一种包载重组抗肿瘤蛋白TmSm的mPEG‑PLGA纳米颗粒及其制备方法和应用与流程
本发明涉及医药技术领域,更具体地讲,涉及重组抗肿瘤蛋白TmSm的单甲氧基聚乙二醇聚乳酸乙醇酸共聚物(mPEG-PLGA)纳米颗粒的制备以及其作为肿瘤治疗药物的用途。
背景技术:
癌症是目前人类死亡率最高的疾病之一,由于癌细胞对放疗和化疗产生耐药性,以及容易转移等特性,使得癌症难以根治。在癌症的发生过程中,常伴有某些有利于癌细胞生长的蛋白过量表达。Survivin基因编码的survivin蛋白,是哺乳动物细胞中凋亡抑制因子(inhibitor of apoptosis protein,IAP)家族最小的成员,在正常细胞中不表达,而在胚胎干细胞和肿瘤细胞中异常表达,并以同源二聚体形式存在。不同于IAP其他家族成员,survivin N端有一杆状病毒IAP重复序列(BIR),由3股反平行β片层和4个α螺旋组成,C末端包含42个氨基酸,形成一个α螺旋。Survivin前体mRNA分子成熟结构包含4个主要功能区域和3个隐含外显子,通过不同的选择性剪接形成6种剪接异构体,编码6种不同的蛋白质,分别为Survivin、Survivin2B、Survivin3B、SurvivinΔEx3、Survivin3α和Survivin2α。
Survivin参与多种细胞进程,它的BIR区域在细胞有丝分裂过程中起重要作用。Survivin是迄今所发现的最强的凋亡抑制因子,它可通过抑制癌细胞中不同效应蛋白酶caspase的活性来干扰细胞凋亡。Survivin可结合X染色体连锁凋亡抑制蛋白(XIAP),形成复合物以抑制XIAP通过泛素蛋白酶体通路降解,并且能抑制Capase-9的活性。更多研究表明,survivin参与细胞自体吞噬,其下调会引起细胞凋亡。且survivin可与多种DNA损伤修复相关因子作用,从而协助核中DNA修复。Survivin蛋白具有细胞周期依赖性,其在G1期表达最低,S期含量上调,在G2/M期达最大值,提示Survivin在细胞分裂中发挥了一定的作用。核定位的Survivin与Aurora B kinase、INCENP和Borealin共同形成CPC(chromosomal passenger complex,染色体通道蛋白复合体),在有丝分裂中,定位于染色体的着丝粒和中心纺锤体上,促进姐妹染色单体的分离并在分裂后期维持微管的稳定性,维持和促进了细胞的增殖。
利用survivin突变体的形式靶向survivin的方式称为survivin负显性突变体治疗。将survivin的第34位磷酸化位点由苏氨酸(Thr)突变为丙氨酸(Ala)后,即survivin(T34A),废除survivin的磷酸化,导致survivin-caspase-9复合物的解离,从而激活caspase活性,导致细胞凋亡。本实验室利用HIV-TAT可以转导蛋白的特性设计了TATm-Survivin(T34A)融合蛋白即TmSm。目前,TmSm注射剂多采用冻干技术以提高药物的稳定性,但是TmSm粉针剂用药剂量较大,需要频繁用药。关键因素是生物活性物质稳定性差,难以大量富集到达肿瘤部位发挥作用。因此,要解决这个问题需引入控、缓释制剂的应用。目前,主要包括脂质体、微球、微囊注射剂、皮下埋植给药剂型、智能型给药系统和纳米技术。可生物降解的合成聚合物材料聚乳酸-羟基乙酸共聚物(poly(lactic-co-glycolic acid),PLGA]是以聚乙醇酸(PGA)和聚乳酸(PLA)为原料合成的,PLA的亲水性较差,降解慢,PGA的亲水性好,降解快,而PLGA可把两种原料的性质加以综合完善,是目前最好的生物可降解聚合物之一,因其制备的纳米颗粒具有良好的性能而备受关注,如:1)生物降解和生物相容性;2)良好的配方和生产方法,适应不同种类的药物,如疏水或亲水性小分子或大分子药物;3)在注射给药的药物传输系统方面得到美国食品及药物管理局(FDA)和欧洲药物管理局(EMA)的批准;4)可保护药物不被降解;5)可持续缓释;6)可进行表面修饰定向传递等。为了控制药物释放,可以使聚合物混合或使用不同的添加剂。例如,结构中包括至少两种不同族群的二聚体和三聚体,既有疏水性又有亲水性,这种结构在控制药物释放中医已赢得越来越多的关注。其中PEG-PLGA显示了很好的特性。聚乙二醇(PEG)的加入,会抑制血清蛋白的吸附,降低细胞识别的程度,改进载药系统的生物相容性,同时有效减少免疫原性和毒性。
PLGA纳米粒的制备方法有:乳化-蒸发法、盐析法、纳米沉淀法、错流过滤法、乳化-溶剂挥发法等。然而当采用某些方法制备纳米粒子时,却遇到一些问题,如使用有毒性的有机溶剂(乳化-蒸发法)、使用与生物不相容的活性化合物稳定剂和盐(盐析法)以及较低的产率和包封率(纳米沉淀法),同时,这些方法也很难降低纳米粒子的大小,无法制备出粒径小于100nm的纳米粒子。通过高效和可再生的方式将预成型的聚合物用乳化-溶剂挥发法成功制备出纳米颗粒。目前,制备PLGA纳米粒最常用的方法是乳化-溶剂挥发法,该方法主要分两步进行,即乳液的制备和溶剂的挥发。常常是根据包载药物的性质制成W/O、O/W、W/O/W、O/W/O型乳状液。W/O/W双乳化溶剂挥发是目前制备水溶性药物、多肽、蛋白质等常用的方法。将水溶性药物分散于含PLGA等载体的有机相中,形成W/O型初乳,再分散外水相中形成W/O/W型乳,最后再除去有机溶剂即可。对O/W乳化方法进行改进的双乳化技术(W/O/W),可用来包裹一些亲水性药物,如多肽类、蛋白质、核酸。首先,配制含有有机相溶剂的常见O/W乳化液体系,继续向体系中加入水使有机溶剂扩散到外相水中,形成纳米颗粒。
技术实现要素:
本发明的第一个目的在于提供一种包载重组抗肿瘤蛋白TmSm的mPEG-PLGA纳米颗粒。
本发明的第二个目的在于提供包载重组抗肿瘤蛋白TmSm的mPEG-PLGA纳米颗粒的制备方法。
本发明的第三个目的在于提供包载重组抗肿瘤蛋白TmSm的mPEG-PLGA纳米颗粒的应用。
为实现以上目的,本发明公开以下技术方案:一种包载重组抗肿瘤蛋白TmSm的mPEG-PLGA纳米颗粒,其特征在于,用下述制备方法制备获得:
(1)将mPEG-PLGA溶于乙酸乙酯中,将TmSm 1-10%PEG水溶液加入到PLGA溶液中,冰浴条件下超声,形成油包水型乳液;
(2)将获得的油包水型乳液加入到PVA水溶液中,将上述混合液引入高压均质机进行2-4个周期,即形成水包油包水型复乳液;
(3)在室温常压下磁力搅拌,挥发有机溶剂乙酸乙酯;
(4)将固化的纳米粒通过低温高速离心收集,超纯水清洗,洗净纳米粒表面的PVA,将离心的上清液进行收集;
(5)将上述清洗的沉淀用蔗糖重悬,先-20℃冷冻半小时,再置于-80℃过夜冷冻,最后置于冷冻干燥机中冷冻干燥24h,去除纳米粒中的水分后,得到纳米粒成品,4℃环境下冷藏。
作为一个优选方案,TmSm与mPEG-PLGA的质量比为1:62.5。
作为一个优选方案,纳米粒粒径为181.0±0.8nm,Zeta电位分别为-26.3±0.4mV,多分散指数为0.122±0.016。
作为一个优选方案,纳米粒包封率为53.61±2.08%,载药量为8.58±0.33μg/mg。
为实现本发明第二个目的,本发明公开以下技术方案:一种包载重组抗肿瘤蛋白TmSm的mPEG-PLGA纳米颗粒的制备方法,其特征在于,所述制备方法包括如下步骤:
(1)将mPEG-PLGA溶于乙酸乙酯中,将TmSm 1-10%PEG水溶液加入到PLGA溶液中,冰浴条件下超声,形成油包水型乳液;
(2)将获得的油包水型乳液加入到PVA水溶液中,将上述混合液引入高压均质机进行2-4个周期,即形成水包油包水型复乳液;
(3)在室温常压下磁力搅拌,挥发有机溶剂乙酸乙酯;
(4)将固化的纳米粒通过低温高速离心收集,超纯水清洗,洗净纳米粒表面的PVA,将离心的上清液进行收集;
(5)将上述清洗的沉淀用蔗糖重悬,先-20℃冷冻半小时,再置于-80℃过夜冷冻,最后置于冷冻干燥机中冷冻干燥24h,去除纳米粒中的水分后,得到纳米粒成品,4℃环境下冷藏。
作为一个优选方案,TmSm与mPEG-PLGA的质量比为1:62.5。
为实现本发明第三个目的,本发明公开以下技术方案:包载重组抗肿瘤蛋白TmSm的mPEG-PLGA纳米颗粒在制备抗肿瘤药物中的应用。
作为一个优选方案,所述肿瘤是指胰腺癌。
作为一个优选方案,当肿瘤是胰腺癌时,有效剂量为3-100mg mg/mLTmSm-PLGA。
本发明制备获得的纳米粒的包封率为53.61±2.08%,载药量为8.58±0.33μg/mg。;该纳米粒粒径为181.0±0.8nm,Zeta电位分别为-26.3±0.4mV,多分散指数为0.122±0.016。
本发明载体材料为mPEG-PLGA。其中,PLGA是可生物降解的合成聚合物材料,由于其无毒、成球性好、化学稳定性高、具有控制释放能力等特点被广泛用作纳米给药系统的载体。在美国PLGA通过FDA认证,被正式作为药用辅料收录进美国药典。mPEG亲水基团可以有效避免内皮网状系统的捕获,体内循环系统中的滞留时间明显延长,能稳定地释放药物。
本发明有机溶剂为乙酸乙酯。作为注射颗粒,利用的乙酸乙酯相对于二氯甲烷,毒性较低。
本发明的优点在于:兼具TmSm的抗肿瘤功效和mPEG-PLGA的生物降解性优良、组织相容性好、降解产物不会在重要器官聚集的特点,促进肿瘤细胞凋亡;同时本发明可以有效提高药物疗效,减少药物刺激,降低毒副作用,延长药物作用时间实现长效缓释,增加药物的稳定性。因此,作为生物大分子药物的靶向递送有着更为广阔的应用前景。本发明的mPEG-PLGA纳米颗粒制备方法简单,适宜大规模连续生产。
附图说明
图1为重组蛋白TmSm纯化SDS-PAGE图。其中,泳道M:Marker;泳道1:诱导前菌液;泳道2:诱导后菌液;泳道3:细胞破碎后上清;泳道4:细胞破碎后沉淀;泳道5:超声洗涤后包涵体;泳道6:一次洗涤后包涵体;泳道7:二次洗涤后包涵体;泳道8:三次洗涤后包涵体;泳道9:溶解包涵体。
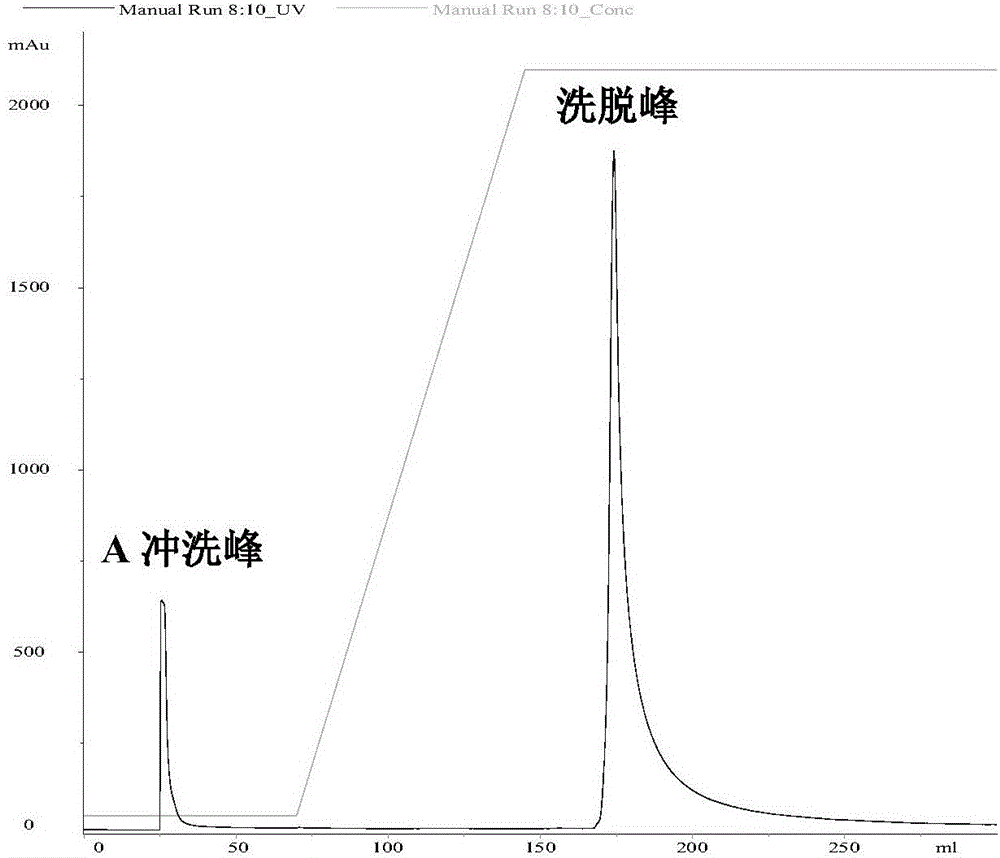
图2为阳离子交换柱SP Sepharose FF柱复性色谱图。
图3为重组蛋白TmSm包涵体柱上复性SDS-PAGE图。其中,泳道M:Marker;泳道1:SP上柱前(溶解后包涵体);泳道2:流穿液;泳道3:A冲洗峰;泳道4:SP下柱后。
图4为RP-HPLC法检测TmSm蛋白纯度色谱图。
图5为复乳-溶剂挥发法结合高压均质制备TmSm-PLGA-NPs流程图。
图6为载TmSm的mPEG-PLGA纳米颗粒的SDS-PAGE图。其中,泳道M:Marker;泳道1:空白PLGA纳米粒;泳道2:33.3mg/mL TmSm-PLGA纳米粒;泳道3:66.7mg/mL TmSm-PLGA纳米粒;泳道4:0.150mg/mL TmSm。
图7为空白PLGA-NPs和TmSm-PLGA-NPs冻干图。其中,A:空白PLGA-NPs冻干前;B:空白PLGA-NPs冻干后;C:TmSm-PLGA-NPs冻干前;D:TmSm-PLGA-NPs冻干后,每一组第一个瓶子为超纯水,后面四个瓶子为1%蔗糖。
图8为空白PLGA-NPs和TmSm-PLGA-NPs透射电镜图。其中,A:空白PLGA-NPs透射电镜图;B:TmSm-PLGA-NPs透射电镜图。
图9为空白PLGA-NPs和TmSm-PLGA-NPs粒径分布和Zeta电位图。其中,A:空白PLGA-NPs粒径分布图;B:空白PLGA-NPs Zeta电位图;C:TmSm-PLGA-NPs粒径分布图;D:TmSm-PLGA-NPs Zeta电位图。
图10为TmSm-PLGA-NPs的载药量和包封率的测定结果。
图11为37℃时PLGA载药体系中TmSm在不同pH条件下(7.4,6.5,5.0)的释放情况。
图12为Blank PLGA和TmSm-PLGA纳米粒对SW1990的细胞存活。
具体实施方式
下面结合具体实施例,进一步阐述本发明。下述实施例中所使用的实验方法如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。
实施例1.重组蛋白TmSm的表达、纯化及指标检测
试剂与试剂盒:异丙基-B-D-硫代吡喃半乳糖苷(Isopropyl B-D-I-Thiogalactopyranoside,IPTG)、氨苄青霉素购自Sigma-Aldrich公司(美国),SP Sepharose FF阳离子交换柱购置于GE公司(美国),蛋白分子量标准品Premixed Protein Marker(Low),蛋白上样缓冲液4×Protein SDS PAGE Loading Buffer,购置于宝生物工程有限公司(大连)。其他的生化试剂属于国产常规的分析纯试剂。
菌株和质粒:
E.coli BL21(DE3)(Invirogen,USA)作为质粒表达菌株。重组质粒pRSET-B-TATm-Survivin(T34A)为本实验室所构建。
试剂配制:
氨苄青霉素(Amp)母液(50mg/mL)配制:用电子天平准确称量氨苄青霉素粉剂0.5g,加入10mL超纯水定容,充分溶解,将溶液用0.22μm无菌滤膜过滤除菌后,每1mL分装1支,-20℃保存,备用。培养大肠杆菌时,每1mLLB培养基中加入1μL氨苄青霉素溶液(1:1000)。终浓度为50μg/mL。
异丙基-β-D-硫代吡喃半乳糖苷(IPTG)母液(1M)配制:用电子天平准确称量2.38g IPTG溶解于10mL超纯水中,配制成238mg/mL的水溶液,用0.22μm无菌滤膜过滤除菌后,分装1mL/支,-20℃保存,备用。诱导时每1mL LB培养基中加入1μL IPTG母液。终浓度为1mmol。
LB培养基的配制:根据《分子克隆实验指导》配制每升培养基中加入950mL去离子水,10g蛋白胨,10g NaCl,5g酵母膏,摇动容器直至溶质溶解。用5mol/L NaOH调节pH至7.0,用去离子水定容至1L。随后121℃高压灭菌20min。
5×蛋白电泳缓冲液(Tris-甘氨酸):称取Tris碱15.1g,甘氨酸94g,SDS 5g,加双蒸水定容至1L。
15%蛋白电泳分离胶:量取ddH2O 2.3mL,30%丙烯酰胺5.0mL,Tris-HCI(pH 6.8)2.5mL,10%SDS 100μL,10%APS 100μL,TEMED 100μL,混合均匀。
5%蛋白电泳浓缩胶:量取ddH2O 3.15mL,30%丙烯酰胺0.75mL,Tris-HCI(pH 6.8)0.57mL,10%SDS 45μL,10%APS 45μL,TEMED 4.5μL,混合均匀。
考马斯亮蓝染色液:称取考马斯亮蓝(R-250)2g,量取乙醇200mL,冰醋酸100mL,去离子水750mL,滤纸过滤,室温保存。
PBS缓冲液:8g NaCl,0.2g KCl,0.24g KH2PO4,1.44g Na2HPO4,定容至1L,用NaOH溶液或浓HCl溶液调pH至7.4,倒入HT瓶中,盖上盖子(但不拧紧),在121℃下灭菌20min,拧紧封口,备用。
0.2M pH6.5磷酸盐缓冲母液(PB):根据《分子克隆第二版》配制,先配制0.2mol/L Na2HPO4 1L,称取28.392g Na2HPO4(Mw=141.96),用超纯水定容至1L。配制0.2mol/L NaH2PO4 1L,称取27.598g NaH2PO4.H2O(Mw=137.99),用超纯水定容至1L。Na2HPO4和NaH2PO4按31.5mL:68.5mL的比例混合。
20mmol/L pH6.5磷酸盐缓冲液(PB):取100mL 0.2mol/L pH6.5磷酸盐缓冲母液(PB),用超纯水定容至900mL,通过pH仪用NaOH溶液或H3PO4溶液调节pH至6.5(初始为pH 6.47),倒入HT瓶中,盖上盖子(但不拧紧),在121℃下灭菌20min,拧紧封口,备用。
超声裂解液:20mmol/L PB,50mmol/L NaCl。量取500mL 20mmol/L PB,称取1.461g NaCl溶解,通过pH仪用NaOH溶液或H3PO4溶液调节pH至6.5。
包涵体洗涤液:20mmol/L PB,50mmol/L NaCl,2mol/L尿素,pH 6.5。量取500mL 20mmol/L PB,称取1.461g NaCl,60.06g尿素溶解。通过pH仪用NaOH溶液或H3PO4溶液调节pH至6.5。
包涵体洗涤液:20mmol/L甘氨酸-氢氧化钠缓冲液,0.5%Triton X-100,0.5mol/L尿素,1mol/L氯化钠,2.5mmol/L EDTA,pH10.0。
包涵体溶解液:20mmol/L PB,50mmol/L NaCl,8mol/L尿素,pH 6.5。量取500mL 20mmol/L PB,称取1.461g NaCl,240.24g尿素溶解。通过pH仪用NaOH溶液或H3PO4溶液调节pH至6.5。
SP Sepharose FF阳离子交换层析缓冲液:
缓冲液A:20mmol/L pH6.50磷酸盐,8mol/L尿素,pH6.50。100mL配制方法:取10mL的0.2mol/L pH6.50的磷酸盐缓冲液,加入双蒸水40mL,加入48g的尿素充分溶解后,定容至100mL。而后加入微量的磷酸调节pH至6.50,过微滤膜后使用。
缓冲液B:20mmol/L pH6.50磷酸盐,2mol/L尿素,pH6.50。100mL配制方法:取10mL 0.2mol/L pH6.50的磷酸盐缓冲液,加入双蒸水30mL,再加入12g的尿素溶解后定容至100mL,并用少量的浓NaOH溶液调节pH至6.50,过微滤膜后使用。
缓冲液C:20mmol/L pH6.50磷酸盐,2mol/L尿素,1mol/L NaCl缓冲液,pH6.50。100mL配制方法:取10mL的0.2mol/L pH6.50的磷酸盐缓冲液,加入双蒸水30mL,而后加入5.844g NaCl溶解后,再加入12g尿素溶解,定容至100mL后用少量的浓NaOH溶液调节pH至6.50,过膜后使用。
透析液A:20mmol/L PB,2mol/L尿素,1mol/L NaCl,10%甘油,1mmol/L精氨酸,pH 6.5。
透析液B:20mmol/L PB,1mol/L尿素,0.5mol/L NaCl,10%甘油,1mmol/L精氨酸,pH 6.5。
透析液C:20mmol/L PB,0.25mol/L尿素,0.5mol/L NaCl,10%甘油,1mmol/L精氨酸,pH 6.5。
Bradford法测蛋白浓度相关溶液:
BSA蛋白标准液(mg/mL):将10mg BSA溶解于10mL无菌水中,分装成1mL每管。
Bradford工作液:准确称取100mg考马斯亮蓝G-250,加入50mL 95%的乙醇,100mL 85%的磷酸,用去离子水定容至1L,棕色瓶避光保存。常温可放置一个月。
RP-HPLC法测蛋白纯度:
缓冲液A:0.08%TFA乙腈:水=5:95,过微滤膜。
缓冲液B:0.1%TFA乙腈:水=95:5。
缓冲液C:纯乙腈。三种缓冲液进行超声,除去气泡。其中TFA为三氟乙酸。
重组蛋白TATm-Survivin(T34A)在大肠杆菌中的表达:从-20℃中取出保藏的菌BL21/pRSET-B-TATm-Survivin(T34A),取50μL菌液置于5mL的含10μL50mg/mL Amp(100μg/mL)LB培养基的LB培养基中,37℃,200rmp,过夜培养;取过夜培养的菌液1mL于100mL含200μL 50mg/mL Amp(100μg/mL)的LB培养基摇瓶中,37℃,200rmp培养3.5h左右,(取1mL菌液用于SDS-PAGE)至OD600=0.6左右,加IPTG(75μL)(终浓度为0.75mmol/mL)后转入30℃,200rmp培养4h;4h后,收集菌液(1mL),用SDS-PAGE电泳鉴定产物表达情况,证明目的蛋白成功表达,结果如图1所示。
菌体破碎及包涵体洗涤:将培养液4℃,8000rmp离心10min,获得湿菌体;湿菌体用10mL细胞破碎液重悬,4℃,8000rmp离心10min;将湿菌体用细胞破碎液重悬,倒入高压均质机中,1000mpa压力下破碎菌体(破碎3次);将破碎的菌体4℃,10000rmp离心15min,弃去上清,收集沉淀,得到的沉淀为粗包涵体(在上清液和沉淀中分别取样,用于SDS-PAGE);将粗包涵体重悬于10mL包涵体洗涤液中洗涤,冰浴下400rmp搅拌10min。洗涤后于12000rpm,4℃下离心10min,弃去上清,收集沉淀。重复此步3次,收集沉淀为精制包涵体;将精制包涵体溶解于10mL包涵体溶解液中,冰浴下400rmp搅拌4h,于12000rpm,4℃下离心20min,去掉少量不溶物,上清即为溶解的包涵体,于4℃保存。
SP Sepharose FF柱上复性:将5mL SP Sepharose FF预装柱,以阳离子上柱缓冲液A冲洗5个柱体积,速率3mL/min;将0.22μm膜过滤的TmSm包涵体溶液8mL上柱,用缓冲液A继续冲洗直至吸光值回到基线,去除未结合的杂蛋白,速率3mL/min;以缓冲液A和缓冲液B组成的线性梯度缓冲液(0%-100%B)冲洗15个柱体积,速率2mL/min;用缓冲液B继续冲洗SP柱直至吸光值回到基线;而后则更换阳离子柱洗脱液C进行洗脱,速率2mL/min,收集洗脱峰,SDS-PAGE检测纯度。
将透析袋按照通用流程预处理,将收集的蛋白放入透析袋中。透析袋按照1:10的体积放入透析液A中,透析6h。随后取出,放入透析液B中,透析6h。随后取出,放入透析液C中,透析6h。以上都在冰浴或者4℃下进行。将透析的溶液于12000rpm,4℃下离心20min,随后取上清用3KDa的超滤管4℃,5000rmp离心超滤,倒去滤液重复数次浓缩蛋白,随后-20℃保存蛋白。将浓缩的TmSm蛋白利用Bradford法测蛋白浓度。
SDS-PAGE蛋白电泳:
(1)将玻璃板用蒸馏水洗净晾干,准备2个干净的50mL烧杯。
(2)把玻璃板在灌胶支架上固定好。
(3)按比例配好15%分离胶,用移液枪快速加入,大约5cm左右,之后加少许20%乙醇,静置30min。凝胶配制过程要迅速,催化剂TEMED要在注胶前再加入,否则凝结无法注胶。注胶过程最好一次性完成,避免产生气泡。
(4)倒出20%乙醇并用滤纸把剩余的20%乙醇吸干,按比例配好5%浓缩胶,连续平稳加入浓缩胶至离边缘5mm处,迅速插入样梳,静置30min。
(5)拔出样梳后,在上槽内加入1×蛋白缓冲液(用5×蛋白缓冲液稀释),没过锯齿。
(6)样品准备:取蛋白样品30μL与4×上样缓冲液10μL(最终稀释成1×上样缓冲液loading buffer)(当需要进行非还原电泳时用非还原的上样缓冲液)混合,煮沸10min,离心。
(7)上样根据蛋白浓度可选择5-30μL,蛋白样品上样20μL,蛋白marker上样10μL。
(8)接通电源,恒压80V跑30min。
(9)接着换电压到120V,待溴酚蓝跑出胶后(40-50min左右)关闭电源。
(10)凝胶板剥离与染色:电泳结束后,撬开玻璃板,将凝胶板做好标记后放在大培养皿内,加入染色液,染色30h左右。
(11)脱色:染色后的凝胶板用水漂洗数次,再加入脱色液或水置于微波炉中高火加热15min进行快速脱色。
(12)脱色后,将凝胶板用水漂洗数次,置于生物电泳图像分析系统中进行蛋白电泳图像拍摄。
RP-HPLC法检测TmSm蛋白纯度:
色谱柱:C18色谱柱(4.6mm×150mm,5μm)
流动相:缓冲液A:0.08%TFA乙腈:水=5:95。缓冲液B:0.1%TFA乙腈:水=95:5。缓冲液A:B=60%:40%
柱温:25℃
检测波长:280nm
检测流速:0.2mL/min
上样量:20μL
洗脱方式:在室温条件下,进行梯度洗脱。
纯度计算:按面积归一化法计算,以目标峰的峰面积占总积分面积的百分比作为纯度数据。
(1)过SP FF阳离子柱收集的洗脱液进行透析,将透析上清超滤脱盐,再加入PBS缓冲液进行换液,换液后的TmSm蛋白溶液测定蛋白浓度。
(2)将换液后的TmSm蛋白溶液用石英比色皿进行全波长扫描(200-400nm),记录最大吸收峰处的吸收波长。
(3)利用C18柱进行纯度测定,记录色谱图。
实施例2.TmSm-PLGA-NPs的制备及指标检测
试剂:单甲氧基聚乙二醇聚乳酸乙醇酸共聚物mPEG5000-PLGA50/5028000购置于济南岱罡生物工程有限公司,乙酸乙酯购置于上海化学试剂有限公司,PEG8000购置于北京拜尔迪生物技术有限公司,聚乙烯醇PVA购置于上海凌峰化学试剂有限公司,BCA法微量蛋白质浓度测定试剂盒购置于生工生物工程有限公司(上海)。其他的生化试剂属于国产常规的分析纯试剂。
试剂配制:
1%PVA溶液:称取1g PVA,用超纯水加热溶解,定容至100mL。
5%PEG8000水溶液:称取0.5g PEG8000,用超纯水溶解,定容至10mL。
10%蔗糖:称取1g蔗糖,用超纯水溶解,定容至10mL。
PLGA纳米颗粒制备:25mg mPEG-PLGA溶于2mL乙酸乙酯中,将200μL 1mg/mL TmSm 5%PEG水溶液加入到PLGA溶液中,冰浴条件下超声l min(功率为100W,超1s,停1s,60次),形成油包水(W1/O)型乳液。将获得的(W1/O)型乳液加入到20mL 1%PVA水溶液中,将上述混合液引入高压均质机(100MPa)进行两个周期,即形成水包油包水(W1/O/W2)型复乳液。在室温常压下800rmp磁力搅拌4h,挥发有机溶剂乙酸乙酯。然后,将固化的纳米粒通过低温高速离心(4℃,12100r/min,20min)收集,用5mL超纯水清洗3次,洗净纳米粒表面的PVA。其中,将离心的上清液进行收集。将上述清洗的沉淀用2mL 1%蔗糖重悬,置于西林瓶中,先-20℃冷冻半小时,再置于-80℃过夜冷冻,最后置于冷冻干燥机中冷冻干燥24h,去除纳米粒中的水分后,得到纳米粒成品,4℃环境下冷藏。其中,利用加超纯水代替1%蔗糖作为对照,进行冷冻干燥。空白PLGA纳米颗粒制备是利用200μL 5%PEG水溶液作为内水相,其他条件不变。
纳米颗粒的载药量和包封率的测定:将离心洗涤过程中的上清液收集,并用50mL容量瓶定容到50mL,采用Micro BCA法测定未被包进纳米粒中的TmSm含量,并计算纳米粒的包封率。由以下公式计算纳米颗粒的载药量和包封率:
BCA法微量蛋白质浓度测定
A试剂准备
(1)按照以下公式计算所需的BCA工作液总体积。
BCA工作液总体积=(标准曲线测定点数+样品数)×重复次数×每个样品所需的BCA工作液体积
(2)根据所需的BCA工作液总体积,定量取溶液A:溶液B:溶液C=25:24:1,混匀,制成BCA工作液。
(3)取一定量的BSA标准溶液,用1×PBS溶液稀释为40μg/mL。
(4)取9支1.5mL离心管,按照下表平行操作,配制BSA浓度梯度。
B酶标板法
(1)选取酶标板上22个孔,分为标准组和样品组。其中16个孔,每个标准品设置1个重复,各孔分别加入100μL相应浓度的标准蛋白质溶液(40μg/mL);剩余4孔,分为2个重复组,重复孔编号相同。其中编号不同的两孔加入浓度不同的100μL样品稀释液。
(2)各酶标孔加入100μL BCA工作液,迅速混匀。
(3)在37℃保温30min。
(4)冷却至室温后,在酶标仪上测各孔的A562值。
(5)以标准组各孔A562平均值为纵坐标,对应的蛋白质浓度为横坐标,在坐标纸上或Microsoft Excel软件中绘制标准曲线。
(7)根据两个相同样品稀释液A562值的平均值,在标准曲线上计算出该样品经过稀释后的蛋白质浓度。选择合适稀释度的样品计算最终的样品蛋白浓度,再由稀释倍数计算原样品蛋白质浓度。
纳米颗粒的体外释放实验:精密称取载药纳米颗粒或空白纳米颗粒10mg置于离心管中,加入1.5mL PBS缓冲液(pH 7.4),置于37℃恒温摇床中,振荡速度100rpm。分别在第l、2、4、6、8、12、24、48h以及第3、4、5、6天取出离心管,置于冷冻离心机中,14000rpm离心10min,将全部的释放介质小心吸出并更换新的PBS缓冲液释放介质,以空白纳米颗粒的释放上清液作为空白对照,按Micro BCA试剂盒要求进行加样,反应显色后于562nm处测得其吸光度,代入药物释放标准曲线,计算求得各个时间点释放的TmSm的药量(C0)。同时,测定释放前纳米颗粒中TmSm的含量(C0)。计算累计释放百分率Ft:Ft=(ΣCt/C0)×100%,以Ft对时间t作图,获得药物释放曲线。
实施例3.TmSm-PLGA-NPs的体外抗肿瘤活性检测
试剂与试剂盒:MTT,青链霉素混合液,DMSO购置于北京索来宝公司,RPMI-1640培养基,胎牛血清,胰酶购自Gibco公司(美国)。其他的生化试剂属于国产常规的分析纯试剂。
细胞株和培养基:
人胰腺癌细胞SW1990,培养条件为37℃,5%CO2浓度,培养基为RPMI1640培养基(含10%胎牛血清)。
试剂配制:
RPMI1640完全培养基:量取90mL RPMI1640培养液(1×),加入10mL胎牛血清,1mL青霉素(100IU/L)和1mL链霉素(100mg/L),充分混匀,置于4℃保存。
5mg/mL MTT溶液:称取250mg MTT溶于50mL PBS中,60℃助溶使其充分溶解,用0.22μm无菌滤膜过滤除菌,-20℃避光保存。
细胞的传代:将超净工作台用紫外灭菌30min;培养基放入37℃培养箱中预热。用倒置显微镜观察细胞生长状态,当细胞铺满瓶底80%-90%时,弃去瓶内培养基,用PBS清洗两遍,洗去残留的培养基,然后加入500μL胰酶,消化1-2min,待细胞变圆并从瓶壁上脱落时,加入4ml已预热的新鲜培养基终止消化,并用无菌的吸管沿着瓶壁吹打细胞,直到形成单细胞悬液,稀释到合适的细胞密度传代。
细胞的冻存:超净台紫外灭菌30min,培养基放入37℃培养箱中预热。将培养瓶中的旧培养基弃去,用PBS清洗两遍;再向培养基中加入500μL胰酶,放入培养箱中消化1-2min,待细胞变圆并有少量细胞漂浮时,加入培养基终止消化;吹打细胞形成单细胞悬液后,将其移入离心管中以1000rpm离心5min,弃去上清,加入1ml细胞冻存液(700μL基础培养基,200μL胎牛血清,100μL DMSO),用吸管吹打均匀,移入冻存管中盖紧盖子。将冻存管于4℃冰箱中放置0.5h,-20℃放置2h,再放入-80℃过夜,最后放入液氮中长期保持。
细胞的复苏:从液氮中取出细胞,迅速将其置于37℃水浴中,不停地摇动,使细胞快速溶解;然后将其移入离心管中以1000rpm离心5min;弃去上清,再加入4ml新鲜培养基,吹打均匀后,移入培养瓶中,置于37℃,5%CO2的培养箱中培养。过夜后,换培养液除去死细胞。
MTT法检测TmSm蛋白对胰腺癌SW1990细胞增殖的影响:接种胰腺癌SW1990细胞:细胞铺满细胞瓶80-90%时,用0.5mL 0.25%胰酶消化后,加入2mL RPMI1640完全培养基(10%胎牛血清,100IU/L青霉素,100mg/L链霉素,pH7.2)终止消化并吹打细胞,使形成细胞悬液。吸取1mL细胞悬液加入15mL的离心管中,并用6mL含10%胎牛血清的培养液配成单个细胞悬液,以2×104个细胞/mL接种到96孔板,每孔加入体积100μL。37℃、5%CO2培养箱中培养24h,观察到孔中细胞长满至70-80%。小心吸取旧的培养液弃去,每孔加入200μL含10%胎牛血清的培养液,最外一圈加入PBS。其中空白对照孔加入200μL RPMI1640完全培养基;其他孔为不同浓度的空白PLGA和TmSm-PLGA纳米颗粒,蛋白浓度分别为2mg/mL、4mg/mL、6mg/mL、8mg/mL、10mg/mL和12mg/mL,分别加入200μL,每个浓度共4个平行孔。将加好样品的96孔板置37℃、5%CO2培养箱中继续培养24h,显微镜下观察胰腺癌SW1990细胞形态是否发生变化。每孔加20μL 5mg/mL MTT溶液。继续孵育4h,终止培养,小心吸去孔内培养上清液。每孔加入150μL DMSO,在振荡仪上充分振荡10min使结晶溶解。在酶标仪上测定各孔的λ=490nm处吸光值,计算细胞存活率。细胞存活率=待测组吸光值/阴性对照组吸光值×100%。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!