环糊精‑金属有机骨架材料复合微球及其制备方法与流程
本发明涉及生物材料领域,更具体地涉及一种环糊精-金属有机骨架材料复合微球及其制备方法。
背景技术:
环糊精-金属有机骨架主要是利用环糊精在水溶液中能与第一、二主族金属离子以一种有机配位的方式形成一种新的晶体,这种晶体具有多孔、表面积大、储存气体等特点。这种绿色、多孔材料能够吸附一些结构不稳定的药物,其巨大的空腔能够对药物起到保护作用,这使得其用于商业发展成为可能,尤其是由于环糊精-金属有机骨架为可食用衍生物,适于人类食用。将环糊精作为有机配体,金属离子作为无机金属中心,可形成新的、安全性较高、可药用的环糊精-金属有机骨架,即cd-mof。
与现有mof类似,cd-mof也存在生物体内稳定性的问题,即遇到体液结构容易瓦解,在到达靶部位之前无法确保多孔晶体结构的稳定。目前虽有mof复合材料报道,如膜、纤维和微球等,以改善mof的机械、热、化学稳定性和成形性。但所用材料的生物相容性较差,如聚甲基丙烯酸甲酯、聚酰亚胺、聚乙酸乙烯酯、聚砜、聚二甲硅氧烷、聚乙烯吡咯烷酮和苯乙烯等。同时cd-mof存在遇水容易崩溃、结构瓦解的问题,用于药物载体易造成药物的突释。
因此,本领域迫切需要开发提升cd-mof生物体内稳定性的材料和方法。
技术实现要素:
本发明的目的在于提供一种环糊精-金属有机骨架材料复合微球及其制备方法。
在本发明的第一方面,提供了一种制备环糊精-金属有机骨架材料复合微球的方法,所述方法包括步骤:
(i)提供一环糊精-金属有机骨架材料;
(ii)在所述的环糊精-金属有机骨架材料上负载药物,得到载药的环糊精-金属有机骨架材料;
(iii)在所述载药的环糊精-金属有机骨架材料的表面包裹聚丙烯酸树脂,从而得到所述的复合微球。
在另一优选例中,所述的步骤(iii)包括步骤:
(iiia)将所述的载药的环糊精-金属有机骨架材料与聚丙烯酸树脂、分散剂、任选地增塑剂、以及有机溶剂b混合,得到一混合溶液c;
(iiib)将混合溶液c与液体石蜡混合,从而得到一混合溶液d;
(iiic)去除混合溶液d中的有机溶剂,从而得到所述的复合微球。
在另一优选例中,所述的步骤(iiia)是将所述的载药的环糊精-金属有机骨架材料和分散剂加入到一混合溶液b中,从而得到所述的混合溶液c,其中,所述的混合溶液b含有聚丙烯酸树脂、增塑剂、有机溶剂b和水;
在另一优选例中,所述的步骤(iiia)包括步骤:
(a)将所述的载药的环糊精-金属有机骨架材料溶解于一有机溶剂b中,从而得到一混合溶液e;
(b)向所述的混合溶液d中加入聚丙烯酸树脂、分散剂、和任选的增塑剂,从而得到所述的混合溶液c;
在另一优选例中,所述的聚丙烯酸树脂包括均聚物和/或共聚物树脂。
在另一优选例中,所述共聚物中源自丙烯酸(酯)单体的单元含量为30-80mol%。
在另一优选例中,在步骤(ii)中,所述的负载药物包括孵育载药和共结晶载药。
在另一优选例中,所述的孵育载药是将所述的环糊精-金属有机骨架材料混悬于含有药物和有机溶剂a的混合溶液a中。
在另一优选例中,所述的有机溶剂a包括但不限于甲醇、乙醇、丙酮、异丙醇、乙酸乙酯、氯仿、正己烷、和/或二甲基甲酰胺,较佳地为乙醇。
在另一优选例中,所述的共结晶载药是在所述环糊精-金属有机骨架材料制备的过程中加入药物。
在另一优选例中,所述的步骤(i)和步骤(ii)同时进行。
在另一优选例中,在步骤(iiia)中,将聚丙烯酸树脂溶解于有机溶剂b中,再加入增塑剂,从而得到混合溶液b。
在另一优选例中,所述的有机溶剂b选自下组:甲醇、乙醇(95%)、异丙醇、丙酮、或其组合。
在另一优选例中,所述混合溶液b中水的含量为2%-5%,较佳地为2-3%,以混合溶液b的总重量计。
在另一优选例中,所述混合溶液b中聚丙烯酸树脂的含量为1.5%-20%,较佳地为2-5%,以混合溶液b的总重量计
在另一优选例中,所述混合溶液b中增塑剂与聚丙烯酸树脂的比例为1:(5-10)。
在另一优选例中,所述混合溶液c中载药cd-mof粉末与聚丙烯酸树脂的重量比为1:(3-10)。
在另一优选例中,所述混合溶液c中分散剂浓度(w/v)为1%-5%,以混合溶液c的总体积计。
在另一优选例中,在步骤(iiib)中,混合溶液c与液体石蜡的混合比例(v/v)为1:3-1:10。
在另一优选例中,在步骤(iiib)中,将混合溶液c和液体石蜡冷却至0-15℃后,再进行混合。
在另一优选例中,在步骤(iiib)中,混合后还包括分散机分散的步骤。
在另一优选例中,在步骤(iiic)中,通过搅拌加热,去除有机溶剂b。
在另一优选例中,在步骤(iiic)中,通过离心和有机溶剂c的洗涤,去除液体石蜡。
在另一优选例中,所述的有机溶剂c包括正己烷。
在另一优选例中,在步骤(i)中,所述的环糊精-金属有机骨架材料的制备包括步骤:
(1)提供第一混合溶液,所述第一混合溶液为含有金属离子和环糊精的溶液;
(2)向所述的第一混合溶液中加入第一有机溶剂,获得第二混合溶液,
其中,所述第一有机溶剂与所述第一混合溶液的体积比为(0.01-5):1,较佳地为(0.1-2):1,最佳地为(0.5-1):1;
(3)对所述第二混合溶液进行预处理,获得经预处理的第一混合物,其中所述的预处理选自下组:溶剂热处理、微波处理、超声波处理、或其组合,
(4)任选地,当第一混合物中含有析出的环糊精-金属有机骨架材料时,从所述第一混合物中分离获得析出的环糊精-金属有机骨架材料;
(5)当从所述第一混合物中分离出部分或全部的溶液,作为第三混合溶液;并向所述第三混合溶液中加入第二有机溶剂和/或尺寸调节剂,从而析出环糊精-金属有机骨架材料;和
(6)任选地对步骤(5)中析出的环糊精-金属有机骨架材料进行分离和/或干燥。
在另一优选例中,所述的预处理为微波处理、或超声波处理。
在另一优选例中,步骤(3)和步骤(5)的总时间t为1分钟-12小时,更佳地为1分钟-3小时,最佳地为1分钟-1小时,或1-30分钟,或5-30分钟、或2-25分钟。
或者,步骤(3)和步骤(5)的总时间t为5分钟-12小时,更佳地为5分钟-3小时,最佳地为10分钟-1小时。
在另一优选例中,所述的尺寸调节剂选自下组:聚乙二醇、聚维酮、聚山梨醇、失水山梨醇单月桂酸酯、聚氧乙烯月桂醇醚、乳化剂op(壬烷基酚聚氧乙烯醚缩合物)、乳百灵a(聚氧乙烯脂肪醇醚)、普流罗尼(聚氧乙烯聚丙二醇缩合物)、十二烷基硫酸钠、十二烷基苯磺酸钠、十二烷基二甲基苄基溴化铵(苯扎溴铵)、或其组合。
在另一优选例中,所述的尺寸调节剂为聚乙二醇。
在另一优选例中,所述的聚乙二醇包括peg200、peg400、peg600、peg800、peg1000、peg1500、peg2000、peg4000、peg6000、peg8000、peg10000、peg20000、或其组合。
在另一优选例中,所述的聚维酮包括pvpk12、pvpk15、pvpk17、pvpk25、pvpk30、pvpk60、pvpk90、pvpk120、或其组合。
在另一优选例中,所述的聚山梨醇包括吐温20、吐温40、吐温60、吐温80、吐温85、或其组合。
在另一优选例中,所述的失水山梨醇单月桂酸酯包括司盘20、司盘40、司盘60、司盘80、或其组合。
在另一优选例中,所述的尺寸调节剂包括peg2000、peg4000、peg6000、peg8000、peg10000、peg20000、或其组合,较佳地为peg20000。
在另一优选例中,所述预处理的温度为25-100℃,较佳地为30-80℃,更佳地为40-60℃。
在另一优选例中,所述预处理的时间为10min-24h,较佳地为15min-1h,更佳地为20-30min。
在另一优选例中,所述的溶剂热处理是对混合溶液进行水浴加热、或油浴加热。
在另一优选例中,所述微波处理的功率为20-1000w,较佳地为25-100w。
在另一优选例中,所述微波处理的辐射频率为916-2450mhz,较佳地为2450mhz。
在另一优选例中,所述超声波处理的功率为20-1000w,较佳地为40w。
在另一优选例中,所述超声波处理的辐射频率为22-100khz,较佳地为30-50khz。
在另一优选例中,所述的第一有机溶剂和第二有机溶剂各自独立地选自下组:甲醇、乙醇、异丙醇、丙酮、乙腈、或其组合。
在另一优选例中,所述的第一有机溶剂和第二有机溶剂是相同或不同的。
在另一优选例中,所述的第一有机溶剂和第二有机溶剂为甲醇。
在另一优选例中,所述的步骤(4)可进行,也可不进行。
在另一优选例中,所述的制备的环糊精-金属有机骨架材料具有选自下组的一个或多个特征:
(i)平均粒径:50nm-50微米,较佳地为100-1000纳米(纳米级)或1-10微米(微米级);
(ii)所述环糊精-金属有机骨架材料中,cd与金属离子的摩尔比为1~1.2:6-10(如1:6-10,或约1:8);
(iii)所述的环糊精-金属有机骨架材料为药学上可接受的载体;
(iv)所述的环糊精-金属有机骨架材料能够对热不稳定药物有较好的保护作用。
在另一优选例中,在步骤(5)中,所述第二有机溶剂与第三混合液的体积比为(0.01-5):1,较佳地为(0.5-2):1,更佳地为1:1。
在另一优选例中,所述的第三混合溶液为上清液。
在另一优选例中,在步骤(5)中,加入的尺寸调节剂的量为1-20mg/ml,较佳地为5-10mg/ml。
在另一优选例中,在步骤(5)中,对第一混合物进行离心处理,从而从所述第一混合物中分离出部分溶液。
在另一优选例中,所述离心处理的转速为1000-5000rpm,较佳地为2000-3000rpm。
在另一优选例中,所述离心处理的时间为3-10min,较佳地为5-8min。
在另一优选例中,在步骤(6)中,包括步骤:
(a)对预处理后的混合溶液进行离心,从未获得沉淀物;
(b)对所述沉淀物进行洗涤;和
(c)对洗涤后的沉淀物进行真空干燥,从而获得结晶的环糊精-金属有机骨架材料。
在另一优选例中,在步骤(b)中,用乙醇对所述沉淀物进行洗涤。
在另一优选例中,在步骤(c)中,所述真空干燥的温度为40-60℃。
在另一优选例中,在步骤(c)中,所述真空干燥的时间为6-24h。
在另一优选例中,在步骤(1)中,将金属化合物的水溶液和环糊精水溶液混合,从而获得所述的第一混合溶液。
在另一优选例中,在步骤(1)中,将金属化合物和环糊精溶解于水中,从而获得所述的第一混合溶液。
在另一优选例中,所述的金属化合物包括金属盐和金属碱。
在另一优选例中,所述的金属化合物为koh。
在另一优选例中,所述的第一混合溶液中金属离子的浓度为0.05-0.4m,较佳地为0.1-0.3m,更佳地为0.2m。
在另一优选例中,所述的第一混合溶液中环糊精的浓度为0.013-0.05m,较佳地为0.02-0.03m,更佳地为0.025m。
在另一优选例中,所述的第一混合溶液中环糊精与金属离子的摩尔比为1:(6-10),较佳地为1:8。
在另一优选例中,所述的金属离子选自下组:li+、k+、rb+、cs+、na+、mg2+、cd2+、sn2+、ag+、yb+、ba2+、sr2+、ca2+、pb2+、la3+、或其组合。
在另一优选例中,所述金属离子为k+。
在另一优选例中,所述的环糊精选自下组:α-环糊精、β-环糊精、γ-环糊精、羟丙基-β-环糊精、磺丁基-β-环糊精、甲基-β-环糊精、羧甲基-β-环糊精、或其组合。
在另一优选例中,所述的环糊精为γ-环糊精。
在另一优选例中,在步骤(i)中,所述的环糊精-金属有机骨架材料的制备包括步骤:
(1)提供第一混合溶液,所述第一混合溶液为含有金属离子和环糊精的溶液;
(2)向所述的第一混合溶液中加入第一有机溶剂,获得第二混合溶液,
其中,所述第一有机溶剂与所述第一混合溶液的体积比为(0.01-0.5):1,较佳地为(0.03-0.3):1,最佳地为(0.05-0.2):1;
(3)对所述第二混合溶液进行预处理,获得经预处理的第一混合物,其中所述的预处理选自下组:
(a)溶剂热挥发处理;
(b)溶剂热挥发处理与选自a组的任一处理方式的组合,其中a组包括溶剂热处理、微波处理、超声波处理、或其组合;
(4)当第一混合物中含有析出的环糊精-金属有机骨架材料时,从所述第一混合物中分离获得析出的环糊精-金属有机骨架材料;
或者从所述第一混合物中分离出部分或全部的溶液,作为第三混合溶液;并向所述第三混合溶液中加入第二有机溶剂和/或尺寸调节剂,从而析出环糊精-金属有机骨架材料;和
(5)任选地对步骤(4)中析出的环糊精-金属有机骨架材料进行分离和/或干燥。
在另一优选例中,在步骤(3)中,所述的溶剂热挥发处理包括步骤:
(i)将混合溶液置于一开口容器i中;
(ii)提供一装有有机溶剂的开口容器ii,将所述开口容器i和开口容器ii共同置于一封闭体系内;和
(iii)对所述开口容器ii中的有机溶剂进行加热/保温处理,使得所述有机溶剂蒸发扩散至混合溶液中。
在另一优选例中,在步骤(iii)中,对所述封闭体系进行整体加热处理,从而加热所述开口容器ii中的有机溶剂
在另一优选例中,在步骤(iii)中,所述加热处理包括水浴加热、和油浴加热。
在另一优选例中,在步骤(iii)中,所述加热处理的温度为25-100℃,较佳地为30-80℃,更佳地为40-60℃。
在另一优选例中,在步骤(iii)中,所述加热处理的时间为4-48h,较佳地为6-24h。
在本发明的第二方面,提供了一种环糊精-金属有机骨架材料复合微球,所述的复合微球含有:
组分(a):载药的环糊精-金属有机骨架材料;和
组分(b):包裹组分(a)的包裹层;
并且,所述的环糊精-金属有机骨架材料具备选自下组的一个或多个特征:
(i)平均粒径:50纳米-50微米,较佳地为100-1000纳米(纳米级)或1-10微米(微米级);
(ii)所述环糊精-金属有机骨架材料中,cd与金属离子的摩尔比为1~1.2:6-10;
(iii)所述的环糊精-金属有机骨架材料为药学上可接受的载体。
在另一优选例中,所述复合微球的粒径为50μm-500μm,较佳地50μm-200μm,更佳地50μm-100μm。
在另一优选例中,所述复合微球中组分(a)和组分(b)的重量比为1:(1-50),较佳地为1:9。
在另一优选例中,所述复合微球中组分(a)和组分(b)的重量之和占复合微球重量的80-100wt%,较佳地90-100wt%,更佳地95-100wt%。
在另一优选例中,所述的包裹层含有聚丙烯酸树脂。
在另一优选例中,所述的包裹层还含有增塑剂和分散剂。
在另一优选例中,所述的聚丙烯酸树脂、增塑剂和分散剂处于混合状态。
在另一优选例中,所述的聚丙烯酸树脂包括ph依赖型聚丙烯酸树脂和/或ph非依赖型聚丙烯酸树脂,较佳地为ph依赖型聚丙烯酸树脂。
在另一优选例中,所述的ph依赖型聚丙烯酸树脂包括:甲基丙烯酸与丙烯酸丁酯(35:65)的共聚物、甲基丙烯酸与甲基丙烯酸甲酯(1:1)的共聚物、和/或甲基丙烯酸与甲基丙烯酸甲酯(1:2)的共聚物。
在另一优选例中,所述的ph非依赖型聚丙烯酸树脂包括:丙烯酸乙酯、甲基丙烯酸甲酯、甲基丙烯酸氯化三甲氨基乙酯(1:2:0.2)的共聚物,和/或丙烯酸乙酯、甲基丙烯酸甲酯、甲基丙烯酸氯化三甲氨基乙酯(1:2:0.1)的共聚物。
在另一优选例中,如果组分(a)负载的药物为酸不稳定药物,所述的组分(b)(聚丙烯酸树脂)中不含有羧基丙烯酸树脂。
在另一优选例中,所述的聚丙烯酸树脂为固体颗粒和/或粉末。
在另一优选例中,所述的增塑剂选自下组:柠檬酸三乙酯、柠檬酸三丁酯、乙酰柠檬酸三乙酯、邻苯二甲酸二丁酯、邻苯二甲酸二乙酯、癸二丁酸二丁酯、癸二丁酸二辛酯、甘油三醋酸酯、甘油单醋酸酯、聚乙二醇、丙二醇、甘油、椰子油、蓖麻油、玉米油、或其组合;较佳地为柠檬酸三乙酯。
在另一优选例中,所述的分散剂包括接触角大于90°的固体微粒乳化剂或w/o型乳化剂。
在另一优选例中,所述的分散剂选自下组:氢氧化钙、氢氧化锌、硬脂酸钠、硬脂酸钾、硬脂酸钙、硬脂酸铝、硬脂酸镁、司盘-80、蔗糖脂肪酸酯、或其组合;较佳地为硬脂酸铝。
在另一优选例中,所述载药的环糊精-金属有机骨架材料是指负载药物的环糊精-金属有机骨架材料。
在另一优选例中,所述药物包括兰索拉唑、布洛芬、芬布芬、地西泮、甲硝唑、硝苯地平、泼尼松龙、双氯芬酸钠、对乙酰氨基酚、甲苯磺丁脲、美洛昔康、克伦特罗、氟康唑、卡托普利、水杨酸、土槿皮乙酸、吲达帕胺、普罗西康、咖啡因、酮洛芬、吲哚美辛、萘普生、白消安、阿霉素、顺铂前药、拓扑替康、5-氟尿嘧啶、单/三磷酸-叠氮胸苷、西多福韦、尼美舒利、盐酸普鲁卡因胺,优选含有羧基的药物和烯醇基的药物,包括兰索拉唑、布洛芬、芬布芬。
在另一优选例中,所述的复合微球用本发明第一方面所述的方法制备。
在本发明的另一个方面,提供了一种环糊精-金属有机骨架材料复合微球,其结构特征在于具有立方晶体结构的环糊精-金属有机骨架被包裹于微球内,内部为具有立方晶体结构的环糊精-金属有机骨架,外部为球形结构;微球基质作为一级药物储库,环糊精-金属有机骨架作为二级药物储库。该特征能防止环糊精-金属有机骨架材料作为药物储库的遇水崩解问题,保护cd-mof多孔骨架结构的完整性。环糊精-金属有机骨架材料用于药物载体储库,载药后的环糊精-金属有机骨架材料作为二级结构,结合一级结构微球,防止药物的突释、实现药物的缓慢释放。主要由载药环糊精-金属有机骨架、聚丙烯酸树脂、增塑剂及分散剂组成。
所述的环糊精-金属有机骨架材料与微球主要形成材料聚丙烯酸树脂的比例为1:1-1:50,优选为1:9。
本发明所述的一种环糊精-金属有机骨架材料复合微球,由以下组分组成(未标明之处均为重量百分比):
所述的环糊精-金属有机骨架材料为γ-环糊精与碱金属盐形成的骨架材料:
碱金属包括但不限于li+、k+、rb+、cs+、na+、mg2+、cd2+、sn2+、ag+、yb+、ba2+、sr2+、ca2+、pb2+、la3+,优选k+。
与碱金属成盐的阴离子包括但不限于oh-、no3-、hco3-、ch3coo-、scn-、c6h5cooh=c6h5coo-、cl-、br-、i-、o2-、s2-、hs-、、hso4-、clo-、clo3-、mno4-,优选oh-。
环糊精-金属有机骨架材料的平均粒径在50纳米-50微米,较佳地为100-1000纳米(纳米级)或1-10微米(微米级)。
所述药物包括兰索拉唑、布洛芬、芬布芬、地西泮、甲硝唑、硝苯地平、泼尼松龙、双氯芬酸钠、对乙酰氨基酚、甲苯磺丁脲、美洛昔康、克伦特罗、氟康唑、卡托普利、水杨酸、土槿皮乙酸、吲达帕胺、普罗西康、咖啡因、酮洛芬、吲哚美辛、萘普生、白消安、阿霉素、顺铂前药、拓扑替康、5-氟尿嘧啶、单/三磷酸-叠氮胸苷、西多福韦、尼美舒利、盐酸普鲁卡因胺,优选含有羧基的药物和烯醇基的药物,包括兰索拉唑、布洛芬、芬布芬。
所述的聚丙烯酸树脂作为微球的基质成分,具有特定的化学结构,是以下成分的一种或两种以上。包括ph依赖型聚丙烯酸树脂和ph非依赖型聚丙烯酸树脂的固体颗粒或粉末。ph依赖型聚丙烯酸树脂包括甲基丙烯酸与丙烯酸丁酯(35:65)的共聚物(ph>5.5时溶解)、甲基丙烯酸与甲基丙烯酸甲酯(1:1)的共聚物(ph>7.0时溶解)、甲基丙烯酸与甲基丙烯酸甲酯(1:2)的共聚物(ph>6.0时溶解),且不溶于水、乙醚,溶于极性有机溶剂中,在丙酮中膨胀。ph非依赖型聚丙烯酸树脂包括丙烯酸乙酯、甲基丙烯酸甲酯、甲基丙烯酸氯化三甲氨基乙酯(1:2:0.2,高渗透性)的共聚物,以及丙烯酸乙酯、甲基丙烯酸甲酯、甲基丙烯酸氯化三甲氨基乙酯(1:2:0.1,低渗透性)的共聚物。优选不含羧基丙烯酸树脂。
所述的增塑剂为柠檬酸三乙酯、柠檬酸三丁酯、乙酰柠檬酸三乙酯、邻苯二甲酸二丁酯、邻苯二甲酸二乙酯、癸二丁酸二丁酯、癸二丁酸二辛酯、甘油三醋酸酯、甘油单醋酸酯、聚乙二醇、丙二醇、甘油、椰子油、蓖麻油、玉米油中的一种或者两种以上,优选柠檬酸三乙酯。
所述的分散剂为接触角大于90°的固体微粒乳化剂或w/o型乳化剂,包括氢氧化钙、氢氧化锌、硬脂酸钠、硬脂酸钾、硬脂酸钙、硬脂酸铝、硬脂酸镁、司盘-80、蔗糖脂肪酸酯中的一种或者两种以上。优选硬脂酸铝。
在本发明的其他方面,还提供了一种制备所述环糊精-金属有机骨架复合微球的方法,以聚丙烯酸树脂为基质材料,用s/o/o复乳溶剂挥发法将载药cd-mof制成微球,将复乳法、相分离法和溶剂挥发法的优势相结合制备环糊精-金属有机骨架复合微球,无需超声、高压均质等特殊乳化工艺,制备工艺简单可控,得到的微球具有表面圆整、粒径均匀可控、释药行为可控的特点。该方法包括:
(1)cd-mof的制备:所述制备方法包括将金属盐溶液与环糊精水溶液混合后,预加一部分有机溶剂,一定温度下,通过溶剂蒸汽扩散方法,反应一定时间,再加入尺寸调节剂,从而得到所述基于环糊精的金属有机骨架材料;或将金属盐溶液与环糊精水溶液混合,预加一部分有机溶剂,用溶剂热/微波/超声波振动反应介质,使得反应物快速反应,反应一定时间后加入尺寸调节剂,从而得到所述基于环糊精的金属有机骨架材料。
所述金属盐溶液中金属盐的浓度为0.05-0.4m。优选0.2m。
所述环糊精水溶液中环糊精的浓度为0.013-0.05m。优选0.025m。
所述尺寸调节剂包括聚乙二醇(peg200、400、600、800、1000、1500、2000、4000、6000、8000、10000、20000)、聚维酮(pvpk12、k15、k17、k25、k30、k60、k90、k120)、聚山梨醇(吐温20、40、60、80、85)、失水山梨醇单月桂酸酯(司盘20、40、60、80)、聚氧乙烯月桂醇醚、乳化剂op(壬烷基酚聚氧乙烯醚缩合物)、乳百灵a(聚氧乙烯脂肪醇醚)、普流罗尼(聚氧乙烯聚丙二醇缩合物)、十二烷基硫酸钠、十二烷基苯磺酸钠、十六烷基三甲基溴化铵(ctab)、十二烷基二甲基苄基溴化铵(苯扎溴铵)及它们的衍生物中的一种或几种,以及几种尺寸调节剂的组合。优选药用辅料peg2000,4000,6000,8000,10000,20000,具体可为peg20000。
所述有机溶剂包括但不限于甲醇、乙醇、丙酮、异丙醇、乙腈,具体可为甲醇。
所述cd-mof制备包括在所得上清液中按照0.05-10ml有机溶剂和/或peg20000/5ml上清液比例加入对应尺寸调节剂。所述peg20000加入量包括1-16mgpeg20000/ml上清液,优选8mgpeg20000/ml上清液。
所述环糊精与金属盐水溶液的摩尔比为0.06:0.5-0.25:2,优选0.125:1。
所述溶剂热挥发方法,温度包括室温-100℃,反应时间4-24h,优选50℃,6h。
所述溶剂热方法,温度包括室温-100℃,反应时间1min-24h,优选50℃,20min。
所述微波辐射频率采用916-2450mhz,功率为20-1000w,温度设置25-100℃,反应时间1min-24h,优选2450mhz,25w,50℃,20min。
所述超声波辐射频率采用22-40khz,功率为100-1000w,温度设置25-100℃,反应时间1min-24h,优选30khz,300w,50℃,20min。
(2)载药cd-mof的制备:所述的载药方法包括但不限于孵育载药和共结晶载药,具体而言,孵育载药是将适量cd-mof粉末混悬于药物的有机溶剂溶液,有机溶剂包括但不限于甲醇、乙醇、丙酮、异丙醇、乙酸乙酯、氯仿、正己烷、二甲基甲酰胺等,优选乙醇。共结晶载药是在cd-mof制备过程中加入药物,包括但不限于将药物直接溶解于γ-cd-koh水溶液、将药物溶解于甲醇加入γ-cd-koh水溶液中、将药物溶解于甲醇加入完成甲醇扩散的γ-cd-koh水溶液中。
(3)内油相聚丙烯酸树脂溶液制备:将聚丙烯酸树脂或其混合物溶解于有机溶剂中,再加入增塑剂,得到有机溶液。其中,所用的溶剂有甲醇、乙醇(95%)、异丙醇、丙酮及它们的混合物,使用无水溶剂易使所述的聚丙烯酸树脂结块,因此需加入2%~5%的水,优选2%水;聚丙烯酸树脂有机溶液浓度为1.5%~20%;增塑剂与聚丙烯酸树脂的比例为1:5~1:10。
(4)s/o初乳制备:将载药cd-mof粉末及分散剂加到步骤(2)得到的有机溶液中,超声形成s/o初乳。其中,载药cd-mof粉末与聚丙烯酸树脂的重量比为1:3~1:10;分散剂在初乳中的浓度为1%-5%(w/v)。
(4)s/o/o复乳制备:将步骤(3)的s/o型初乳与液体石蜡分别冷却至一定温度,然后将两者混合,分散机分散,形成s/o/o型复乳。其中s/o型初乳与液体石蜡的体积比为1:3~1:10。其中,冷却至一定温度,该温度为0℃~15℃,优选10℃。
(5)溶剂挥发:边搅拌边将步骤(4)所得s/o/o复乳缓慢升温;然后将所得液体旋蒸,除去剩余有机溶剂。其中,升温起始温度为0℃~15℃,优选10℃;升温终点温度为25℃~45℃,优选35℃;升温速率为0.5~1.5℃/min;搅拌转速为600rpm-1000rpm。
(6)离心收集、洗涤干燥:将(5)所得液体离心,收集微球,经正己烷洗涤、真空干燥后即得载药cd-mof的微球。其中,除使用正己烷洗涤微球外,还可用石油醚、氯仿洗涤。
本发明的技术特点是:提供了一种由药物辅料γ-环糊精和丙烯酸树脂组成的金属有机骨架复合微球,其安全性高、生物相容性好(附图5)。采用cd-mof作为药物储库,将药物以分子形式均匀填充于cd-mof晶体中,再分散于含高分子材料的内相,再加入连续相外相制备s/o/o型乳剂,蒸发除去内相溶剂,分离干燥得到微球。与w/o/o、w/o/w等常规方法相比,这种方法避免了cd-mof溶解于水、多孔骨架结构瓦解,在保持cd-mof多孔骨架结构完整性方面具有独特的优势。此外,与药物微球、药物-γ-环糊精包合物微球相比,本发明中cd-mof作为药物的二级储库,可以实现药物的缓慢释放(附图3和附图4)。本发明制备工艺简单可控、无需昂贵的设备,得到的微球具有表面圆整、粒径均匀可控(50~300μm,附图1和附图2)的特点。还可通过控制丙烯酸树脂的类型,控制药物的胃溶和肠溶释放行为。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
图1显示了实施例1制备的布洛芬cd-mof微球的sem图。
图2显示了实施例2制备的兰索拉唑cd-mof微球的sem图。
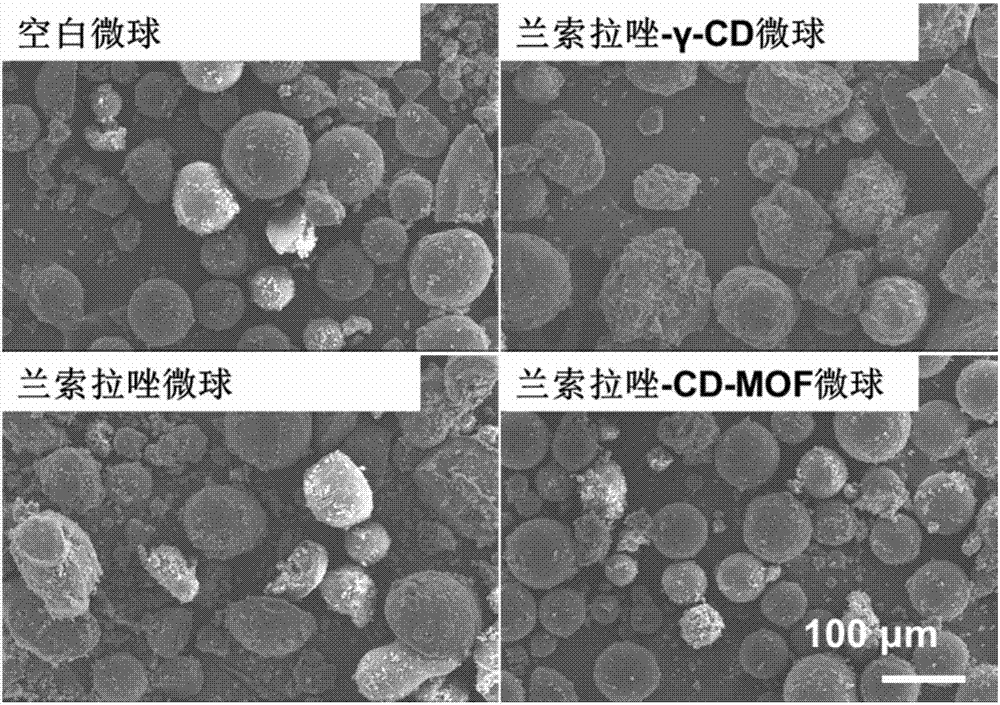
图3显示了实施例1制备的布洛芬cd-mof微球的释药曲线(其中,■:布洛芬-γ-环糊精微球;▲:布洛芬微球;●:布洛芬-cd-mof微球)。
图4显示了实施例2制备的兰索拉唑cd-mof微球的释药曲线(其中,■:兰索拉唑-γ-环糊精微球;▲:兰索拉唑微球;●:兰索拉唑-cd-mof微球)。
图5显示了实施例1制备的布洛芬cd-mof微球的细胞毒性结果。
图6显示了实施例3微球中提取得到的布洛芬cd-mof及兰索拉唑cd-mof的sem图。
图7为实施例4中溶剂挥发法制备的cd-mofi光学显微图。
图8为实施例5中溶剂挥发法所得cd-mofii的光学显微图。
图9为实施例5中溶剂挥发法所得cd-mofii的扫描电镜图。
图10为实施例6中溶剂挥发法所得cd-mofnano的扫描电镜图。
图11为实施例4中溶剂挥发法所得cd-mofi的x-射线粉末衍射图。
图12为实施例5中溶剂挥发法所得cd-mofii的x-射线粉末衍射图。
图13为实施例6中溶剂挥发法所得cd-mofnano的x-射线粉末衍射图。
图14为实施例5中溶剂挥发法所得cd-mofii的粒径分布图。
图15为实施例7中溶剂热法所得cd-mofii的光学显微图。
图16为实施例8中溶剂热法所得cd-mofii的光学显微图。
图17为实施例9中溶剂热法所得cd-mofii的光学显微图。
图18为实施例10中溶剂热法所得cd-mofii的光学显微图。
图19为实施例7中溶剂热法所得cd-mofii的扫描电镜图。
图20为实施例11中溶剂热法所得cd-mofnano的扫描电镜图。
图21为实施例7中溶剂热法所得cd-mofii的x-射线粉末衍射图。
图22为实施例11中溶剂热法所得cd-mofnano的x-射线粉末衍射图。
图23为实施例7中溶剂热法所得cd-mofii的粒径分布图。
图24为实施例8中溶剂热法所得cd-mofii的粒径分布图。
图25为实施例9中溶剂热法所得cd-mofii的粒径分布图。
图26为实施例10中溶剂热法所得cd-mofii的粒径分布图。
图27为实施例12中微波法所得cd-mofii的光学显微图。
图28为实施例12中微波法所得cd-mofii的扫描电镜图。
图29为实施例13中微波法所得cd-mofnano的扫描电镜图。
图30为实施例12中微波法所得cd-mofii的x-射线粉末衍射图。
图31为实施例13中微波法所得cd-mofnano的x-射线粉末衍射图。
图32为实施例14中超声波法所得cd-mofii的光学显微图。
图33为实施例14中超声波法所得cd-mofii的扫描电镜图。
图34为实施例15中超声波法所得cd-mofnano的扫描电镜图。
图35为实施例14中超声波法所得cd-mofii的x-射线粉末衍射图。
图36为实施例15中超声波法所得cd-mofnano的x-射线粉末衍射图。
具体实施方式
本发明人经过广泛而深入地研究,首次意外地发现一种环糊精-金属有机骨架材料(cd-mof)复合微球及其制备方法。本发明的复合微球含有具有立方晶体结构的载药环糊精-金属有机骨架组分(a)和包裹组分(a)的组分(b),能够避免cd-mof作为药物储库的遇水崩解问题,保护cd-mof多孔骨架结构完整的同时,防止药物的突释、实现药物的缓慢释放。在此基础上,完成本发明。
复合微球
如本文所用,术语“环糊精-金属有机骨架材料复合微球”、“cd-mof复合微球”、“cd-mof/paa复合微球”、“复合微球”可互换使用,是指本发明第一方面所述的复合微球。
具体地,本发明提供了由cd-mof和药物辅料聚丙烯酸树脂(paa)组成的复合微球,其中微球基质(聚丙烯酸树脂)为药物的一级储库,cd-mof为药物的二级储库。在保证微球制备过程中cd-mof多孔骨架结构完整的同时,具有生物安全性好,缓慢释放药物的特点。目前尚未有报道采用类似的技术和策略对载药后的cd-mof的结构进行保护并实现药物的缓慢释放。
本发明所述的载药mof复合微球不同于已有的mof微球报道,manju等制备的聚硅氧烷mof5微球是以聚二甲基硅氧烷(pdms)球形结构为核,以mof为壳结构。feike等报道的fe3o4磁性微球也是以fe3o4为核,以mof为壳。而以mof为核,pb等纳米颗粒为壳,合成的结构为杂合物,亦不同于本发明所述的载药mof复合微球中mof单独成体系作为药物的二级储库。
制备方法
本发明还提供了cd-mof/paa复合微球的制备方法,具体而言,采用可药用的γ-cd作为有机连接体,k+作为无机金属中心,在制备形态规则的载药cd-mof立方晶体的基础上,将载药cd-mof分散于含paa有机溶液的内油相;将内油相与外油相混合形成油包油包固体(s/o/o)乳液;搅拌蒸发除去内油相溶剂,洗涤除去外油相溶剂,干燥、收集微球(50-300μm)。
金属有机骨架材料
金属-有机骨架(metal-organicframeworks,mofs)是一种由金属(金属离子、金属离子簇或金属链)和有机桥连配体在较温和的条件下以配位键方式通过自组装形成的无机-有机杂化材料。由于mofs超高的孔隙率和巨大的比表面积,且无机和有机等多种不同成分的组合使得其结构及组成多样,为mofs在气体储存、吸附和分离、电和磁、催化、药物输送等领域的应用提供了新的研究方向。对于药物输送,mofs由于具有较大的孔径和比表面积大、结构和组成多样、载药量高且能实现缓释等独特优势。近年来,mofs已经用于多种药物的输送,如抗肿瘤药物(白消安、阿霉素、顺铂前药、拓扑替康、5-氟尿嘧啶)、抗病毒药物(单/三磷酸-叠氮胸苷、西多福韦)、抗心律失常药物(盐酸普鲁卡因胺)以及抗炎药(布洛芬、尼美舒利)等。作为药物输送载体,某些金属如铬、镉、钴、镍、钆等毒性较大,不适合用来合成作为药用载体的mofs,而应该选择毒性较低的钾、钙、铁、锌、镁等生物体中存在的金属元素,有机桥连配体也应选择毒性较小可药用材料或生物体内存在的内源性化合物,如氨基酸或核苷碱基等。
环糊精
环糊精是由直链淀粉经葡萄糖基转移酶作用下生成的一系列环状低聚糖的总称,通常含有6~12个d-吡喃葡萄糖单元。其中研究得较多并且具有重要实际意义的是含有6、7、8个葡萄糖单元的分子,分别称为α、β-和γ-环糊精。环糊精是迄今所发现的类似于酶的理想宿主分子,并且其本身就有酶模型的特性。
环糊精-金属有机骨架材料
如本文所用,术语“基于环糊精的金属有机骨架材料”、“环糊精-金属有机骨架材料”、“环糊精-金属有机骨架化合物”可互换使用,是利用环糊精在水溶液中能与第一、二主族金属离子以一种有机配位的方式形成一种新的晶体,这种晶体具有多孔、表面积大、储存气体等特点。这种绿色、多孔材料能够吸附一些结构不稳定的药物,其巨大的空腔能够对药物起到保护作用,这使得其用于商业发展成为可能,尤其是由于环糊精-金属有机骨架为可食用衍生物,适于人类食用。将环糊精作为有机配体,金属离子作为无机金属中心,可形成新的、安全性较高、可药用的环糊精-金属有机骨架,即cd-mofs(cd-mof)。
与现有mof类似,cd-mof也存在生物体内稳定性的问题,即遇到体液结构容易瓦解,在到达靶部位之前无法确保多孔晶体结构的稳定。目前虽有mof复合材料报道,如膜、纤维和微球等,以改善mof的机械、热、化学稳定性和成形性。但所用材料的生物相容性较差,如聚甲基丙烯酸甲酯、聚酰亚胺、聚乙酸乙烯酯、聚砜、聚二甲硅氧烷、聚乙烯吡咯烷酮和苯乙烯等。同时cd-mof存在遇水容易崩溃、结构瓦解的问题,用于药物载体易造成药物的突释。
如本文所用,术语“cd-mofi”是指第一阶段cd-mof晶体,指将γ-cd与koh混合,通过甲醇蒸汽蒸发,经过一定时间,直接析出所得到的晶体;本发明方法制得的第一阶段cd-mof晶体的尺寸约为40-500μm。
如本文所用,术语“cd-mofii”是指第二阶段cd-mof晶体,是指将γ-cd与koh混合,通过甲醇蒸汽蒸发,在还未产生或只产生少量第一阶段晶体时,将上清液取出,加入尺寸调节剂,然后再析出所得到的晶体;本发明方法制得的第二阶段cd-mof晶体的尺寸约为1-10μm。
如本文所用,术语“cd-mofnano”是指纳米尺寸的cd-mof晶体,指将γ-cd与koh混合,通过甲醇蒸汽蒸发,在还未产生或只产生少量第一阶段晶体时,将上清液取出,按照上清液体积加入大量甲醇,再加入尺寸调节剂,然后再析出所得到的晶体;本发明方法制得的cd-mofnano的尺寸约为200-500nm。
如本文所用,术语“碱性环糊精-金属有机骨架材料”是以碱金属和环糊精为原料制备的环糊精-金属有机骨架材料,呈碱性,将其溶解于水中制成10mg/ml的水溶液时,其ph约为11-13。
如本文所用,术语“中性环糊精-金属有机骨架材料”、“经酸化的环糊精-金属有机骨架材料”可互换使用,是将碱性环糊精-金属有机骨架材料进行酸化处理得到的近中性的环糊精-金属有机骨架材料,将其溶解于水中制成10mg/ml的水溶液时,其ph约为5-8。一种优选的酸化处理的方法如下:称取一定量的环糊精-金属有机骨架置于乙醇中,加入一定量的冰醋酸,25℃下,振摇孵育一定时间后,所得固体用乙醇洗涤,即得近中性的环糊精-金属有机骨架。
预加有机溶剂
在本发明的环糊精-金属有机骨架材料制备方法中,在进行反应之前,在反应体系中预加一定量的有机溶剂,使所得cd-mofs晶体能够更快析出,同时又不能加入过量有机溶剂,否则容易使已溶解的环糊精直接析出来,最终所得的cd-mofs就会掺杂有一部分环糊精。
预处理
在本发明的环糊精-金属有机骨架材料制备方法中,为了达到快速反应的目的,对含有金属盐和环糊精,并预加有有机溶剂的混合液进行了预处理,所述预处理包括溶剂热处理、微波处理、和/或超声波处理。
溶剂热法是水热法的优化,微波处理能够使得物质分子发生高频振动,不仅产生热量,使温度快速升高,同时增强了物质传递,降低了反应活化能,促进氢氧化钾与γ-环糊精发生反应,使得加热均匀,缩短热传导的时间,并且不会出现传统方法加热不均的弊端。超声波处理主要是利用超声波空化作用使反应溶液出现膨胀、压缩、溃陷等一系列动作,所产生的化学效应和机械效应能够改善反应条件,加快反应速度。微波及超声波能的产生和关闭是瞬时的,没有热惯性,安全可靠,便于自动化控制。
本发明的主要优点包括:
(a)本发明的复合微球安全性高、生物相容性好。
(b)本发明的复合微球可以实现药物的缓慢释放。
(c)本发明的制备方法可以保持cd-mof的结构完整性。
(d)本发明的制备方法工艺简单可控、成本低、效果好。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。
通用材料
下述实施例中所用“聚丙烯酸树脂”是指丙烯酸乙酯、甲基丙烯酸甲酯和甲基丙烯酸氯化三甲胺基乙酯(1:2:0.1)共聚物(赢创工业基团)。
液体石蜡为分析纯,购自国药集团化学试剂有限公司。
实施例1
处方如下表所示:
制备工艺如下:
称取50mg载布洛芬cd-mof分散于3ml丙酮中,超声使之分散均匀,称取450mg聚丙烯酸树脂(mof:聚丙烯酸树脂为1:9,w/w)溶解于其中,再加入120mg硬脂酸铝(分散剂)超声5min分散均匀,为分散相。将含硬脂酸铝的分散相,加入至液体石蜡中(已冰浴至10℃),将上述混悬液于10℃冰水浴条件下,磁力搅拌(500rpm,30s),再用分散机分散(10000rpm,5min),得s/o/o型乳液。将上述乳液置于磁力搅拌器上,500rpm搅拌条件下,由10℃缓慢升温至35℃(磁力搅拌加热的设置为65,升温速度为10℃·min-1),再继续搅拌3h(500rpm,35℃),去除大部分丙酮。将上述液体转移至50ml离心管中,离心(2000rpm,5min),弃去液体石蜡(上层);下层固体用正己烷洗涤2次,每次用30ml正己烷,离心(2000rpm,5min)。洗涤后,放入通风橱中干燥过夜。将所得干燥微球粉末称重,过80目筛。作为比较,制备了空白微球,制备方法同载药cd-mof微球,但不同的是制备过程中不加入载布洛芬cd-mof;还制备了布洛芬微球、布洛芬-γ-cd微球,制备方法同布洛芬cd-mof微球,只是将其中的载布洛芬cd-mof分别改为布洛芬、布洛芬-γ-cd包合物。
制备的布洛芬cd-mof微球的sem图结果见图1,释药曲线见图3(布洛芬cd-mof微球缓释效果显著),细胞毒性见图5(布洛芬cd-mof微球对细胞毒性较低)。
实施例2
处方如下表所示:
制备工艺如下:
称取50mg载兰索拉唑cd-mof分散于3ml丙酮中,超声使之分散均匀,称取450mg聚丙烯酸树脂(mof:聚丙烯酸树脂为1:9,w/w)溶解于其中,再加入120mg硬脂酸铝(分散剂)超声5min分散均匀,为分散相。将含硬脂酸铝的分散相,加入至液体石蜡中(已冰浴至10℃),将上述混悬液于10℃冰水浴条件下,磁力搅拌(500rpm,30s),再用分散机分散(10000rpm,5min),得s/o/o型乳液。将上述乳液置于磁力搅拌器上,500rpm搅拌条件下,由10℃缓慢升温至35℃(磁力搅拌加热的设置为65,升温速度为10℃·min-1),再继续搅拌3h(500rpm,35℃),去除大部分丙酮。将上述液体转移至50ml离心管中,离心(2000rpm,5min),弃去液体石蜡(上层);下层固体用正己烷洗涤2次,每次用30ml正己烷,离心(2000rpm,5min)。洗涤后,放入通风橱中干燥过夜。将所得干燥微球粉末称重,过80目筛。作为比较,制备了空白微球,制备方法同载兰索拉唑cd-mof微球,但不同的是制备过程中不加入载兰索拉唑cd-mof;还制备了兰索拉唑微球、兰索拉唑-γ-cd微球,制备方法同兰索拉唑cd-mof微球,只是将其中的载兰索拉唑cd-mof分别改为兰索拉唑、兰索拉唑-γ-cd包合物。制备的兰索拉唑cd-mof微球的sem结果见图2,释药曲线见图4(兰索拉唑cd-mof微球缓释效果显著)。
实施例3
微球中cd-mof立方晶体结构表征
称取100mg复合微球置于10ml离心管中,加入8ml乙醇溶解微球中的paa聚合物,离心(12000rpm,5min),所得沉淀物主要含有cd-mof和硬脂酸铝。由于cd-mof和硬脂酸铝的密度不同,将混合物分散于8ml二氯甲烷中,离心(12000rpm,5min),下层沉淀物主要是cd-mof晶体,硬脂酸铝由于密度小,漂浮于上层。将上层固体用吸管吸出,收集下层固体,扫描电子显微镜下观察样品的形态(图6),结果表明cd-mof以立方晶体形式存在于微球中,s/o/o的无水微球制备技术完全避免了cd-mof与水的接触,确保了cd-mof结构的完整性。
实施例4
溶剂热挥发法制备第一阶段cd-mof晶体
将163.0mgγ-cd和56.0mgkoh混合物(γ-cd和koh摩尔比为0.125)溶解于5ml水中,超声10分钟使其充分溶解,0.45μm滤膜过滤。然后预加0.5ml甲醇至γ-cd与koh混合溶液内,在密闭容器中50℃条件下加热甲醇(密闭容器整体加热),使甲醇蒸汽蒸发至γ-cd与koh混合体系内。反应6h时即开始有少量晶体产生,反应24h后,得到大量无色透明晶体,弃去上清液,3000rpm离心5min,用乙醇(10ml×3)洗涤,将所得晶体50℃真空干燥12h,即得可长期保存的第一阶段cd-mof晶体(cd-mofi),尺寸为40-500μm,如图7和图11,产率为76.3%。
实施例5
溶剂热挥发法制备第二阶段微米级cd-mof晶体
称取163.0mgγ-cd和56.0mgkoh混合物(γ-cd和koh摩尔比为0.125)溶解于5ml水中,超声10分钟使其充分溶解,0.45μm滤膜过滤。然后预加0.5ml甲醇至γ-cd与koh混合溶液内,在密闭容器中50℃条件下加热甲醇(密闭容器整体加热),使甲醇蒸汽蒸发至γ-cd与koh混合体系内。反应6小时后,取出上清液,按8mg/ml上清液的比例加入peg20000,静置半小时后,3000rpm离心5min,分别用乙醇(10ml×2)、二氯甲烷(10ml×2)洗涤,将所得晶体50℃真空干燥12h,即得可长期保存的第二阶段微米级cd-mof晶体(cd-mofii),尺寸为1-10μm,如图8、图9、图12和图14,产率为85.1%。
实施例6
溶剂热挥发法制备第二阶段纳米级cd-mof晶体
将163.0mgγ-cd和56.0mgkoh混合物(γ-cd和koh摩尔比为0.125)溶解于5ml水中,超声10分钟使其充分溶解,0.45μm滤膜过滤。然后预加0.5ml甲醇至γ-cd与koh混合溶液内,在密闭容器中50℃条件下加热甲醇(密闭容器整体加热),使甲醇蒸汽蒸发至γ-cd与koh混合体系内。反应6小时后,取出上清液,加入等体积甲醇,再按8mg/ml上清液的比例加入peg20000,静置半小时后,3000rpm离心5min,分别用乙醇(10ml×2)、二氯甲烷(10ml×2)洗涤,将所得晶体50℃真空干燥1h,即得第二阶段纳米级cd-mof晶体(cd-mofnano),尺寸为200-500nm,如图10和图13,产率为90.3%。
实施例7
溶剂热法制备第二阶段微米级cd-mof晶体
使用溶剂热的方式,直接对γ-环糊精与koh水溶液与一部分有机溶剂混合体系进行加热。称取163.0mgγ-cd和56.0mgkoh混合物(γ-cd和koh摩尔比为0.125)溶解于5ml水中,预加3ml甲醇至混合溶液内,50℃水浴加热20min后,取出溶液,再加入64mgpeg20000,静置半小时后,3000rpm离心5min,分别用乙醇(10ml×2)、二氯甲烷(10ml×2)洗涤,将所得晶体50℃真空干燥12h,即得第二阶段微米级cd-mof晶体(cd-mofii),尺寸为1-10μm,如图15,图19,图21和图23,产率为87.0%。
实施例8
溶剂热法制备第二阶段微米级cd-mof晶体
使用溶剂热的方式,直接对γ-环糊精与koh水溶液与一部分有机溶剂混合体系进行加热。称取163.0mgγ-cd和56.0mgkoh混合物(γ-cd和koh摩尔比为0.125)溶解于5ml水中,预加3ml甲醇至混合溶液内,50℃水浴加热20min后,取出溶液,再加入16mgpeg20000,静置半小时后,3000rpm离心5min,分别用乙醇(10ml×2)、二氯甲烷(10ml×2)洗涤,将所得晶体50℃真空干燥12h,即得第二阶段微米级cd-mof晶体(cd-mofii),尺寸为1-10μm,如图16和图24,产率为58.3%。
实施例9
溶剂热法制备第二阶段微米级cd-mof晶体
使用溶剂热的方式,直接对γ-环糊精与koh水溶液与一部分有机溶剂混合体系进行加热。称取163.0mgγ-cd和56.0mgkoh混合物(γ-cd和koh摩尔比为0.125)溶解于5ml水中,预加3ml甲醇至混合溶液内,50℃水浴加热20min后,取出溶液,再加入64mgpeg2000,静置半小时后,3000rpm离心5min,分别用乙醇(10ml×2)、二氯甲烷(10ml×2)洗涤,将所得晶体50℃真空干燥12h,即得第二阶段微米级cd-mof晶体(cd-mofii),尺寸为1-10μm,如图17和25,产率为83.0%。
实施例10
溶剂热法制备第二阶段微米级cd-mof晶体
使用溶剂热的方式,直接对γ-环糊精与koh水溶液与一部分有机溶剂混合体系进行加热。称取163.0mgγ-cd和56.0mgkoh混合物(γ-cd和koh摩尔比为0.125)溶解于5ml水中,预加3ml甲醇至混合溶液内,50℃水浴加热20min后,取出溶液,再加入64mgpeg10000,静置半小时后,3000rpm离心5min,分别用乙醇(10ml×2)、二氯甲烷(10ml×2)洗涤,将所得晶体50℃真空干燥12h,即得第二阶段微米级cd-mof晶体(cd-mofii),尺寸为1-10μm,如图18和图26,产率为87.4%。
实施例11
溶剂热法制备第二阶段纳米级cd-mof晶体
使用溶剂热的方式,直接对γ-环糊精与koh水溶液与一部分有机溶剂混合体系进行加热。称取163.0mgγ-cd和56.0mgkoh混合物(γ-cd和koh摩尔比为0.125)溶解于5ml水中,预加3ml甲醇至混合溶液内,50℃水浴加热20min后,取出溶液,加入等体积的甲醇,再加入64mgpeg20000,静置半小时后,3000rpm离心5min,分别用乙醇(10ml×2)、二氯甲烷(10ml×2)洗涤,将所得晶体50℃真空干燥12h,即得第二阶段纳米级cd-mof晶体(cd-mofnano),尺寸为200-500nm,如图20和图22,产率为90.5%。
实施例12
微波法制备第二阶段微米级cd-mof晶体
使用微波的方式,对γ-环糊精与koh水溶液与一部分有机溶剂混合体系进行微波加热。称取163.0mgγ-cd和56.0mgkoh混合物(γ-cd和koh摩尔比为0.125)溶解于5ml水中,预加3ml甲醇至混合溶液内,2450mhz的微波反应器,功率设置25w,温度设置50℃,反应20min后,取出溶液,再加入64mgpeg20000,静置半小时后,3000rpm离心5min,分别用乙醇(10ml×2)、二氯甲烷(10ml×2)洗涤,将所得晶体50℃真空干燥12h,即得第二阶段微米级cd-mof晶体(cd-mofii),尺寸为1-10μm,如图27,图28和图30,产率为82.2%。
实施例13
微波法制备第二阶段纳米级cd-mof晶体
使用微波的方式,对γ-环糊精与koh水溶液与一部分有机溶剂混合体系进行微波加热。称取163.0mgγ-cd和56.0mgkoh混合物(γ-cd和koh摩尔比为0.125)溶解于5ml水中,预加3ml甲醇至混合溶液内,2450mhz的微波反应器,功率设置25w,温度设置50℃,反应20min后,取出溶液,加入等体积的甲醇,再加入64mgpeg20000,静置半小时后,3000rpm离心5min,分别用乙醇(10ml×2)、二氯甲烷(10ml×2)洗涤,将所得晶体50℃真空干燥12h,即得第二阶段纳米级cd-mof晶体(cd-mofnano),尺寸为200-500nm,如图31和图29,产率为90.1%。
实施例14
超声波法制备第二阶段微米级cd-mof晶体
使用超声波的方式,对γ-环糊精与koh水溶液与一部分有机溶剂混合体系进行超声加热。称取163.0mgγ-cd和56.0mgkoh混合物(γ-cd和koh摩尔比为0.125)溶解于5ml水中,预加3ml甲醇至混合溶液内,用40khz的超声波反应器,功率设置40w,温度50℃,反应20min后取出上清液,再加入64mgpeg20000,静置半小时后,3000rpm离心5min,分别用乙醇(10ml×2)、二氯甲烷(10ml×2)洗涤,将所得晶体50℃真空干燥12h,即得第二阶段微米级cd-mof晶体(cd-mofii),尺寸为1-10μm,如图32,33和图35,产率为79.7%。
实施例15
超声波法快速合成纳米级cd-mof晶体
使用超声波的方式,对γ-环糊精与koh水溶液与一部分有机溶剂混合体系进行超声加热。称取163.0mgγ-cd和56.0mgkoh混合物(γ-cd和koh摩尔比为0.125)溶解于5ml水中,预加3ml甲醇至混合溶液内,用40khz的超声波反应器,功率设置40w,温度50℃,反应20min后取出上清液,加入8ml甲醇,再加入64mgpeg20000,静置半小时后,3000rpm离心5min,分别用乙醇(10ml×2)、二氯甲烷(10ml×2)洗涤,将所得晶体50℃真空干燥12h,即得第二阶段纳米级cd-mof晶体(cd-mofnano),尺寸为200-500nm,如图34和图36,产率为85.2%。
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!