑并二氮杂化合物与PVP的重量比为1:3至1:1。在一个实施方案中,至少一部分噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在另一个实施方案中,噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在一些实施方案中,固体分散体表现出单一拐点的玻璃化转变温度(Tg)。在一些实施方案中,单一的Tg在175℃至约185℃之间出现。在其他这样的实施方案中,单一的Tg在约179℃出现。在一些这样的实施方案中,固体分散体在40℃、相对湿度为75%的环境下暴露至少1个月。在一些实施方案中,在固体分散体的X射线粉末衍射图案中,基本上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。就本申请的目的而言,“基本上不存在”是指在约21°2θ下,在无定形晕上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。
在一个实施方案中,本发明的药物组合物包含无定形形式的式(1)所示的噻吩并三唑并二氮杂化合物或其可药用的盐、溶剂化物(包括水合物)、消旋物、对映异构体、异构体或同位素标记形式与可药用的聚合物的固体分散体。在一个实施方案中,可药用的聚合物为醋酸羟丙甲纤维素琥珀酸酯。在一个实施方案中,式(1)所示的噻吩并三唑并二氮杂化合物与醋酸羟丙甲纤维素琥珀酸酯的重量比为1:3至1:1。在一个实施方案中,至少一部分噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在另一个实施方案中,噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在一些实施方案中,固体分散体表现出单一拐点的玻璃化转变温度(Tg)。在一些实施方案中,单一的Tg在130℃至140℃之间出现。在其他这样的实施方案中,单一的Tg在约135℃出现。在一些这样的实施方案中,固体分散体在40℃、相对湿度为75%的环境下暴露至少1个月。在一些实施方案中,在固体分散体的X射线粉末衍射图案中,基本上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。就本申请的目的而言,“基本上不存在”是指在约21°2θ下,在无定形晕上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。
在一个实施方案中,本发明的药物组合物包含无定形形式的式(1)所示的噻吩并三唑并二氮杂化合物或其可药用的盐、溶剂化物(包括水合物)、消旋物、对映异构体、异构体或同位素标记形式与可药用的聚合物的固体分散体。在一个实施方案中,可药用的聚合物为聚乙烯吡咯烷酮。在一个实施方案中,式(1)所示的噻吩并三唑并二氮杂化合物与聚乙烯吡咯烷酮的重量比为1:3至1:1。在一个实施方案中,至少一部分噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在另一个实施方案中,噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在一些实施方案中,固体分散体表现出单一拐点的玻璃化转变温度(Tg)。在一些实施方案中,单一的Tg在175℃至约185℃之间出现。在其他这样的实施方案中,单一的Tg在约179℃下出现。在一些这样的实施方案中,固体分散体在40℃、相对湿度为75%的环境下暴露至少1个月。在一些实施方案中,在固体分散体的X射线粉末衍射图案中,基本上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。就本申请的目的而言,“基本上不存在”是指在约21°2θ下,在无定形晕上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。
在一个实施方案中,本发明的药物组合物包含结晶形式的式(1)所示的噻吩并三唑并二氮杂化合物或其可药用的盐、溶剂化物(包括水合物)、消旋物、对映异构体、异构体或同位素标记形式与可药用的聚合物的固体分散体。在一个实施方案中,可药用的聚合物为醋酸羟丙基甲基纤维素琥珀酸酯。在一个实施方案中,式(1)所示的噻吩并三唑并二氮杂化合物与醋酸羟丙基甲基纤维素琥珀酸酯的重量比为1:3至1:1。
在一个实施方案中,本发明的药物组合物包含结晶形式的式(1)所示的噻吩并三唑并二氮杂化合物或其可药用的盐、溶剂化物(包括水合物)、消旋物、对映异构体、异构体或同位素标记形式与可药用的聚合物的固体分散体。在一个实施方案中,可药用的聚合物为聚乙烯吡咯烷酮。在一个实施方案中,式(1)所示的噻吩并三唑并二氮杂化合物与聚乙烯吡咯烷酮的重量比为1:3至1:1。
在一些实施方案中,通过喷雾干燥来制备包含固体分散体的药物组合物。
在一个实施方案中,本发明的药物组合物包含喷雾干燥的固体分散体,该固体分散体由以下形成:式(1)所示的噻吩并三唑并二氮杂化合物或其可药用的盐、溶剂化物(包括水合物)、消旋物、对映异构体、异构体或同位素标记形式,和可药用的聚合物。在一个实施方案中,可药用的聚合物为醋酸羟丙基甲基纤维素琥珀酸酯。在一个实施方案中,化合物(1)与醋酸羟丙基甲基纤维素琥珀酸酯的重量比为1:3至1:1。在一个实施方案中,至少一部分噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在另一个实施方案中,噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在一些实施方案中,固体分散体表现出单一拐点的玻璃化转变温度(Tg)。在一些实施方案中,单一的Tg在130℃至140℃之间出现。在其他这样的实施方案中,单一的Tg在约135℃出现。在一些这样的实施方案中,固体分散体在40℃、相对湿度为75%的环境下暴露至少1个月。在一些实施方案中,在固体分散体的X射线粉末衍射图案中,基本上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。就本申请的目的而言,“基本上不存在”是指在约21°2θ下,在无定形晕上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。
在一个实施方案中,本发明的药物组合物包含喷雾干燥的固体分散体,该固体分散体由以下形成:式(1)所示的噻吩并三唑并二氮杂化合物或其可药用的盐、溶剂化物(包括水合物)、消旋物、对映异构体、异构体或同位素标记形式和可药用的聚合物。在一个实施方案中,可药用的聚合物为聚乙烯吡咯烷酮。在一个实施方案中,化合物(1)与聚乙烯吡咯烷酮的重量比为1:3至1:1。在一个实施方案中,至少一部分噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在另一个实施方案中,噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在一些实施方案中,固体分散体表现出单一拐点的玻璃化转变温度(Tg)。在一些实施方案中,单一的Tg在175℃至185℃之间出现。在其他这样的实施方案中,单一的Tg在约179℃出现。在一些这样的实施方案中,固体分散体在40℃、相对湿度为75%的环境下暴露至少1个月。在一些实施方案中,在固体分散体的X射线粉末衍射图案中,基本上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。就本申请的目的而言,“基本上不存在”是指在约21°2θ下,在无定形晕上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。
在一个实施方案中,本发明的药物组合物包含喷雾干燥的固体分散体,该固体分散体由以下形成:无定形的式(1)所示的噻吩并三唑并二氮杂化合物或其可药用的盐、溶剂化物(包括水合物)、消旋物、对映异构体、异构体或同位素标记形式和可药用的聚合物。在一个实施方案中,可药用的聚合物为醋酸羟丙基甲基纤维素琥珀酸酯。在一个实施方案中,式(1)所示的噻吩并三唑并二氮杂化合物与醋酸羟丙基甲基纤维素琥珀酸酯的重量比为1:3至1:1。在一个实施方案中,至少一部分噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在另一个实施方案中,噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在一些实施方案中,固体分散体表现出单一拐点的玻璃化转变温度(Tg)。在一些实施方案中,单一的Tg在130℃至140℃之间出现。在一些这样的实施方案中,固体分散体在40℃、相对湿度为75%的环境下暴露至少1个月。在其他这样的实施方案中,单一的Tg在约135℃出现。在一些实施方案中,在固体分散体的X射线粉末衍射图案中,基本上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。就本申请的目的而言,“基本上不存在”是指在约21°2θ下,在无定形晕上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。
在一个实施方案中,本发明的药物组合物包含喷雾干燥的固体分散体,该固体分散体由以下形成:无定形的式(1)所示的噻吩并三唑并二氮杂化合物或其可药用的盐、溶剂化物(包括水合物)、消旋物、对映异构体、异构体或同位素标记形式和可药用的聚合物。在一个实施方案中,可药用的聚合物为聚乙烯吡咯烷酮。在一个实施方案中,式(1)所示的噻吩并三唑并二氮杂化合物与聚乙烯吡咯烷酮的重量比为1:3至1:1。在一个实施方案中,至少一部分噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在另一个实施方案中,噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在一些实施方案中,固体分散体表现出单一拐点的玻璃化转变温度(Tg)。在一些实施方案中,单一的Tg在175℃至185℃之间出现。在其他这样的实施方案中,单一的Tg在约179℃出现。在一些这样的实施方案中,固体分散体在40℃、相对湿度为75%的环境下暴露至少1个月。在一些实施方案中,在固体分散体的X射线粉末衍射图案中,基本上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。就本申请的目的而言,“基本上不存在”是指在约21°2θ下,在无定形晕上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。
在一个实施方案中,本发明的药物组合物包含喷雾干燥的固体分散体,该固体分散体由以下形成:结晶形式的式(1)所示的噻吩并三唑并二氮杂化合物或其可药用的盐、溶剂化物(包括水合物)、消旋物、对映异构体、异构体或同位素标记形式和可药用的聚合物。在一个实施方案中,可药用的聚合物为醋酸羟丙基甲基纤维素琥珀酸酯。在一个实施方案中,式(1)所示的噻吩并三唑并二氮杂化合物与醋酸羟丙基甲基纤维素琥珀酸酯的重量比为1:3至1:1。
在一个实施方案中,本发明的药物组合物包含喷雾干燥的固体分散体,该固体分散体由以下形成:结晶形式的式(1)所示的噻吩并三唑并二氮杂化合物或其可药用的盐、溶剂化物(包括水合物)、消旋物、对映异构体、异构体或同位素标记形式和可药用的聚合物。在一个实施方案中,可药用的聚合物为聚乙烯吡咯烷酮。在一个实施方案中,式(1)所示的噻吩并三唑并二氮杂化合物与聚乙烯吡咯烷酮的重量比为1:3至1:1。
在一个优选的实施方案中,本发明提供了一种药物组合物,其包含固体分散体,该固体分散体由以下形成:2-[(6S)-4-(4-氯代苯基)-2,3,9-三甲基-6H-噻吩并[3,2-f]-[1,2,4]三唑并[4,3-a][1,4]二氮杂-6-基]-N-(4-羟苯基)乙酰胺二水合物(化合物(1-1))或其可药用的盐、溶剂化物(包括水合物)、消旋物、对映异构体、异构体或同位素标记形式;以及可药用的聚合物,其中所述化合物(1-1)如下:
在一个实施方案中,可药用的聚合物为HPMCAS。在一个实施方案中,分散体中化合物(1-1)和HPMCAS的重量比为1:3至1:1。在一个实施方案中,至少一部分噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在另一个实施方案中,噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在一个实施方案中,固体分散体是喷雾干燥的。在一些实施方案中,固体分散体表现出单一拐点的玻璃化转变温度(Tg)。在一些实施方案中,单一的Tg在130℃至140℃之间出现。在其他这样的实施方案中,单一的Tg在约135℃出现。在一些这样的实施方案中,固体分散体在40℃、相对湿度为75%的环境下暴露至少1个月。在一些实施方案中,在固体分散体的X射线粉末衍射图案中,基本上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。就本申请的目的而言,“基本上不存在”是指在约21°2θ下,在无定形晕上不存在与结晶的噻吩并三唑并二氮杂化合物(1-1)有关的衍射线。
在另一个实施方案中,所述药物组合物包含化合物(1-1)或其可药用的盐、溶剂化物(包括水合物)、消旋物、对映异构体、异构体或同位素标记形式;以及可药用的聚合物的固体分散体。在一个实施方案中,可药用的聚合物为PVP。在一个实施方案中,分散体中化合物(1-1)和PVP的重量比为1:3至1:1。在一个实施方案中,至少一部分噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在另一个实施方案中,噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在一个实施方案中,固体分散体是喷雾干燥的。在一些实施方案中,固体分散体表现出单一拐点的玻璃玻璃化转变温度(Tg)。在一些实施方案中,单一的Tg在175℃至185℃之间出现。在其他这样的实施方案中,单一的Tg在约179℃出现。在一些这样的实施方案中,固体分散体在40℃、相对湿度为75%的环境下暴露至少1个月。在一些实施方案中,在固体分散体的X射线粉末衍射图案中,基本上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。就本申请的目的而言,“基本上不存在”是指在约21°2θ下,在无定形晕上不存在与结晶的噻吩并三唑并二氮杂化合物(1-1)有关的衍射线。
在一个实施方案中,本发明的药物组合物包含无定形形式的噻吩并三唑并二氮杂化合物(1-1)或其可药用的盐、溶剂化物(包括水合物)、消旋物、对映异构体、异构体或同位素标记形式;以及可药用的聚合物。在一个实施方案中,可药用的聚合物为HPMCAS。在一个实施方案中,分散体中化合物(1-1)与HPMCAS的重量比为1:3至1:1。在一个实施方案中,至少一部分噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在另一个实施方案中,噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在一个实施方案中,固体分散体是喷雾干燥的。在一些实施方案中,固体分散体表现出单一拐点的玻璃化转变温度(Tg)。在一些实施方案中,单一的Tg在130℃至140℃之间出现。在其他这样的实施方案中,单一的Tg在约135℃出现。在一些这样的实施方案中,固体分散体在40℃、相对湿度为75%的环境下暴露至少1个月。在一些实施方案中,在固体分散体的X射线粉末衍射图案中,基本上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。就本申请的目的而言,“基本上不存在”是指在约21°2θ下,在无定形晕上不存在与结晶的噻吩并三唑并二氮杂化合物(1-1)有关的衍射线。
在一个实施方案中,本发明的药物组合物包含无定形形式的噻吩并三唑并二氮杂化合物(1-1)或其可药用的盐、溶剂化物(包括水合物)、消旋物、对映异构体、异构体或同位素标记形式与可药用的聚合物的固体分散体。在一个实施方案中,可药用的聚合物为PVP。在一个实施方案中,分散体中化合物(1-1)与PVP的重量比为1:3至1:1。在一个实施方案中,至少一部分噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在另一个实施方案中,噻吩并三唑并二氮杂化合物均匀地分散于整个固体分散体中。在一个实施方案中,固体分散体是喷雾干燥的。在一些实施方案中,固体分散体表现出单一拐点的玻璃化转变温度(Tg)。在一些实施方案中,单一的Tg在175℃至185℃之间出现。在其他这样的实施方案中,单一的Tg在约189℃出现。在一些这样的实施方案中,固体分散体在40℃、相对湿度为75%的环境下暴露至少1个月。在一些实施方案中,在固体分散体的X射线粉末衍射图案中,基本上不存在与结晶的式(1)所示的噻吩并三唑并二氮杂化合物有关的衍射线。就本申请的目的而言,“基本上不存在”是指在约21°2θ下,在无定形晕上不存在与结晶的噻吩并三唑并二氮杂化合物(1-1)有关的衍射线。
在一个实施方案中,本发明的药物组合物包含结晶形式的噻吩并三唑并二氮杂化合物(1-1)或其可药用的盐、溶剂化物(包括水合物)、消旋物、对映异构体、异构体或同位素标记形式与可药用的聚合物的固体分散体。在一个实施方案中,可药用的聚合物为HPMCAS。在一个实施方案中,分散体中化合物(1-1)与HPMCAS的重量比为1:3至1:1。在一个实施方案中,固体分散体是喷雾干燥的。
在一个实施方案中,本发明的药物组合物包含结晶形式的噻吩并三唑并二氮杂化合物(1-1)或其可药用的盐、溶剂化物(包括水合物)、消旋物、对映异构体、异构体或同位素标记形式与可药用的聚合物的固体分散体。在一个实施方案中,可药用的聚合物为PVP。在一个实施方案中,分散体中化合物(1-1)与PVP的重量比为1:3至1:1。在一个实施方案中,固体分散体是喷雾干燥的。
当经口施用本发明所述的固体分散体时,其表现出特别有利的性质。当在动物或人类的标准生物利用度试验中施用时,所述固体分散体的有利性质的示例包括但不限于一致的且高水平的生物利用度。本发明的固体分散体可以包括含有式(1)所示的噻吩并三唑并二氮杂化合物、聚合物以及添加剂的固体分散体。在一些实施方案中,固体分散体可以实现使式(1)所示的噻吩并三唑并二氮杂化合物吸收入血流中,而这是不可能通过仅仅将式(1)所示的噻吩并三唑并二氮杂化合物与添加剂混合而获得的,这是因为式(1)所示的噻吩并三唑并二氮杂化合物药物在水和大多数水性介质中的溶解度可忽略不计。可以使用多种体外和/或体内研究来测量式(1)所示的噻吩并三唑并二氮杂化合物或者噻吩并三唑并二氮杂化合物(1-1)的生物利用度。体内研究可以使用例如大鼠、狗或人类来进行。
相对于时间横坐标(X轴),以纵坐标(Y轴)绘制式(1)所示的噻吩并三唑并二氮杂化合物或者噻吩并三唑并二氮杂化合物(1-1)的血清或血浆浓度,可以通过由此获得的曲线下面积(AUC)值测量生物利用度。然后,将所述固体分散体中式(1)所示的噻吩并三唑并二氮杂化合物或者噻吩并三唑并二氮杂化合物(1-1)的AUC值与等价浓度的不包含聚合物的结晶的式(1)所示的噻吩并三唑并二氮杂化合物或结晶性的噻吩并三唑并二氮杂化合物(1-1)的AUC值比较。在一些实施方案中,当将所述固体分散体经口施用给狗时,该固体分散体的曲线下面积(AUC)值选自:由静脉施用给狗的对照组合物的相应AUC值的至少0.4倍、0.5倍、0.6倍、0.8倍、1.0倍,其中所述对照组合物包含等价量的结晶的式(1)所示的噻吩并三唑并二氮杂化合物。
可以通过模拟胃环境和肠环境的pH值的体外测试来测量生物利用度。可以通过以下方法进行测量:将式(1)所示的噻吩并三唑并二氮杂化合物或噻吩并三唑并二氮杂化合物(1-1)的固体分散体悬浮在pH为1.0至2.0之间的水性体外测试介质中,随后将对照体外测试介质的pH值调节为5.0至7.0。可以在pH值调节后的最初2个小时中的任何时间来测量无定形的式(1)所示的噻吩并三唑并二氮杂化合物或无定形噻吩并三唑并二氮杂化合物(1-1)的浓度。在一些实施方案中,在pH值为5.0至7.0之间的水性体外测试介质中,与不包含聚合物的结晶的式(1)所示的噻吩并三唑并二氮杂化合物或者结晶的噻吩并三唑并二氮杂化合物(1-1)的浓度相比,固体分散体使无定形的式(1)所示的噻吩并三唑并二氮杂化合物或者无定形的噻吩并三唑并二氮杂化合物(1-1)的浓度高至少5倍、至少6倍、至少7倍、至少8倍、至少9倍或者至少10倍。
在其他的实施方案中,在pH值为1.0至2.0的水性体外测试介质中,与不包含聚合物的结晶的式(1)所示的噻吩并三唑并二氮杂化合物的浓度相比,固体分散体中无定形的式(1)所示的噻吩并三唑并二氮杂化合物或无定形噻吩并三唑并二氮杂化合物(1-1)的浓度高至少40%、至少50%、至少60%、至少70%、至少80%。在一些这样的实施方案中,固体分散体的聚合物为HPMCAS。在一些这样的实施方案中,固体分散体的聚合物为PVP。
在其他的实施方案中,所述固体分散体中无定形的式(1)所示的噻吩并三唑并二氮杂化合物或者无定形噻吩并三唑并二氮杂化合物(1-1)的浓度是式(1)所示的噻吩并三唑并二氮杂化合物和可药用的聚合物的固体分散体中的式(1)所示的噻吩并三唑并二氮杂化合物的浓度的至少40%、至少50%以上、至少60%、至少70%、至少80%,其中所述可药用的聚合物选自以下物质组成的组中:羟丙基甲基纤维素邻苯二甲酸酯和丙烯酸乙酯-甲基丙烯酸甲酯-三甲基铵乙基甲基丙烯酸酯氯化物共聚物,其中各固体分散体被置于pH值为1.0至2.0的水性体外测试介质中。在一些这样的实施方案中,固体分散体的聚合物为HPMCAS。在一些这样的实施方案中,固体分散体的聚合物为PVP。
在一些实施方案中,当在一定的湿度和温度下暴露一段时间时,本发明所述的固体分散体表现出抵抗式(1)所示的噻吩并三唑并二氮杂化合物或噻吩并三唑并二氮杂化合物(1-1)的重结晶的稳定性。在一个实施方案中,无定形的式(1)所示的噻吩并三唑并二氮杂化合物或噻吩并三唑并二氮杂化合物(1-1)保持无定形的浓度选自:至少90%、至少91%、至少92%、至少93%、至少94%、至少95%、至少96%、至少97%、至少98%和至少99%。
V.剂型:
可以用于本发明的固体分散体的合适的剂型包括但不限于胶囊、片剂、小型片剂、珠、微囊剂(beadlet)、丸剂、颗粒(granule)、粒剂(granulate)和粉末。可以使用例如肠溶包衣来包裹合适的剂型。合适的包衣可以包括但不限于邻苯二甲酸乙酸纤维素、羟丙基甲基纤维素(HPMC)、羟丙基甲基纤维素邻苯二甲酸酯、聚甲基丙烯酸共聚物、或醋酸羟丙基甲基纤维素琥珀酸酯(HPMCAS)。在一些实施方案中,可存在某些组合,例如,在同一样品中,本发明的噻吩并三唑并二氮杂的一些分子可以成簇存在,而一些分子则以分子形式分散于载体中。
在一个实施方案中,本发明的固体分散体可以配制成片剂、囊片或胶囊。在一些实施方案中,本发明的固体分散体可以配制成小型片剂或倒入口中(pour-into-mouth)的颗粒、或者口服粉末的组成(constitution)。在一些实施方案中,本发明的固体分散体与其他的赋形剂(例如,抑制重结晶/沉淀的聚合物、掩味成分等)组合并分散于合适的稀释剂中,从而提供即用型的悬浮制剂。在一些实施方案中,本发明的固体分散体可以配制用于儿科治疗。
在一个实施方案中,本发明的药物组合物被配制用于口服给药。在一个实施方案中,所述的药物组合物包含根据本发明所述的多个实施方案的固体分散体,其包含:式(1)所示的噻吩并三唑并二氮杂化合物、或其可药用的盐、溶剂化物(包括水合物)、消旋物、对映异构体、异构体或同位素标记的形式;以及聚合物载体。在一个实施方案中,所述的药物组合物还包含一种或多种添加剂,例如崩解剂、润滑剂、助流剂、粘结剂和填料。
用于所述的药物组合物含义合适的可药用的润滑剂和可药用的助流剂的实例包括但不限于胶态氧化硅、三硅酸镁、淀粉、滑石、三碱式磷酸钙、硬脂酸镁、硬脂酸铝、硬脂酸钙、碳酸镁、氧化镁、聚乙二醇、粉末状纤维素、甘油二十二烷酸酯、硬脂酸、氢化蓖麻油、单硬脂酸甘油酯和硬脂酰富马酸钠。
用于所述的药物组合物的合适的可药用的粘结剂的实例包括但不限于:淀粉、纤维素及其衍生物,例如微晶纤维素(例如AVICEL PH,得自FMC公司)、羟丙基纤维素、羟乙基纤维素和羟丙基甲基纤维素(HPMC,例如METHOCEL,得自Dow Chemical公司);蔗糖、右旋糖、玉米糖浆;多糖和明胶。
用于所述的药物组合物的合适的可药用的填料和可药用的稀释剂的实例包括但不限于:糖果制造商的糖、可压缩糖、葡萄糖结合剂、糊精、右旋糖、乳糖、甘露醇、微晶纤维素(MCC)、粉末状纤维素、山梨醇、蔗糖和滑石。
在一些实施方案中,在所述的药物组合物中,赋形剂可以发挥多于一种的功能。例如,填料或粘结剂还可以为崩解剂、助流剂、抗粘附剂、润滑剂、甜味剂等。
在一些实施方案中,本发明的药物组合物可以还包含添加剂或配料,例如抗氧化剂(例如抗坏血酸棕榈酸酯、丁基化羟基茴香醚(BHA)、二丁基化羟基甲苯(BHT)、α-生育酚、丙基没食子酸盐和富马酸)、抗微生物剂、酶抑制剂、稳定剂(例如丙二酸)和/或防腐剂。
通常,本发明的药物组合物可以配制成任何合适的固体剂型。在一些实施方案中,本发明的固体分散体混合在单位剂型中,例如作为胶囊或片剂、或者多微粒系统(例如颗粒或粒剂、或者粉末),以用于给药。
在一个实施方案中,药物组合物包含:根据本发明所述的固体分散体的多个实施方案的式(1)所示的噻吩并三唑并二氮杂化合物和醋酸羟丙基甲基纤维素琥珀酸酯(HPMCAS)的固体分散体,其中所述噻吩并三唑并二氮杂化合物在固体分散体中是无定形的,并且噻吩并三唑并二氮杂化合物与醋酸羟丙基甲基纤维素琥珀酸酯(HPMCAS)的重量比为1:3至1:1;45重量%至50重量%的乳糖一水合物;35重量%至40重量%的微晶纤维素;4重量%至6重量%的交联羧甲基纤维素钠;0.8重量%至1.5重量%的胶体二氧化硅;和0.8重量%至1.5重量%硬脂酸镁。
VI.剂量:
在一个实施方案中,本发明提供了可以配制成任何合适的固体剂型的药物组合物。在一个实施方案中,根据本发明的药物组合物包含本发明所述式(1)所示的噻吩并三唑并二氮杂化合物的多种实施方案中的一种或多种,其中所述式(1)所示的噻吩并三唑并二氮杂的剂量为约10mg至约100mg。在一个实施方案中,本发明的药物组合物包含本发明所述式(1)所示的噻吩并三唑并二氮杂的多种实施方案中的一种或多种,其中所述式(1)所示的噻吩并三唑并二氮杂的剂量选自由如下剂量组成的组:约10mg至约100mg、约10mg至约90mg、约10mg至约80mg、约10mg至约70mg、约10mg至约60mg、约10mg至约50mg、约10mg至约40mg、约10mg至约30mg和约10mg至约20mg。在一个实施方案中,本发明的药物组合物包含本发明所述式(1)所示的噻吩并三唑并二氮杂的多种实施方案中的一种或多种,其中所述式(1)所示的噻吩并三唑并二氮杂的剂量选自由约10mg、约50mg、约75mg、约100mg构成的组。
在一个实施方案中,本发明的药物组合物包括向有需要的受试者施用本发明所述多种实施方案中的一种或多种的式(1)所示的噻吩并三唑并二氮杂其剂量选自由如下剂量组成的组:约1mg、约2mg、约2.5mg、约3mg、约4mg、约5mg、约7.5mg、约10mg、约15mg、约20mg、约25mg、约30mg、约35mg、约40mg、约45mg、约50mg、约55mg、约60mg、约65mg、约70mg、约75mg、约80mg、约85mg、约90mg、约95mg、约100mg、约110mg、约120mg、约130mg、约140mg和约150mg;其剂型选自由以下组成的组:每周一次、每六天一次、每五天一次、每四天一次、每三天一次、每隔一天一次、每天一次、每天两次、每天三次、每天四次、每天五次。在另一个实施方案中,任意前述剂量或剂型可以定期减少或增加。在一个实施方案中,本发明的药物组合物包括向有需要的受试者施用选自由以下化合物构成的组中的噻吩并三唑并二氮杂:化合物(1-1)、(1-2)、(1-3)、(1-4)、(1-5)、(1-6)、(1-7)、(1-8)、(1-9)、(1-10)、(1-11)、(1-12)、(1-13)、(1-14)、(1-15)、(1-16)、(1-17)和(1-18);所述噻吩并三唑并二氮杂的施用剂量选自由以下剂量构成的组:约1mg、约2mg、约2.5mg、约3mg、约4mg、约5mg、约7.5mg、约10mg、约15mg、约20mg、约25mg、约30mg、约35mg、约40mg、约45mg、约50mg、约55mg、约60mg、约65mg、约70mg、约75mg、约80mg、约85mg、约90mg、约95mg、约100mg、约110mg、约120mg、约130mg、约140mg和约150mg;其剂型选自由以下组成的组:每周一次、每六天一次、每五天一次、每四天一次、每三天一次、每隔一天一次、每天一次、每天两次、每天三次、每天四次以及每天五次。在另一个实施方案中,任意前述剂量或剂型可以定期减少或增加。
根据特定的治疗目的、治疗阶段等,所述单位剂型适合每日给药1至5次。在一个实施方案中,有需要的受试者可以至少连续两天每日至少1次施用该剂型。在一个实施方案中,有需要的受试者可以至少隔日并每日至少1次施用该剂型。在一个实施方案中,有需要的受试者可以至少每周施用该剂型,并且将该剂型分成相等的和/或不等的剂量。在一个实施方案中,有需要的受试者可以每周施用该剂型,每隔3天和/或每周6次施用。在一个实施方案中,有需要的受试者可以每隔1天、每3天、每4天、每5天、每6天和/或每周以分剂量施用该剂型。在一个实施方案中,有需要的受试者可以每个月服用该剂型的2个或多个等分剂量或非等分剂量。
可以使用例如肠溶包衣来包裹所用的剂型(例如胶囊、片剂、小型片剂、珠、微囊剂、丸剂、颗粒、粒剂或粉末)。合适的包衣可以包括但不限于邻苯二甲酸乙酸纤维素、羟丙基甲基纤维素(HPMC)、羟丙基甲基纤维素邻苯二甲酸酯、聚甲基丙烯酸共聚物、或醋酸羟丙基甲基纤维素琥珀酸酯(HPMCAS)。
VII.工艺:
本发明所公开的噻吩并三唑并二氮杂化合物可以以游离碱或酸加成盐的形式存在,其可以根据美国专利申请公开No.2010/0286127(其全部内容以引用方式并入本文或本申请中)中所述的工序来获得。本发明的噻吩并三唑并二氮杂化合物的各对映异构体和非对映异构体可以由包含不对称中心或立体异构中心的市售可得的起始材料以合成方式制备,或者通过制备外消旋的混合物、然后通过本领域普通技术人员公知的拆分方法来制备。这些拆分方法例如有:(1)使对映异构体的混合物附着在手性助剂上,通过重结晶或色谱法分离所得的非对映异构体的混合物,并从助剂上释放光学纯产物;(2)使用光学活性拆分剂形成盐;(3)在手性液相色谱柱上直接分离光学对映异构体的混合物;或者(4)使用立体选择性化学试剂或酶试剂进行动力学拆分。此外,外消旋的混合物还可以通过公知的方法(例如手性相色谱法或使化合物在手性溶剂中结晶)而拆分成它们的构成成分的对映异构体。
如果需要,本发明所公开的噻吩并三唑并二氮杂化合物的特定对映异构体可以通过不对称合成或者通过使用手性助剂诱导来制备,其中所得的非对映异构体的混合物被分离并且辅助基团被切割,从而提供所需的纯的对映异构体。可选地,在所述分子包含碱性官能团(例如氨基)或酸性官能团(例如羧基)的情况下,非对映异构体的盐是通过以下过程形成的:使用合适的光学活性的酸或碱,然后对非对映异构体(通过本领域公知的分级结晶或色谱手段而由此形成)进行拆分,并在随后回收纯的对映异构体。可使用本领域公知的多种方法来制备式(1)的噻吩并三唑并二氮杂化合物,其中对映异构体的过量百分比通常超过约80%。有利的是,优选的对映异构体的过量百分比大于80%,优选大于90%,更优选大于95%,并且最优选为99%以上。
本发明的固体分散体可以通过多种方法来制备,包括熔融和溶剂蒸发方法。本发明的固体分散体还可以根据如下文中所描述的工序制备:Chiou WL,Riegelman S:“Pharmaceutical applications of solid dispersion systems”,J.Pharm.Sci.1971;60:1281-1302;Serajuddin ATM:“Solid dispersion of poorly water-soluble drugs:early promises,subsequent problems,and recentbreakthroughs”,J.Pharm.Sci.1999;88:1058-1066;Leuner C,Dressman J:“Improving drug solubility for oral delivery using solid dispersions”,Eur.J.Pharm.Biopharm.2000;50:47-60;以及Vasconcelos T,Sarmento B,Costa P:“Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs”,Drug Discovery Today 2007;12:1068-1075,所有这些文献均以引用的方式将其全文并入本文中。
在一个实施方案中,本发明的固体分散体是通过熔融工艺制备的。在一个实施方案中,熔融工艺包括在载体中将式(1)的噻吩并三唑并二氮杂的多个实施方案的一个或多个熔融。在一个实施方案中,熔融工艺包括使本发明的熔融化合物和载体冷却。在一个实施方案中,熔融工艺包括将熔融的化合物和载体粉碎。在一个实施方案中,在冷却步骤之后将本发明的熔融的化合物和载体粉碎。
在一些实施方案中,式(1)的噻吩并三唑并二氮杂或者其药学可接受的盐、溶剂化物(包括水合物)、外消旋物、对映异构体、异构体或同位素标记的形式与载体间不相容,在这种情况中,可以在熔融步骤中加入表面活性剂,从而防止在加热的混合物中形成两个液相或悬液。在一些实施方案中,式(1)的噻吩并三唑并二氮杂的多个实施方案中的一个或多个实施方案悬浮于之前熔融的载体中,而并未使用熔融状态的药品和载体,由此降低了工艺的温度。在一个实施方案中,在冰浴搅拌下使熔融的药品和载体的混合物冷却。在一个实施方案中,通过喷雾冷却(可选的通过喷雾冷凝)使熔融的药品和载体混合物冷却并固化。
在一个实施方案中,通过将熔融物喷雾至冷却室(使环境温度或冷却温度的低温空气通过该冷却室)中以将熔融物形成颗粒,从而使熔融的药品和载体的混合物冷却或固化。在一个实施方案中,通过在合适的流化床处理器中使熔融分散体雾化和再固化,从而使熔融的药品和载体混合物冷却和固化。在一个实施方案中,通过在可加热的高剪切混合器中进行熔融制粒,从而使熔融的药品和载体的混合物冷却并固化。
在一些实施方案中,可以采用热段挤出(hot-stage extrusion)或熔融团聚物来避免药物的熔融局限。热段挤出包括如下过程:在熔融温度下将预先混合的药品和载体以高转速在短时间内挤出;在室温下冷却后收集所得的产物并研磨。
在一个实施方案中,将式(1)的噻吩并三唑并二氮杂的多种具体物质中的一种或多种物质在较低的温度下加工,从而避免任何热不稳定的化合物发生降解。在一个实施方案中,通过将热段挤出与暂时性增塑剂(例如二氧化碳)结合而实现较低的处理温度。在一个实施方案中,在根据本发明的固体分散体的制备中,在常规的高剪切混合器或旋转处理器中使用熔融团聚物。在一个实施方案中,通过将包含根据本发明的噻吩并三唑并二氮杂化合物的熔融载体加入至加热的赋形剂中,从而制备根据本发明的固体分散体。在一个实施方案中,通过将包含根据本发明的噻吩并三唑并二氮杂化合物、载体和一种或多种赋形剂的混合物加热至载体的熔融范围内或以上的温度,从而制备根据本发明的固体分散体。
在一些实施方案中,通过溶剂蒸发方法来制备根据式(1)的噻吩并三唑并二氮杂的制剂的多种具体物质中的一种或多种物质。在一个实施方案中,溶剂蒸发方法包括使根据式(1)的噻吩并三唑并二氮杂和载体溶解于挥发性溶剂中,然后使挥发性溶剂蒸发。在一个实施方案中,挥发性溶剂可以为一种或多种赋形剂。在一个实施方案中,一种或多种赋形剂包括但不限于抗粘剂、惰性填料、表面润湿剂、pH调节剂和添加剂。在一个实施方案中,赋形剂可以溶解于挥发性溶剂中,或者在挥发性溶剂中呈悬浮或膨胀状态。
在一个实施方案中,根据本发明的固体分散体的制备包括使悬浮于挥发性溶剂中的一种或多种赋形剂干燥。在一个实施方案中,所述的干燥包括真空干燥、使挥发性溶剂在低温下缓慢蒸发、使用旋转蒸发仪、喷雾干燥、喷雾制粒、冷冻干燥或使用超临界流体。
在一个实施方案中,使用了喷雾干燥制备根据式(1)的噻吩并三唑并二氮杂组合物的制剂,其包括使所述的组合物的悬液或溶液雾化成小滴,然后快速除去制剂中的溶剂。在一个实施方案中,根据本发明的制剂的制备涉及喷雾制粒,其中将位于溶液中的组合物的溶液或悬液喷雾至合适的化学惰性和/或物理惰性填料上,例如乳糖或甘露醇。在一个实施方案中,通过双通或三通喷嘴对所述的组合物的溶液或悬液进行喷雾制粒。
在一些实施方案中,根据本发明的固体分散体的制备包括使用超临界流体。术语“超临界流体”是指在临界温度和临界压力以上以单一液相存在的物质。在一个实施方案中,根据本发明的制剂的制备包括使用超临界二氧化碳流体。在一个实施方案中,利用超临界流体技术制备根据本发明的制剂,其包括:将根据式(1)的噻吩并三唑并二氮杂组合物和载体溶解于常见溶剂中,该溶剂通过喷嘴与二氧化碳同时被引入至颗粒形成容器中;以及将所述溶液喷雾,从而使溶剂被超临界流体快速萃取,由此使得固体分散体颗粒沉淀在容器的壁上。
在一些实施方案中,根据本发明的固体分散体的制备包括使用共沉淀方法。在一个实施方案中,在持续搅拌下将非溶剂滴加至基于式(1)的噻吩并三唑并二氮杂和载体溶液的组合物中。在一个实施方案中,在添加非溶剂的过程中,使根据式(1)的噻吩并三唑并二氮杂组合物和载体进行共沉淀,从而形成微颗粒。在一个实施方案中,过滤并干燥所得的微颗粒,从而提供所需的固体分散体。
对于式(1)的化合物与聚合物载体(一种或多种)的混合比例没有特别限定,只要其可以改善式(1)的化合物的生物利用率即可,并且该比例根据聚合物的种类而改变。
通过以下非限定性实施例来说明本发明。
VIII.例子:
通过以下非限定性实施例来说明本发明。
实施例1:化合物(1-1)的固体分散体的体外筛选
使用化合物(1-1)以及以下五种聚合物中的一种来制备十种固体分散体,所述聚合物包括醋酸羟丙基甲基纤维素琥珀酸酯(HPMCAS-M)、羟丙基甲基纤维素邻苯二甲酸酯(HPMCP-HP55)、聚乙烯吡咯烷酮(PVP)、PVP-乙酸乙烯酯(PVP-VA)和Eudragit L100-55,并且对于每种聚合物,化合物(1-1)的负载率为25%和50%。使用喷雾干燥,然后在低温对流烘箱中进行二次干燥,从而通过溶剂蒸发法来制备固体分散体。通过非沉降溶解性能测试(non-sink dissolution performance test)来评估各固体分散体的性能,所述测试测量药物的总量和随着时间溶液中所存在的游离药物的量。选择非沉降溶解是由于其能够最好地代表低溶解度化合物在体内的情况。该测试包括在分散体被引入至测试介质后约30分钟至40分钟后由胃pH(0.1N NaCl,pH 1.0)转变至肠pH(FaFSSIF,pH 6.5)的“胃转移”,从而模拟了体内的情况。[FaFSSIF为禁食状态的人工肠液,其由3mM的牛黄胆酸钠、0.75mM的卵磷脂、0.174g NaOH颗粒、1.977g NaH2PO4·H2O、3.093g NaCl和适量纯净水500mL组成]。使用高效液相色谱(HPLC)法和Agilent 1100串联HPLC来定量溶解药物的量。制剂的溶解情况(图1A-1J)表明:相对于在相同介质中的未经配制的化合物而言,在所有的分散体候选物中,药物的溶解度均大幅升高。在所有固体分散体中,基于在肠pH下释放的游离药物的水平较高这一发现可知,与未经配制的化合物相比,25%化合物(1-1)在PVP中的分散体、25%化合物(1-1)在HPMCAS-M中的分散体、以及50%化合物(1-1)在HPMCAS-M中的分散体提供了增强的经口吸收。
实施例2:化合物(1-1)的固体分散体的体内筛选
以更大的规模制备化合物(1-1)的固体分散体,即25%化合物(1-1)在PVP中的分散体、25%化合物(1-1)在HPMCAS-MG中的分散体、以及50%化合物(1-1)在HPMCAS-M中的分散体,以用于体内研究。各制剂均经过了实施例1所述的体外溶解测试的评估。为了确保这些分散体是无定形且均质的,通过X射线粉末衍射法(PXRD)和修正的差示扫描量热法(mDSC)来评估各分散体。X射线衍射计为Bruker D-2 Phaser。另外,为了了解水对各种分散体的玻璃化转变温度(Tg)的影响,对首先在设定的相对湿度(即,25%、50%和75%RH)下平衡至少18小时的样品进行mDSC。[水可以用作固体分散体的增塑剂,并且由活性化合物或聚合物形成的体系的吸湿性可以影响被这些体系吸收的水的量]。
非沉降溶解结果(图2A-2C)与实施例1中分散体的结果相当。PXRD结果(图3)显示没有证据表明任意一种分散体中存在结晶化合物,并且mDSC结果(图4A-4C)显示各分散体均存在单一的玻璃化转变温度(Tg),这表明各分散体是均匀的。对于各分散体,观察到Tg与相对湿度之间逆相关(图5)。值得注意的是,对于在75%RH下平衡的25%化合物(1-1)在PVP中的固体分散体,出现了两个Tg,这表明发生了相分离,并且该分散体还显示出在75%RH下发生了熔化,这表明在RH平衡过程中发生了结晶(图6)。该发现表明25%化合物(1-1)在PVP中的分散体的稳定性弱于HPMCAS-M分散体。
为了评估这三种分散体的生物利用度,向多组雄性猎兔犬(每组3只)给予剂量为3mg/kg的化合物(1-1)的固体分散体的水性悬液(通过口服填喂法给药),或者给予剂量为1mg/kg的溶解于水:乙醇:聚乙二醇(PEG)400(60:20:20)中的化合物(1-1),并以静脉推注给药至头静脉中。在静脉给药后的0分钟(服药之前)、5分钟、15分钟和30分钟、和1小时、2小时、4小时、8小时、12小时和24小时,以及在口服填喂给药后的0分钟(服药之前)、15分钟和30分钟、和1小时、2小时、4小时、8小时、12小时和24小时由各动物的颈静脉收集血液样品。使用合格的LC-MS/MS法检测各样品中存在的化合物(1-1)的量,并且量化下限为0.5ng/mL。通过使用线性梯形法则(直至最终可测的浓度,而未将终末消除相外推至无穷大)来测定血浆浓度-时间曲线的曲线下面积(AUC)。通过对对数浓度-时间曲线的终末线性部分进行最小二乘回归分析来计算消除半衰期(t1/2)。由血浆浓度数据直接得到最大血浆浓度(Cmax)和达到Cmax的时间(tmax)。通过用口服给药后的剂量归一化的AUC除以静脉给药后的剂量归一化的AUC来计算口服生物利用度(F),并以百分数(%)形式报告。下表1中总结的结果给出了25%化合物(1-1)在PVP中的固体分散体、25%化合物(1-1)在HPMCAS-M中的固体分散体和50%化合物(1-1)在HPMCAS-M中的固体分散体的平均口服生物利用度,分别为58%、49%和74%。
表1:狗口服(po)和静脉(iv)给药后的化合物(1-1)的药物动力学参数(数值为3只狗的平均值)
AUC:血浆浓度-时间曲线下的面积;Cmax:最大血浆浓度;F:生物利用度;HPMCAS:醋酸羟丙基甲基纤维素钠;IV:静脉内;PEG:聚乙二醇;PO:经口,口服;PVP:聚乙烯吡咯烷酮;tmax:达到Cmax的时间;t1/2:血浆消除半衰期
实施例3:包含化合物(1-1)的固体分散体的胶囊的制备和临床应用
制备10mg强度的明胶胶囊,在患有恶性血液病的患者中进行初步的临床研究。基于实施例1和2中所述的化合物(1-1)的固体分散体的体外和体内测试结果,选择50%化合物(1-1)在HPMCAS-M中的固体分散体用于胶囊的研发。以3号尺寸的硬明胶胶囊,190mg的填充量为目标来开始胶囊的研发,因为这样的构造可以通过填充更大尺寸的胶囊而潜在地增加胶囊的强度,同时保持药物组合物。根据经验,设计了4种胶囊制剂,其中具有不同量的崩解剂并且具有或不具有润湿剂。由于所有4种制剂都显示出相似的崩解测试和溶解测试结果,所以选择最简单的制剂(不具有润湿剂,并具有最少量的崩解剂)用于制造。进行制造工艺的研发和规模扩大的研究,以确定固体分散体的喷雾干燥工艺和干燥后的时间;共混的参数;为获得约0.60g/cc的目标堆密度的共混物的辊压和研磨;以及胶囊填充的条件。
将结晶化合物(1-1)和聚合物醋酸羟丙基甲基纤维素琥珀酸酯(HPMCAS-M)溶解于丙酮中,并喷雾干燥,从而生产包含50%化合物(1-1)载量的固体分散体中间体(SDI)颗粒。PXRD分析显示SDI是无定形的,mDSC分析显示SDI是均匀的(即在环境条件下具有单一的Tg)。在V型搅拌机的多个段中将50%化合物(1-1)在HPMCAS-M中的固体分散体(1000g)和赋形剂(包括微晶纤维素填料-粘结剂(4428g)、交联羧甲基纤维素钠崩解剂(636g)、胶体二氧化硅分散剂/润滑剂(156g)、硬脂酸镁分散剂/润滑剂(156g)和乳糖一水合物填料(5364g))共混。然后将混合物压制并制粒,从而得到约0.6g/mL的堆密度。使用自动化的填充机将混合物分配至3号尺寸的硬明胶胶囊(目标填充量:190mg)中,并使用胶囊抛光机将制得的胶囊抛光。
在口服给药10mg胶囊(包含50%化合物(1-1)在HPMCAS中的固体分散体)后进行药物动力学的评估,并将结果与对健康志愿者口服给药4x10mg胶囊(其包含化合物(1-1)的Eudragit固体分散体)后进行的药物动力学评估加以对比。
在下表2A和2B中提供了这两种药物组合物的比较。Eudragit制剂之前在2009年1月8日公开的美国专利申请2009/0012064 A1的实施例5中有所描述。该申请记载:通过在水和乙醇的混合物中溶解和/或分散式(A)所示的噻吩并三唑并二氮杂和包裹用赋形剂(包含氨溶甲基丙烯酸酯共聚物B型(Eudragit RS)、甲基丙烯酸共聚物C型(Eudragit L100-55)、滑石和硅酸铝镁)来制备Eudragit固体分散体制剂。然后,使用离心流化床制粒机将上述的非均匀混合物施加在微晶纤维素球(Nonpareil 101,Freund)上,从而生产分配至2号尺寸的羟丙基甲基纤维素胶囊中的颗粒。
在这两个临床研究中,使用经验证的LC-MS/MS方法来测定化合物(1-1)的血液水平,并根据在胶囊给药后24小时内各时间点测量的化合物(1-1)的血浆浓度来进行药物动力学分析。下表3中总结的结果显示,根据AUC(924*4/1140,由于给药剂量的差异而改变),HPMCAS-M固体分散体制剂在人体中的生物利用度比Eudragit固体分散体制剂高3倍以上。此外,根据观察到的Tmax,HPMCAS制剂的吸收速度快于Eudragit制剂(Tmax为1小时vs 4-6小时)。HPMCAS-M固体分散体制剂的在系统暴露方面的明显改善是意料之外的。
表2A:用于临床应用的化合物(1-1)的固体分散体胶囊
包含化合物(1-1)的50%HPMCAS固体分散体的药物组合物:10mg强度,3号尺寸的硬明胶胶囊
表2B:包含化合物(1-1)的Eudragit L100-55固体分散体的药物组合物:10mg强度,2号尺寸的硬明胶胶囊
*为无水形式
表3:在将化合物(1-1)的固体分散体经口施用给人后的药物动力学参数
AUC0-24h:24小时内化合物(1-1)血浆浓度对时间曲线下的面积
Cmax:最大血浆浓度
hr:小时
HPMCAS:醋酸羟丙基甲基纤维素琥珀酸酯
mL:毫升
ng:纳克
PO:经口,口服
Tmax:达到Cmax的时间
实施例4:在大鼠中的口服暴露
在大鼠中测定化合物(1-1)的固体分散体的3种制剂的口服生物利用度。所选的3种分散体为25%化合物(1-1)在PVP中的分散体、25%化合物(1-1)在HPMCAS-MG中的分散体和50%化合物(1-1)在HPMCAS-MG中的分散体。在该研究中使用的动物为得自土尔库大学(芬兰)的中央动物实验室(Central Animal Laboratory)的无特定病体(SPF)的Hsd:Sprague Dawley大鼠。大鼠最初购自位于荷兰的Harlan。大鼠为雌性,并且为10周龄,12只大鼠用于该研究中。在聚碳酸酯Makrolon II笼中饲养动物(每个笼中3只),动物室温为21+/-3℃,动物室内的相对湿度为55+/-15%,并且动物室内照明为人工照明,而且为12小时明暗周期的循环(暗周期为18:00至06:00之间)。白杨木屑(Tapvei Oy,Estonia)用于垫料,并且每周至少更换1次垫料。在对动物定量给药之前提供食物和水,但是在定量给药后最初的2个小时中移除食物和水。
将预先计算的量的注射用无菌水加入至装有采用合适的量的分散体的容器中,获得化合物(1-1)的浓度为0.75mg/mL,由此制备包含25%化合物(1-1)在PVP中的分散体的口服定量给药溶液、包含25%化合物(1-1)在HPMCAS-MG中的分散体的口服定量给药溶液和包含50%化合物(1-1)在HPMCAS-MG中的分散体的口服定量给药溶液。在每次给药之前,将口服定量给药溶液进行涡旋混合20秒。用于静脉给药的定量给药溶液包含0.25mg/mL的化合物(1-1),该定量给药溶液是通过将5mg的化合物(1-1)溶解于这样的混合物中来制备的,所述混合物包含4mL的平均分子量为400Da的聚乙二醇(PEG400)、4mL的乙醇(96%纯度)和12mL的注射用无菌水。在加入水后的30分钟之内使用含有25%化合物(1-1)在PVP中的分散体的定量给药溶液。在加入水后的60分钟之内使用含有25%化合物(1-1)在HPMCAS-MG中的分散体的定量给药溶液和含有50%化合物(1-1)在HPMCAS-MG中的分散体的定量给药溶液。使用4mL/kg定量给药量,从而使化合物(1-1)的静脉给药的给药水平为1mg/kg,口服给药的给药水平为3mg/kg。定量给药方案在表4中给出。
表4.用于大鼠口服暴露研究的定量给药方案
在定量给药后的0.25小时、0.5小时、1小时、2小时、4小时、8小时、12小时和24小时的时间点,将约50μL的血液样品收集至包含5μL乙二胺四乙酸(EDTA)溶液的Eppendorf管中,并且每个样品都是在距所述时间点5分钟时间窗内收集的。由各样品获得20μL血浆并储存在干冰温度下以用于分析。使用经验证的液相色谱串联质谱(LC-MS/MS)法对各样品中化合物(1-1)的浓度进行分析,其定量下限为0.5ng/mL。
使用Phoenix WinNonlin软件包(6.2.1版,Pharsight Corp公司,CA,USA),采用标准的非房室模型方法来计算药物动力学参数。通过对对数浓度-时间曲线的终末线性部分进行最小二乘回归分析来计算消除半衰期(t1/2)。通过使用线性梯形法则直至最终可测的浓度,此后将终末消除相外推至无穷大来测定血浆浓度-时间曲线的曲线下面积(AUC)。通过将药物浓度曲线外推至无穷大来计算平均保留时间(MRT),其代表了化合物在房室或系统中保留的平均时间量。由血浆浓度数据直接得到最大血浆浓度(Cmax)和达到Cmax的时间(tmax)。通过用口服给药后的剂量归一化的AUC除以静脉给药后的剂量归一化的AUC来计算试验性口服生物利用度(F),即F=(AUC(口服)/剂量(口服))/(AUC(静脉)/剂量(静脉))],并以百分数(%)形式报告。
药物动力学参数在表5中给出,血浆浓度对时间的图示于图7和8中。
表5.口服和静脉给药后的化合物(1-1)的药物动力学参数。值为得自3只动物的平均值
实施例5.喷雾干燥分散体的制备
使用五种所选的聚合物来制备化合物(1-1)的喷雾干燥的分散体,这5种聚合物为:HPMCAS-MG(Shin Etsu Chemical公司)、HPMCP-HP55(Shin Etsu Chemical公司)、PVP(ISP,Ashland公司的部门)、PVP-VA(BASF公司)和Eudragit L100-55(Evonik Industries AG)。使用每种聚合物以25%和50%制备所有的喷雾干燥溶液。除了PVP溶液以外,所有的溶液都是在丙酮中制备的,PVP溶液是在乙醇中制备的。对于各溶液而言,在10g溶剂中制备1.0g固体(聚合物和化合物(1-1))。使用具有1.5mm喷嘴的Büchi B-290,PE-024喷雾干燥器和Büchi B-295,P-002冷凝器将溶液喷雾干燥。将喷雾干燥器的喷嘴压力设定为80psi,目标出口温度设定为40℃,冷却器的温度设定至-20℃,泵速设定为100%,并且抽吸器设定为100%。在喷雾干燥后,收集固体分散体,并在低温对流烘箱中过夜干燥,从而除去残余的溶剂。
实施例6:湿度和温度的稳定性
通过在高温下暴露于水分来评估化合物(1-1)在HPMCAS-MG中的喷雾干燥分散体的稳定性。通过在75%相对湿度和40℃下放置1、2和3个月,来测定作为相对湿度的函数的玻璃化转变温度(Tg)。在HDPE瓶中将喷雾干燥的分散体储存在LDPE袋内,从而模拟散装物品包装。结果总结于表6。在时间0时,Tg为134℃,在1个月时Tg为134℃,在2个月时Tg为135℃,在3个月时Tg为134℃,并且在各测量中仅观测到单一的拐点。对每种样品还获得了X射线衍射图谱。图9示出了在稳定性测试的时间0时,化合物(1-1)在HPMCAS-MG中的固体分散体的X射线粉末衍射图谱。图10、11和12示出了分别在暴露于40℃和75%相对湿度1个月、2个月和3个月后,化合物(1-1)在HPMCAS-MG中的固体分散体的X射线粉末衍射图谱。图案未显示与化合物(1-1)有关的任何衍射线。
实施例7:NSCLC细胞系的体外处理
将5种已建立的NSCLC细胞系(即H2228、H3122、A549、HOP62和HOP92)暴露于剂量增加的化合物(1-1)(OncoEthix SA,瑞士)。暴露72小时后通过MTT试验确定对细胞存活力的作用。用GraphPad Prism 5.0软件计算生长抑制(GI)50%值。并使用市售抗体通过蛋白质印迹分析蛋白质水平。按照制造商的说明,用Qiagen RNAEasy试剂盒提取RNA,并用RT-PCR试剂盒的Superscript第一链合成体系反转录。在StepOnePlus实时PCR系统中利用Fast SYBR Green Master Mix进行RT-PCR。
在暴露72小时后,化合物(1-1)在两种EML4-ALK-阳性NSCLC细胞中示出了抗增殖作用,在H2228和H3122细胞中的GI50值分别为629nM和627nM。令人关注地,化合物(1-1)在EML4-ALK-阴性(A549)细胞系中具有活性,GI50值为432nM。在所有细胞系中以蛋白质和mRNA水平表征BRD4/3/2、c-MYC、BCL-2、p21和细胞周期蛋白D1的表达。对化合物(1-1)敏感和抵抗的细胞系中前述蛋白均表现出类似的基础表达水平。在H3122和H2228细胞中分别鉴定了EML4-ALK变体1和3。对抗增殖活性的细胞信号传导途径评价表明,化合物(1-1)诱导STAT3的暂时上调,随后在暴露24小时至至多72小时下调,该途径是抗克唑替尼细胞系中ALK频繁上调的关键下游效应子。令人关注地,C-MYC蛋白和mRNA水平似乎没有被25化合物(1-1)改变。在化合物(1-1)处理后,EML4-ALK-阳性H3122细胞示出了N-MYC mRNA水平的下调。
这些结果表明,具有基因组ALK改变的NSCLC细胞系对通过化合物(1-1)引起的BET-BRD抑制是敏感的,随着下游信号传导途径的抑制伴随明显的抗增殖作用,这表明化合物(1-1)在ALK阳性NSCLC患者中能够作为抗癌剂进行临床开发。
实施例8:NSCLC细胞系的体外处理
在5种已建立的NSCLC细胞系(H2228、H3122、A549、HOP62和HOP92)中暴露72小时后,用MTT测定法测定化合物(1-1)生长抑制50%(GI50)值。利用商用抗体通过蛋白质印迹分析蛋白质水平;以基线水平,在StepOnePlus实时PCR系统上用Fast SYBR Green Master Mix进行RT-PCR,然后进行4小时和24小时后处理。对于细胞周期分析,用碘化丙啶染色细胞,并用FACScan流式细胞仪分析DNA含量。将化合物(1-1)与靶向剂依维莫司和克唑替尼组合,通过Chou-Talalay方法测定联合指数(CI)。
化合物(1-1)在5种NSCLC细胞系(2个含有融合蛋白EML4-ALK+)中的4种中具有GI50值为110至940nM的抗增殖作用。A549细胞中,KRAS和LKB1基因的同时突变消除了OTX015效应,如针对化合物(1-1)类似物JQ1所描述的那样,而HOP92和HOP62是LKB1野生型的。在500nM下暴露24小时后,HOP92、H2226和H3122中S期的细胞百分比分别减少了40%、42%和52%。化合物(1-1)与ALK+抑制剂克唑替尼的联合在H2228细胞中共同暴露48小时后具有累加效应(CI=0.98),化合物(1-1)与依维莫司的联合在HOP62、HOP92、A549和H2228中具有累加/协同效应。化合物(1-1)-敏感性和-抗性细胞系具有相似的基础mRNA和BRD2/3/4、HEXIM BCL-2和P21的蛋白表达,在HOP92和H3122中,CMYC和NMYC分别过表达。暴露于化合物(1-1)(500nM)24小时后,在HOP92细胞中CMYC蛋白和mRN下调,在H3122和HOP62细胞中NMYC下调,而在化合物(1-1)-敏感性细胞系中HEXIM mRNA上调。
NSCLC细胞系对化合物(1-1)BET抑制敏感,下调MYC家族成员并上调HEXIM(包括具有EML4-ALK融合基因或KRAS突变的NSCLC细胞),这表明了其在NSCLC中的治疗潜力。
实施例9:具有致癌基因突变的NSCLC
材料和方法:将5种已建立的具有KRAS、LKBI、TP53和ALK不同致癌突变的NSCLC细胞系(即HOP62、HOP92、A549、H2228和H3122)(图13)暴露于剂量增加的化合物(1-1)(OncoEthix SA,瑞士)72小时,并通过MTT试验评价细胞增殖。结果表示以一式三份进行的至少3次独立试验的平均值±95%CI。使用市售抗体通过Western Blot分析蛋白质水平。对于细胞周期分析,细胞用PI染色并在处理24小时后用FACScan流式细胞仪分析。在StepOnePlus实时PCR系统中利用Fast SYBR Green Master Mix进行RT-PCR。采用Anova之后进行Dunnett's多重比较检验,结果表示至少2次独立试验的平均值±SD,其中相对于对照细胞(0.1%DMSO)*p<0.05。在细胞系中进行联合化合物(1-1)的组合研究,所述细胞系单独暴露于增加剂量的化合物(1-1)或暴露于增加剂量的化合物(1-1)与依维莫司或克唑替尼的联合,并利用Chou&Talalay方法评价。利用CalcuSyn软件通过中效图(median effect plot)分析确定联合指数(CI)。CI<1,协同作用;CI=1,累加作用;以及CI>1,拮抗作用。结果表示三次进行的3个独立试验的中值和范围。
体内研究:将5x106个H3122细胞注入无胸腺的NMRI雄性裸鼠的侧腹部,当肿瘤体积达到100mm3时随机分成3组(8只小鼠/组);对照(PBS,每天1次,口服,连续)、化合物(1-1)(50mg/kg/每天2次,灌胃,连续)或克唑替尼(25mg/kg/每天2次,灌胃,连续)。
结果:化合物(1-1)在5种NSCL细胞系的4种中显示出抑制细胞生长的作用,其中2种具有融合蛋白EML4-ALK+。在A549细胞中,KRAS和LKB1基因同时突变消除了化合物(1-1)的作用。在我们的细胞系组中,暴露于化合物(1-1)72小时后的GI50值的范围在0.1-0.7μM之间,表明化合物(1-1)在NSCLC细胞中具有广泛的活性(图13)。化合物(1-1)治疗也导致S期细胞百分比的减少(图14)。在NSCLC细胞系中,BRD4/3/2、c-MYC、BCL-2、p21和细胞周期蛋白D1的蛋白质水平和mRNA水平表达的表征未显示出这些蛋白的基础水平和对化合物(1-1)敏感性之间的任何关系(图15A和15B)。即,对化合物(1-1)敏感性与NSCLC细胞中BRDs、C-MYC、N-25MYC、BCL2或P21的基础水平无关。在对化合物(1-1)敏感的细胞系中,我们观察到HOP92细胞中C-MYC mRNA水平和蛋白质水平的快速下调,以及在4小时后HOP62和H3122细胞中N-MYC mRNA水平的快速下调,在24小时后仍然维持这样(图16A至16C)。化合物(1-1)与ALK+-抑制剂克唑替尼联合,在H2228细胞中共暴露48小时后具有累加作用(CI=0.9),在HOP62、HOP92、A549和H2228细胞系中与依维莫司具有累加/协同作用(图17)。在具有H3122肿瘤的小鼠中,化合物(1-1)抑制肿瘤生长的程度类似于EML4-ALK抑制剂克唑替尼(图18A至B)。我们的数据表明包括具有EML4-ALK融合基因或KRAS突变的NSCLC细胞系,在体外和体内对化合物(1-1)BET抑制是敏感的。
实施例10.SCLC细胞系的体外研究
在4种已建立的SCLC细胞系(H82、H69、DMS79、DMS114)中暴露72小时后,用MTT测定法测定化合物(1-1)生长抑制50%(GI50)的值。利用商用抗体通过蛋白质印迹分析蛋白质水平,以基线水平,在StepOnePlus实时PCR系统上用Fast SYBR Green Master Mix进行RT-PCR,然后进行4和24小时后处理。对于细胞周期分析,用碘化丙啶染色细胞,并用FACScan流式细胞仪分析DNA含量。
DMS114细胞对化合物(1-1)[GI50=120(84-172)nM]显示出剂量依赖性敏感性,而H82、H69,DMS79细胞是抗性的[GI50>6μM],尽管它们过表达CMYC和/或NMYC蛋白。四种SCLC细胞系都是KRAS野生型和TP53突变的,化合物(1-1)-抗性SCLC细胞系在RB1中具有纯合突变,而DMS144细胞表达RB1野生型。所有细胞系显示出相似的BRD2/3/4、CMYC、NMYC、HEXIM、BCL-2和p21的基础mRNA水平。在500nM的化合物(1-1)处理后,CMYC和NMYC mRNA水平未发生改变,而HEXIM和编码组蛋白的基因(HIST1H2BK、HIST2H2BJ)的mRNA水平在所有四种细胞系中均上调。在DMS114细胞中,化合物(1-1)以时间依赖性方式使得G1中的细胞周期停滞,不受理论束缚,其可以通过细胞周期抑制剂p27的蛋白水平上调来解释。
不受特定理论的束缚,MYC家族在SCLC细胞系中没有介导化合物(1-1)的抗肿瘤作用。功能性RB1蛋白的存在,控制细胞在G1的进展,这可以解释化合物(1-1)的细胞抑制作用。
实施例11.SCLC细胞系的体外研究
选择四种已建立的SCLC细胞系以评价化合物(1-1),即NCI-H69、NCI-H82、DMS79和DMS114,它们具有KRAS、PI3KCA、TP53和RB1的不同致癌突变,并具有表7所示的C-MYC和N-MYC的不同表达水平。
将这些细胞系暴露于剂量增加的化合物(1-1)中72小时,并用MTT测定法评价细胞增殖。使用Windows用Prism 5.00,通过S形剂量响应的方程式计算GI50和Emax值。结果表示以一式三份进行的至少3次独立实验的平均值±95%CI。
使用商用抗体通过蛋白质印迹分析蛋白质水平。对于细胞周期分析,细胞用PI染色,并在72小时暴露后使用FACScan流式细胞仪分析。在StepOnePlus实时PCR系统上使用Fast SYBR Green Master Mix进行RT-qPCR。细胞周期结果表示至少3次独立实验的平均值±SD。RT-qPCR结果以荧光强度表示,标准化为两个持家基因,并且表示以一式两份或一式三份进行的一个实验的平均值±SD。
化合物(1-1)在4种SCLC细胞系中的1个中显示出细胞抑制作用。DMS114细胞显示对化合物(1-1)[GI50=120(84-172)nM]的剂量依赖性敏感性,而H82、H69、DMS79是细胞抗性的[GI50>6μM]。参见图19。4种SCLC细胞系都是KRAS野生型和TP53突变的,化合物(1-1)-抗性SCLC细胞系在RB1中具有纯合突变,而DMS144细胞表达RB1野生型。
化合物(1-1)诱导C-MYC表达的下调和HEXIM1以及编码组蛋白的基因的上调。图20A示出了当用化合物(1-1)以500nM处理NCI-H69、NCI-H82、DMS79和DMS114细胞系4和24小时时所观察到的与C-MYC相关的荧光强度。在化合物(1-1)敏感性细胞系DMS114中,C-MYC mRNA快速下调。图20B示出了当用化合物(1-1)以500nM处理NCI-H69、NCI-H82、DMS79和DMS114细胞系4和24小时时所观察到的与N-MYC相关的荧光强度。
图20C示出了当以500nM的化合物(1-1)处理4和24小时的DMS114细胞系观察到的与HEXM1、HIST2H2BE和HIST1H2BK相关的荧光强度。在DMS114细胞系中HEXIM1和编码组蛋白的基因(HIST2H2BE、HIST1H2BK)的mRNA水平上调。
图20D示出了当以500nM化合物(1-1)处理NCI-H69、NCI-H82细胞系0、4、24、48和72小时时,C-MYC和β-肌动蛋白水平的蛋白质印迹结果。图20E示出了当以500nM的化合物(1-1)处理DMS79和DMS114细胞系0、4、24、48和72小时时,C-MYC和β-肌动蛋白水平的蛋白质印迹结果。在24小时暴露后,在DMS114细胞中观察到C-MYC蛋白下调。
在4种SCLC细胞系中,以500nM的化合物(1-1)处理后,没有在任一细胞系中观察到敏感性与BRD2/3/4、C-MYC、N-MYC、HEXIM1、Bcl-2和p21的基础mRNA水平之间的相关性。图21A-21G分别示出了用化合物(1-1)处理72小时的NCI-H69、NCI-H82、DMS79和DMS114细胞系中的BRD2、BRD3、BRD4、C-MYC、N-MYC、Bc12和p21水平的荧光强度,其按照GI50值排序。图21H示出了用化合物(1-1)处理4和24小时的NCI-H69、NCI-H82、DMS79和DMS114细胞系中,BRD2、BRD3、C-MYC、N-MYC、Bc12、p21和β-肌动蛋白水平的基础蛋白表达的蛋白质印迹结果。
在DMS114细胞中,化合物(1-1)以时间依赖性方式使得G1中的细胞周期停滞,不受理论束缚,其可以通过细胞周期抑制剂p27的蛋白水平上调来解释。图22A示出了用化合物(I-1)和对照物处理72小时的NCI-H69、NCI-H82、DMS79和DMS114细胞系在G1期的细胞数目。化合物(1-1)在72小时暴露后显著增加了DMS114细胞中的G1期细胞分数。图22B示出了用化合物(I-1)和对照物处理72小时的NCI-H69、NCI-H82、DMS79和DMS114细胞系的p27和β-肌动蛋白水平的蛋白质印迹。
不受特定理论的束缚,MYC家族在SCLC细胞系中没有介导化合物(1-1)的抗肿瘤作用。功能性RB1蛋白的存在,控制细胞在G1的进展,这可以解释化合物(1-1)的细胞抑制作用。在携带野生型RB1的敏感性细胞系中,化合物(1-1)降低C-MYC mRNA和蛋白质表达,并诱导G1期细胞周期停滞。这些发现表明,MYC家族在介导OTX015细胞抑制作用中发挥作用,该抑制作用出现在表达非突变RB1蛋白的SCLC细胞中,其发生在10%的SCLC患者中。
实施例12:在NSCLC和SCLC的临床前模型中的化合物(1-1)
在5种NSCLC细胞系(H2228、H3122、A549、HOP62、HOP92)和4种SCLC细胞系(H82、H69、DMS79、DMS114)中暴露72小时后,用MTT测定法测定化合物(1-1)(OncoEthix SA,瑞士)的生长抑制50%(GI50)值。对于细胞周期分析,用碘化丙啶染色细胞并用FACScan流式细胞仪分析。在用DMSO或OTX015(500nM)处理2、4、8或12和24小时后分离RNA,并在所有NSCLC和2种SCLC(H82和DMS114)细胞系中用Illumina HumanHT-12 Expression BeadChips获得基因表达谱(GEP)。使用LIMMA、GSEA、Metacore进行数据挖掘。
化合物(1-1)在5种NSCLC细胞系的4种中具有GI50值为110至940nM的抗增殖作用,而A549细胞(在KRAS和LKB1基因中具有突变)对化合物(1-1)具有抗性,如JQ1所描述的那样。在SLCL模型中,DMS114细胞对化合物(1-1)[GI50=120nM显示出剂量依赖性敏感性,而H82、H69和DMS79细胞是抗性的[GI50>6μM],尽管它们过表达CMYC和/或NMYC。在敏感性-NSCLC模型中,我们观察到化合物(1-1)处理后S期细胞增加,而在敏感性DMS114细胞系中观察到G1期增加。为了确定在SCLC细胞中化合物(1-1)处理后的转录变化,进行GEP分析以鉴定在敏感或抗性细胞系中,差异表达的总转录物以及早期(2-8h)或晚期(12-24h)期转录物。这表明EFR3B和与RNA生物合成有关的基因与对化合物(1-1)具有降低的敏感性相关。另一方面,在敏感性和抗性细胞系中,与组蛋白和染色质结构相关的基因在化合物(1-1)暴露后差异表达最大。在NSCLC模型中,EFR3B、IRF7和FOS基因与更高的化合物(1-1)敏感性相关,在化合物(1-1)治疗后观察到EFR3B和FOS基因上调以及IL7R和IL6相关基因下调。与化合物(1-1)抗性相关的转录物是转录因子FOXD1和与NSCLC细胞中的RNA加工、炎症和染色质结构相关的基因。
本领域技术人员应该理解的是,在不脱离本发明的宽泛的发明构思的条件下可以对上文所示的并描述的示例性实施方案进行修改。因此,应该理解的是本发明不限于所示的并描述的示例性实施方案,其旨在涵盖在权利要求书所限定的本发明的精神和范围内的修改。例如,示例性实施方案的特定特征可以为所要求保护的发明的一部分或不是其一部分,并且所公开的实施方案的特征可以结合。除非本发明作出具体说明,术语“一个”、“一种”和“所述”不限于一个元素,而是应该理解为“至少一个”。
应该理解的是,本发明的至少一些特征和描述被简化为集中于与清楚地理解本发明有关的元素,同时为了清晰起见,除去了本领域普通技术人员将理解的其他元素(也可以包含本发明的一部分)。但是由于此类元素是本领域公知的,并且由于它们不一定会更有利于更好地理解本发明,所以在本发明中未提供此类元素的描述。
此外,所述的方法并非依赖于本发明列出的步骤的特定顺序,就此而言,步骤的特定顺序不应该解释为对权利要求的限定。涉及本发明的方法的权利要求不应该限于书写顺序的步骤的性能,并且本领域的技术人员可以容易理解这些步骤可以改变,并且仍然保持在本发明的精神和范围内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种紫外线吸收剂uv-234的制备方法
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑5-甲基间苯二甲酸锰配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑镉混配配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑间苯二甲酸镉配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑锌混配配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑三维锌配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑对苯二甲酸铜配合物单晶与应用
- 1,4-二甲基-2,5-二-1h-双三唑均苯四酸铜配合物单晶与应用
- 一种4-苯基-3-((4,6-二甲基嘧啶-2-基硫代)甲基)-5-苄硫基三唑类化合物及其应用
- 1,4-二甲基-2,5-二-1h-1,2,4-双三唑二维铜配合物单晶与应用