一种提高难溶性药物口服吸收的纳晶组合物的制作方法
本发明属于药物制剂领域,涉及一种提高难溶性药物口服吸收的纳晶组合物。
背景技术:
口服是最简单、方便、患者顺应性好的给药方式,尤其对于慢性病的治疗。但是,口服药物需要在肠道溶解、吸收入血后才能发挥疗效。而对于新化合物实体,约70%难溶;对于上市药物,高达40-70%为水不溶或难溶药物,常因吸收少而导致疗效差,只能以增大剂量提高疗效为代价,这会引起毒副作用的增加和用药成本的上升。因此,提高难溶性药物的口服吸收是药剂学领域的研究热点和难点。
多种制剂学方法可提高难溶性药物的水溶性和口服吸收,包括成盐、应用潜溶剂、助溶剂、增溶剂、制备包合物和固体分散体等。然而,这些传统方法存在着各种各样的问题,如药物不一定都有成盐基团、可用的潜溶剂或助溶剂很少、增溶剂和包合物的毒副作用较大、固体分散体易陈化等,因此很难从根本上解决难溶性药物的口服吸收问题。纳米技术的快速发展为难溶性药物口服吸收问题的解决带来了曙光。其中纳米结晶避免了上述方法的弊端,具有明显优势,近来最为引人关注。
纳米结晶是一种不用载体材料,只利用少量表面活性剂或聚合物的稳定作用,将药物颗粒分散在水中,通过自组装技术或破碎技术形成稳定的纳米胶体分散体。体系中纳米级粒径的纯药物颗粒依靠表面活性剂或聚合物的电荷效应和/或立体效应稳定地悬浮在溶液中,其中药物的平均粒径一般为100-1000nm。与其他纳米制剂相比,纳米结晶有其自身的优点:适用于水油都不溶的药物,解决了非溶解必须的药物递送问题;冻干纳米结晶可解决物理或化学不相容问题,再分散性能好;致密的固体粒子载药量大,可以减少用药剂量及次数,降低不良反应,特别适合大剂量口服和注射给药;既可以提高溶解度,也可以提高溶出速率,还可以增强粘附性,能解决许多与口服生物利用度低的相关问题。此外,由于处方中不含载体和共溶剂,注射给药的安全性显著提高,而且也可经多种途径给药。例如,与市售制剂lipidiltmez相比,专利cn102988339a公开的非诺贝特纳米结晶的相对生物利用度为89.6%。
此外,小肠上皮细胞表达有特异性受体,若将纳米结晶表面用这些特异性受体的配体进行修饰,预期可增强小肠上皮细胞对纳米结晶的转运,促进药物吸收,实现提高难溶药物生物利用度的目的。现有技术中并未见有关配体修饰的纳米结晶用于口服的研究报道。
因此,研制一种提高难溶药物的溶解度、增强小肠上皮细胞的摄取以促进难溶药物的口服吸收的纳米结晶,有着广阔的应用前景。
技术实现要素:
本发明的目的在于克服难溶性药物常规增溶方式的不足以及吸收不高的问题。
一方面,本发明提供一种用于提高难溶性药物口服吸收的纳晶组合物。
另一方面,本发明提供难溶性药物纳晶组合物用于口服给药的用途。
根据本发明的一个方面,其提供一种用于口服的难溶性药物纳晶组合物,其包含难溶性药物、稳定剂、小肠上皮细胞表达的特异性受体的配体和/或转运体的底物以及任选的可药用辅料。
本发明还涉及制备难溶性药物纳晶组合物的方法,该方法选自溶剂非溶剂法、介质研磨法、高压均质法、沉淀-高压均质组合法、冻干-高压均质组合法等本领域公知的方法。纳晶粒子的平均粒径在500nm以下,优选300nm以下。
本发明组合物可根据任何常规方法配制成各种口服制剂,例如口服液、片剂、胶囊剂、颗粒剂等。
本发明组合物包括作为活性成分的难溶性药物,还包括稳定剂、小肠上皮细胞表达的特异性受体。所述的难溶性药物选自但不限于紫杉醇、多西他赛、9-硝基喜树碱、10-羟基喜树碱类药物如、伊曲康唑、替尼泊苷、依托泊苷、阿霉素、姜黄素、和厚朴酚、环孢素a、他克莫司、布洛芬、布地奈德、氟米龙、酚丁胺、地塞米松、醋酸可的松、丙酸氟卡替松、水飞蓟宾、水飞蓟素、阿瑞匹坦和非诺贝特,优选选自紫杉醇、多西紫杉醇、和厚朴酚、环孢素a和他克莫司。
所述的稳定剂选自但不限于tween80、十二烷基硫酸钠(sds)、聚氧乙烯蓖麻油、聚氧乙烯氢化蓖麻油、卵磷脂、豆磷脂、dspe-peg、聚乙二醇、普朗尼克、聚乙烯吡咯烷酮(pvp)、聚乙二醇维生素e琥珀酸酯(tpgs)、胆酸、胆酸钠、甲基纤维素(mc)、羟丙基纤维素(hpc)、羟丙基甲基纤维素(hpmc)和聚乙烯醇,优选选自卵磷脂、豆磷脂、dspe-peg、聚乙二醇、普朗尼克、聚乙烯吡咯烷酮(pvp)、聚乙二醇维生素e琥珀酸酯(tpgs)、羟丙基甲基纤维素(hpmc)和聚乙烯醇,更优选选自卵磷脂、豆磷脂、普朗尼克、聚乙二醇维生素e琥珀酸酯(tpgs)、羟丙基甲基纤维素(hpmc)和聚乙烯醇。
所述的难溶性药物与稳定剂的质量比为1:0.1-1:15,优选1:0.5-1:10,更优选1:2-1:6。
所述的小肠上皮细胞表达的特异性受体的配体选自但不限于转铁蛋白受体的配体,例如转铁蛋白、新生儿fc受体(fcrn)的配体,例如fcbp、叶酸受体的配体,例如叶酸、表皮生长因子受体egfr的配体,例如egf,和整合素受体αvβ3的配体,例如rgd。
所述的小肠上皮细胞表达的摄取性转运体的底物选自但不限于寡肽转运体pept1的底物,例如二肽和三肽、有机阳离子转运体oct的底物,例如胆碱、有机阳离子/肉毒碱转运体octns 的底物,例如肉毒碱、有机阴离子转运体oats的底物,例如牛磺胆酸盐、单羧酸转运体mct的底物,例如水杨酸、氨基酸转运体lat的底物,例如各种氨基酸。
所述的纳晶组合物,其中小肠上皮细胞表达的特异性受体的配体和/或转运体的底物的含量为组合物质量的至少5%,优选至少20%,更优选至少50%。
另外,本发明的组合物还可以包含药学上可接受的添加剂。
另一方面,本发明还涉及难溶性药物纳晶组合物用于治疗相关疾病的用途。
另一方面,本发明涉及以下方案:
方案1.一种提高难溶性药物口服吸收的纳晶组合物,其包含难溶性药物、稳定剂、小肠上皮细胞表达的特异性受体的配体和/或转运体的底物以及任选的可药用辅料。
方案2.如方案1所述的纳晶组合物,其中的难溶性药物选自但不限于紫杉醇、多西他赛、9-硝基喜树碱、10-羟基喜树碱类药物如、伊曲康唑、替尼泊苷、依托泊苷、阿霉素、姜黄素、和厚朴酚、环孢素a、他克莫司、布洛芬、布地奈德、氟米龙、酚丁胺、地塞米松、醋酸可的松、丙酸氟卡替松、水飞蓟宾、水飞蓟素、阿瑞匹坦和非诺贝特,优选选自紫杉醇、多西紫杉醇、和厚朴酚、环孢素a和他克莫司。
方案3.如方案1-2任一项所述的纳晶组合物,其中的稳定剂选自但不限于tween80、十二烷基硫酸钠(sds)、聚氧乙烯蓖麻油、聚氧乙烯氢化蓖麻油、卵磷脂、豆磷脂、dspe-peg、聚乙二醇、普朗尼克、聚乙烯吡咯烷酮(pvp)、聚乙二醇维生素e琥珀酸酯(tpgs)、胆酸、胆酸钠、甲基纤维素(mc)、羟丙基纤维素(hpc)、羟丙基甲基纤维素(hpmc)和聚乙烯醇,优选选自卵磷脂、豆磷脂、dspe-peg、聚乙二醇、普朗尼克、聚乙烯吡咯烷酮(pvp)、聚乙二醇维生素e琥珀酸酯(tpgs)、羟丙基甲基纤维素(hpmc)和聚乙烯醇,更优选选自卵磷脂、豆磷脂、普朗尼克、聚乙二醇维生素e琥珀酸酯(tpgs)、羟丙基甲基纤维素(hpmc)和聚乙烯醇。
方案4.如方案1-3任一项所述的纳晶组合物,其中的难溶性药物与稳定剂的质量比为1:0.1-1:15,优选1:0.5-1:10,更优选1:2-1:6。
方案5.如方案1-4任一项所述的纳晶组合物,其中的小肠上皮细胞表达的特异性受体的配体选自但不限于转铁蛋白受体的配体,例如转铁蛋白、新生儿fc受体(fcrn)的配体,例如fcbp、叶酸受体的配体,例如叶酸、表皮生长因子受体egfr的配体,例如egf,和整合素受体αvβ3的配体,例如rgd。
方案6.如方案1-4任一项所述的纳晶组合物,其中的小肠上皮细胞表达的摄取性转运体的底物选自但不限于寡肽转运体pept1的底物,例如二肽和三肽、有机阳离子转运体oct的底物,例如胆碱、有机阳离子/肉毒碱转运体octns的底物,例如肉毒碱、有机阴离子转 运体oats的底物,例如牛磺胆酸盐、单羧酸转运体mct的底物,例如水杨酸,和氨基酸转运体lat的底物,例如各种氨基酸。
方案7.如方案1-6任一项所述的纳晶组合物,其中小肠上皮细胞表达的特异性受体的配体和/或转运体的底物的含量为组合物质量的至少5%,优选至少20%,更优选至少50%。
方案8.如方案1-7任一项所述的纳晶组合物,其中所述纳晶的平均粒径在500nm以下,优选300nm以下。
方案9.如方案1-8任一项所述的纳晶组合物用于制备治疗相关疾病的药物的用途。
为了达到本发明的目的,本发明进行了本发明组合物的体外释放实验、离体小肠吸收实验和药代动力学实验。
附图说明参考附图1-图5,通过本发明的以下描述,本发明的上述及其它目的和特征将是显而易见的。
附图1为本发明实施例2的粒径分布图。
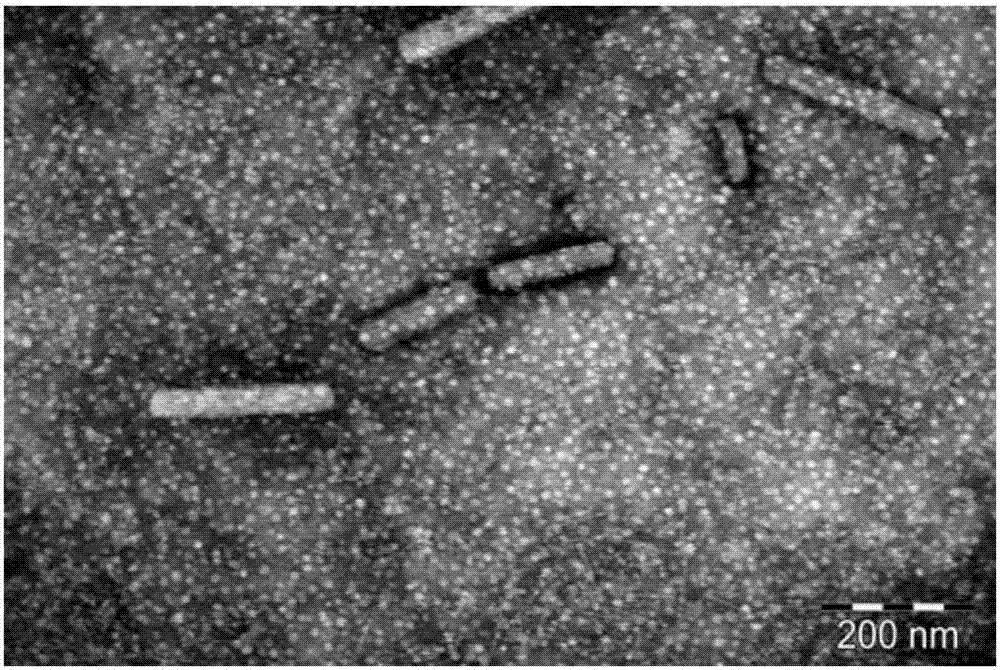
附图2显示了本发明实施例2的透射电镜图。
附图3显示了本发明实施例1和实施例2的体外释放曲线。
附图4显示了本发明实施例1和实施例2的离体小肠吸收百分率。
附图5显示了本发明实施例1和实施例2的药时曲线。
如上所述,本发明的组合物使难溶性药物的口服吸收显著提高。
具体实施方式
以下实施例用于进一步详细说明本发明,但绝对不是对本发明范围的限制。
实施例1紫杉醇纳晶的制备
将10mg紫杉醇和40mgtpgs溶于5ml二氯甲烷中,水浴超声使其全溶解后,氮气吹干。置于干燥器中,减压干燥30min后,加入4ml的蒸馏水,水化2h。涡旋5min,水浴超声15min即得紫杉醇纳晶的混悬液,冻干可得紫杉醇纳晶粉末。
实施例2转铁蛋白修饰的紫杉醇纳晶的制备
将10mg紫杉醇和40mgtpgs溶于5ml二氯甲烷中,水浴超声使其完全溶解后,氮气吹干。置于干燥器中,减压干燥30min后,加入4ml含有22mg转铁蛋白的蒸馏水,水化2h。涡旋5min,水浴超声15min即得转铁蛋白修饰的紫杉醇纳晶的混悬液,冻干可得转铁蛋白修饰的紫杉醇纳晶粉末。
将制得的纳晶混悬液采用malvern使zetasizernanozs仪进行进行动态光散射(dynamiclightscattering,dls)分析,测定胶束的粒径及其分布。仪器的激光束波长设定为525nm,入射光与散射光束的夹角为173°,测定温度为25℃。测定结果见附图1。由图可见,该实 施例的本发明胶束的平均粒径为230nm,粒度分布均匀,多分散性指数小于0.30。
采用醋酸双氧铀负染法,用透射电子显微镜观察纳晶的形态。结果见图2。由图可见,该实施例的本发明纳晶呈棒状,大小较均匀。
实施例3多西他赛纳晶的制备
将16mg多西他赛和80mg普朗尼克f127溶于4ml的氯仿中,超声使其完全溶解,然后用氮气恒速吹干,室温下置于真空干燥箱中12h以除去残余有机溶剂。加入4ml蒸馏水水化30min,涡旋10min,超声15min,即得多西他赛纳晶的混悬液,冻干可得多西他赛纳晶粉末。
实施例4rgd修饰的多西他赛纳晶的制备
将16mg多西他赛和80mg普朗尼克f127溶于4ml的氯仿中,超声使其完全溶解,然后用氮气恒速吹干,室温下置于真空干燥箱中12h以除去残余有机溶剂。加入4ml含有24mgrgd的蒸馏水水化30min,涡旋10min,超声15min,即得rgd修饰的多西他赛纳晶的混悬液,冻干可得rgd修饰的多西他赛纳晶粉末。
实施例59-羟基喜树碱纳晶的制备
将8mg9-羟基喜树碱和32mgtpgs溶于2ml的二氯甲烷中,超声使其完全溶解,旋转蒸发除去溶剂,室温下置于真空干燥箱中12h以除去残余有机溶剂。加入5ml蒸馏水水化40min,涡旋10min,超声15min,即得9-羟基喜树碱纳晶的混悬液,冻干可得9-羟基喜树碱纳晶粉末。
实施例6叶酸修饰的9-羟基喜树碱纳晶的制备
将8mg9-羟基喜树碱和32mgtpgs溶于2ml的二氯甲烷中,超声使其完全溶解,旋转蒸发除去溶剂,室温下置于真空干燥箱中12h以除去残余有机溶剂。加入5ml含有26mg叶酸的ph9的碳酸盐缓冲液水化40min,涡旋10min,超声15min,即得叶酸修饰的9-羟基喜树碱纳晶的混悬液,冻干可得叶酸修饰的9-羟基喜树碱纳晶粉末。
实施例7替尼泊苷纳晶的制备
将8mg替尼泊苷和24mgtween80溶于2ml的无水乙醇中,超声使其完全溶解,氮气恒速吹干,室温下置于真空干燥箱中12h以除去残余有机溶剂。加入5ml蒸馏水水化40min,涡旋10min,超声15min,即得替尼泊苷纳晶的混悬液,冻干可得替尼泊苷纳晶粉末。实施例8l-肉毒碱修饰的替尼泊苷纳晶的制备
将8mg替尼泊苷和24mgtween80溶于2ml的无水乙醇中,超声使其完全溶解,氮气恒速吹干,室温下置于真空干燥箱中12h以除去残余有机溶剂。加入5ml含有8mgl-肉毒碱的蒸馏水水化40min,涡旋10min,超声15min,即得l-肉毒碱修饰的替尼泊苷纳晶 的混悬液,冻干可得l-肉毒碱修饰的替尼泊苷纳晶粉末。
实施例9他克莫司纳晶的制备
将150mg卵磷脂分散于30ml蒸馏水中,加入15mg他克莫司,超声分散均匀,然后用高速剪切机于20000rpm剪切5min。接着用高压均质机分别在200bar循环10次、500bar循环10次、1000bar循环20次,即得他克莫司纳晶的混悬液,冻干可得他克莫司纳晶粉末。
实施例10phe-gly修饰的他克莫司纳晶的制备
将150mg卵磷脂分散于30ml蒸馏水中,加入15mg他克莫司、18mggly-phe-tpgs,超声分散均匀,然后用高速剪切机于20000rpm剪切3min。接着用高压均质机分别在200bar循环10次、500bar循环10次、1000bar循环20次,即得phe-gly修饰的他克莫司纳晶的混悬液,冻干可得phe-gly修饰的他克莫司纳晶粉末。
实施例11阿瑞匹坦纳晶的制备
将0.5ghpmc和0.5g卵磷脂分散于50ml蒸馏水中,加入1g阿瑞匹坦,超声分散均匀,然后用高速剪切机于15000rpm剪切5min。接着用高压均质机分别在500bar循环10次、1500bar循环20次,即得阿瑞匹坦纳晶的混悬液,冻干可得阿瑞匹坦纳晶粉末。
实施例12phe-gly-gly修饰的阿瑞匹坦纳晶的制备
将0.5ghpmc和0.5g卵磷脂分散于50ml蒸馏水中,加入1g阿瑞匹坦、0.25ggly-gly-phe-peg-dspe,超声分散均匀,然后用高速剪切机于15000rpm剪切5min。接着用高压均质机分别在500bar循环10次、1500bar循环20次,即得phe-gly-gly修饰的阿瑞匹坦纳晶的混悬液,冻干可得phe-gly-gly修饰的阿瑞匹坦纳晶粉末。
实施例13水飞蓟宾纳晶的制备
将25mg水飞蓟宾溶于5ml丙酮中,超声5min,得到淡黄色的澄清溶液。再将20mgtpgs和5mgpvp溶于50ml的蒸馏水,超声5min,得到透明的溶液。将丙酮溶液缓慢加入搅拌的水溶液中,加完后,继续搅拌10min。在50℃下,旋转蒸发除去丙酮。然后在高速剪切机中于15000rpm剪切10min,得到水飞蓟宾纳晶的混悬液,冻干可得水飞蓟宾纳晶粉末。
实施例14egf修饰的水飞蓟宾纳晶的制备
将25mg水飞蓟宾溶于5ml丙酮中,超声5min,得到淡黄色的澄清溶液。再将20mgtpgs、5mgpvp和8.8mgegf-tpgs溶于50ml的蒸馏水,超声5min,得到透明的溶液。将丙酮溶液缓慢加入搅拌的水溶液中,加完后,继续搅拌10min。在50℃下,旋转蒸发除去丙酮。然后在高速剪切机中于15000rpm剪切10min,得到egf修饰的水飞蓟宾纳晶的混悬液,冻干可得egf修饰的水飞蓟宾纳晶粉末。
试验例1体外释放试验
为了评价本发明纳晶组合物的体外释放特性,如下对实施例1、2中的本发明组合物在生理ph下的体外释放进行考察。
采用透析袋法。分别吸取1ml的本发明实施例2混悬液(0.1mg/ml)置于活化好的透析袋(mwco:8000-14000)中,并用透析夹密封,然后置于25ml含0.1%tween80的ph7.4的pbs中,于37℃、100rpm的水浴摇床中振荡,每个样品平行操作三份。分别在0.25、0.5、1、2、4、8、12、24和48h取出0.5ml释放介质,同时补充0.5ml新鲜的释放介质。将各时间点取出的释放介质经0.22μm滤膜过滤后,用rp-hplc方法测定紫杉醇的含量,并计算药物的累积释放百分数,绘制释放曲线。并以实施例1和taxol作为对照。
色谱条件如下:diamobsilodsc18色谱柱,250×4.6mm,5μm;流动相:乙腈∶水=2∶1;流速:1ml/min;柱温:50℃;检测波长:227nm;进样量:20μl。
释放曲线见图3。结果显示,在37℃下的48h内,实施例2的累积释放量为42.6±2.3%,实施例1的累积释放量为76.0±2.0%,而taxol的累积释放量为86.9±0.5%,说明本发明制备的纳晶组合物具有缓慢释放的特征。
试验例2离体小肠吸收试验
采用肠囊翻转法。选取健康的sd大鼠,实验前禁食24h(自由饮水)。用20%的乌拉坦腹腔注射麻醉,沿腹中线打开腹腔,取出实验的小肠段。将取出的小肠立即置于预热37℃的krebs液中,用krebs液冲洗小肠表面,并仔细清除浆膜层外面的脂肪组织。剪取大约5cm的一段小肠,用手术线结扎肠段一端,用圆头细玻璃棒将肠段翻转,使粘膜层在外,浆膜层在内,洗净内容物。用手术线结扎肠段另一端,肠囊内注入1.0mlkrebs液,滤纸吸干表面,立即放入含有本发明实施例2的krebs液的小试管中,持续通入氧气,置于37℃水浴中孵育,每个样品平行操作三份。1.5h后,取出肠囊中的200μl肠液于5mlep管中,加入内标液地西泮20μl(300μg/ml)和3ml叔丁基甲醚,涡旋混合5min,1000rpm离心15min。取上清1ml,氮气吹干,加入100μl甲醇复溶。用rp-hplc法测定药物的含量,计算吸收百分数。并以实施例1作为对照。
色谱条件如下:diamobsilodsc18色谱柱,250×4.6mm,5μm;流动相:乙腈∶水=2∶1;流速:1ml/min;柱温:30℃;检测波长:227nm;进样量:20μl。
结果如图4所示。结果表明,供试液与小肠囊共孵育1.5h后,测得实施例1(紫杉醇纳晶)的吸收百分数为5.8±0.6%(n=3);而实施例2(转铁蛋白修饰的紫杉醇纳晶)的吸收百分数为15.0±2.1%(n=3),即实施例2的吸收百分数是实施例1的2.59倍,显示了本发 明组合物的优越性。
试验例3药代动力学试验
为了评价本发明纳晶组合物的口服吸收情况,如下对本发明组合物进行药代动力学试验。
取15只雄性sd大鼠,体重为(300±20)g,随机分为三组:taxol组、本发明实施例1组、本发明实施例2组。实验前禁食12h,可自由饮水。给药剂量为15mg/kg。给药前,大鼠腹腔内注射水合氯醛溶液进行麻醉,剂量为300mg/kg。麻醉后将大鼠仰卧固定,于剑突下方开口,开口尽量与腹中线平行,开口尽量小。确定胃的位置,取出十二指肠,于血管较少处注射给药。给药后肌肉与皮肤分开缝合,皮肤止血。分别于给药前和给药0.5、1、2、4、6、10、24、48h定时从大鼠眼眶后静脉丛取血0.5ml至肝素化的ep管中,8000rpm离心15min得血清。精密吸取100μl于5mlep管中,加入内标液地西泮10μl(300μg/ml)和3ml的叔丁基甲醚,涡旋5min,1000rpm离心15min。取上清1ml,氮气吹干,加入100μl甲醇复溶,用0.2μm滤膜过滤后,采用rp-hplc法测定血中药物浓度。
色谱条件如下:色谱条件如下:diamobsilodsc18色谱柱,250×4.6mm,5μm;流动相:乙腈∶水=1∶1;流速:1ml/min;柱温:50℃;检测波长:227nm;进样量:20μl。
药时曲线如图5所示。结果表明,与taxol组相比,实施例1和实施例2组的血药浓度峰值分别提高了629和5592倍,auc值分别提高了122倍和598倍;而实施例2组的血药浓度峰值又比实施例1组提高了681倍,auc提高了4.8倍。因此,本发明组合物能够明显提高紫杉醇的口服吸收,且配体修饰的纳晶组合物促吸收作用更优,与离体小肠吸收的结果一致,显示了本发明组合物的优越性。
虽然已利用上述具体的实施方案对本发明进行了描述,但应认识到,本领域的技术人员还可进行各种的改进和改变,而且它们也应如权利要求书限定的本发明的范围之内。
- 还没有人留言评论。精彩留言会获得点赞!