一种治疗股骨头坏死的活骨注射液及制备方法与检测方法与流程
本发明属于医药领域,涉及一种治疗股骨头坏死的活骨注射液及制备方法
与检测方法。
背景技术:
骨坏死病是常见的骨科病,尤其以股骨头缺血坏死最为多见。股骨头缺血坏死好发于青壮年,年龄多在20-50岁,致残率高,严重危害人民的身心健康。股骨头缺血坏死发病原因有很多,目前已公认引起股骨头缺血坏死的原因为创伤和激素。据国内外文献报道,近年来本病的发生率不断上升,尤其是因滥用激素与酗酒所致的股骨头缺血坏死为多,其发病率已超过股骨颈骨折所致的股骨头缺血坏死,有学者统计,因激素所致的股骨头缺血坏死已占本病的第一位。随着人口老龄化,每年单纯因骨质疏松所致的股骨颈骨折而并发股骨头缺血坏死就有成千上万之多。目前对该类病的治疗仍是骨科的一大难题,近年来随着生物材料技术与生物力学理论的发展,人工髋关节质量虽然显著提高,全髋置换术能明显提高股骨头缺血坏死患者的生活质量,但由于股骨头缺血坏死患者的年龄相对较轻,目前的人工假体尚不能和患者地寿命相匹配,而早、中期股骨头缺血坏死也不一定需进行全髋置换,因此开发研究阻断或推迟对股骨头缺血坏死进行全髋置换的有效治疗药物仍然迫切需要。
到目前为止,现代医学领域尚无用于治疗骨缺血坏死病的化学药。中药治疗本病的有效作用虽然引起医药学者的广泛关注,但对治疗骨缺血坏死病的新药开发力度却仍然停滞不前,治疗骨缺血坏死病的药品尚少,特别是疗效满意的新药更为缺乏。
申请号为03135340.1、200710055426.7、201410420898.8、200510009962.4、200410069555.8、201410227164.8、201010207274.X、01129030.7、201310137741.X、201510594576.X、200510003123.1、201010279290.X、201110008449.9、201410713897.2、200610159799.4、201410277148.X、200610012248.5、201110439176.3、201010202835.7、200810238738.6、201210575751.7、02125610.1、201510285454.2、201010557465.9、201210182909.4、02110238.4的中国发明专利,公开了多种治疗股骨头坏死的中药组合物,但是,这些药物制剂少则由十几味药味原料制成,多则有二十几味、三十几味,甚至四五十味药味原料制备,其中有的药物组合物还含有一些对人体有毒的药材;而且这些药物多为口服药物制剂,或外用制剂,实际上,这些药物对于股骨头坏死的治疗并没有特别明显的效果;而且由于药味成分复杂,制备工艺非常粗糙,所以它的药物质量难于得到控制。
作为中华人民共和国国家自然科学基金项目——依据补肾/活血法运用中药关节腔灌注对股骨头缺血性坏死修复机制的研究(项目编号:81173276)的项目承担人,发明人对本发明的项目进行了持续的多年研究,得到了本发明的技术方案。
技术实现要素:
本发明的目的提供一种有效治疗股骨头坏死的中药组合物。
本发明的另一目的是提供该治疗股骨头坏死的中药组合物的制备方法。
本发明还有一个目的是提供该治疗股骨头坏死的中药组合物的检测方法。
本发明的目的是通过以下方式实现的:
一种治疗股骨头坏死的药物组合物,该药物组合物由以下重量份的药味原料制成:丹参1~5重量份、川芎1~5重量份、骨碎补1~5重量份。
所述的治疗股骨头坏死的药物组合物,作为优选,该药物组合物由以下重量份的药味原料制成:丹参5重量份、川芎3重量份、骨碎补2重量份。
所述的治疗股骨头坏死的药物组合物,其特征在于该药物组合物与药学可接受的辅料制成注射剂。
所述的治疗股骨头坏死的药物组合物,该药物组合物的制备方法为:取丹参、川芎和骨碎补,混合,粉碎成粗粉,将其进行水蒸气蒸馏提取,从中提取得到水蒸气蒸馏提取物,备用;取水蒸气蒸馏提取后的药渣,加入5~10倍重量比的60~80%乙醇浸渍48~60h,再用60~80%乙醇作溶剂缓慢渗滤至滤液呈微黄色,滤液在60~80℃下减压浓缩得相对密度为1.2~1.3的稠膏;再加8~16倍水充分搅拌并静置12~24h使油水分离,去除沉淀物和油脂,收集水层药液,加入药用活性炭静置1~2h吸附药液至无色,上聚酰胺色谱柱,用水饱和正丁醇洗脱,洗脱液在60~80℃下减压浓缩后加入8~12倍重量的90~95%乙醇进行二次乙醇提取,乙醇提液在4~8℃冷藏12~24h后过滤,滤液经减压浓缩后,再加入8~12倍重量份的水进行水提,经冷藏后12~24h,2~6℃离心过滤得水提液,水提液用0.1~0.5%重量比的活性炭脱色,边加边搅拌,并加热至40~50℃后静止0.5~1h;经膜过滤器过滤,超滤后在40~50℃下减压浓缩,得提取物稠膏;将提取物稠膏、水蒸气蒸馏提取物、药用聚乙二醇、甘露醇和医用氯化钠混合,用注射用水稀释,滤过,灌封、灭菌、灯检、包装,即得药物注射液。
所述的治疗股骨头坏死的药物组合物,作为优选,该药物组合物的制备方法为:取丹参、川芎和骨碎补,混合,粉碎成粗粉,将其进行水蒸气蒸馏提取,从中提取得到水蒸气蒸馏提取物,备用;取水蒸气蒸馏提取后的药渣,加入8倍重量比的70%乙醇浸渍50h,再用70%乙醇作溶剂缓慢渗滤至滤液呈微黄色,滤液在70℃下减压浓缩得相对密度为1.25的稠膏;再加12倍水充分搅拌并静置24h使油水分离,去除沉淀物和油脂,收集水层药液,加入药用活性炭静置2h吸附药液至无色,上聚酰胺色谱柱,用水饱和正丁醇洗脱,洗脱液在70℃下减压浓缩后加入10倍重量的95%乙醇进行二次乙醇提取,乙醇提液在6℃冷藏24h后过滤,滤液经减压浓缩后,再加入10倍重量份的水进行水提,经冷藏后24h,4℃离心过滤得水提液,水提液用0.3%重量比的活性炭脱色,边加边搅拌,并加热至40℃后静止1h;经膜过滤器过滤,超滤后在40℃下减压浓缩,得提取物稠膏;将提取物稠膏、水蒸气蒸馏提取物、药用聚乙二醇、甘露醇和医用氯化钠混合,用注射用水稀释,滤过,灌封、灭菌、灯检、包装,即得药物注射液。
一种治疗股骨头坏死的药物组合物的制备方法,该药物组合物由以下重量份的药味原料制成:丹参1~5重量份、川芎1~5重量份、骨碎补1~5重量份,其特征在于,该药物组合物的制备方法为:取丹参、川芎和骨碎补,混合,粉碎成粗粉,将其进行水蒸气蒸馏提取,从中提取得到水蒸气蒸馏提取物,备用;取水蒸气蒸馏提取后的药渣,加入5~10倍重量比的60~80%乙醇浸渍48~60h,再用60~80%乙醇作溶剂缓慢渗滤至滤液呈微黄色,滤液在60~80℃下减压浓缩得相对密度为1.2~1.3的稠膏;再加8~16倍水充分搅拌并静置12~24h使油水分离,去除沉淀物和油脂,收集水层药液,加入药用活性炭静置1~2h吸附药液至无色,上聚酰胺色谱柱,用水饱和正丁醇洗脱,洗脱液在60~80℃下减压浓缩后加入8~12倍重量的90~95%乙醇进行二次乙醇提取,乙醇提液在4~8℃冷藏12~24h后过滤,滤液经减压浓缩后,再加入8~12倍重量份的水进行水提,经冷藏后12~24h,2~6℃离心过滤得水提液,水提液用0.1~0.5%重量比的活性炭脱色,边加边搅拌,并加热至40~50℃后静止0.5~1h;经膜过滤器过滤,超滤后在40~50℃下减压浓缩,得提取物稠膏;将提取物稠膏、水蒸气蒸馏提取物、药用聚乙二醇、甘露醇和医用氯化钠混合,用注射用水稀释,滤过,灌封、灭菌、灯检、包装,即得药物注射液。
所述的治疗股骨头坏死的药物组合物的制备方法,作为优选,该药物组合物由以下重量份的药味原料制成:丹参5重量份、川芎3重量份、骨碎补2重量份,该药物组合物的制备方法为:取丹参、川芎和骨碎补,混合,粉碎成粗粉,将其进行水蒸气蒸馏提取,从中提取得到水蒸气蒸馏提取物,备用;取水蒸气蒸馏提取后的药渣,加入8倍重量比的70%乙醇浸渍50h,再用70%乙醇作溶剂缓慢渗滤至滤液呈微黄色,滤液在70℃下减压浓缩得相对密度为1.25的稠膏;再加12倍水充分搅拌并静置24h使油水分离,去除沉淀物和油脂,收集水层药液,加入药用活性炭静置2h吸附药液至无色,上聚酰胺色谱柱,用水饱和正丁醇洗脱,洗脱液在70℃下减压浓缩后加入10倍重量的95%乙醇进行二次乙醇提取,乙醇提液在6℃冷藏24h后过滤,滤液经减压浓缩后,再加入10倍重量份的水进行水提,经冷藏后24h,4℃离心过滤得水提液,水提液用0.3%重量比的活性炭脱色,边加边搅拌,并加热至40℃后静止1h;经膜过滤器过滤,超滤后在40℃下减压浓缩,得提取物稠膏;将提取物稠膏、水蒸气蒸馏提取物、药用聚乙二醇和医用氯化钠混合,用注射用水稀释,滤过,灌封、灭菌、灯检、包装,即得药物注射液。
一种治疗股骨头坏死的药物组合物的检测方法,该药物组合物由以下重量份的药味原料制成:丹参1~5重量份、川芎1~5重量份、骨碎补1~5重量份,该药物组合物的制备方法为:取丹参、川芎和骨碎补,混合,粉碎成粗粉,将其进行水蒸气蒸馏提取,从中提取得到水蒸气蒸馏提取物,备用;取水蒸气蒸馏提取后的药渣,加入5~10倍重量比的60~80%乙醇浸渍48~60h,再用60~80%乙醇作溶剂缓慢渗滤至滤液呈微黄色,滤液在60~80℃下减压浓缩得相对密度为1.2~1.3的稠膏;再加8~16倍水充分搅拌并静置12~24h使油水分离,去除沉淀物和油脂,收集水层药液,加入药用活性炭静置1~2h吸附药液至无色,上聚酰胺色谱柱,用水饱和正丁醇洗脱,洗脱液在60~80℃下减压浓缩后加入8~12倍重量的90~95%乙醇进行二次乙醇提取,乙醇提液在4~8℃冷藏12~24h后过滤,滤液经减压浓缩后,再加入8~12倍重量份的水进行水提,经冷藏后12~24h,2~6℃离心过滤得水提液,水提液用0.1~0.5%重量比的活性炭脱色,边加边搅拌,并加热至40~50℃后静止0.5~1h;经膜过滤器过滤,超滤后在40~50℃下减压浓缩,得提取物稠膏;将提取物稠膏、水蒸气蒸馏提取物、药用聚乙二醇、甘露醇和医用氯化钠混合,用注射用水稀释,滤过,灌封、灭菌、灯检、包装,即得药物注射液;
采用如下检测方法对该药物进行检测:
(1)色谱条件:色谱柱:DB-WAX毛细管色谱柱,氢火焰离子化检测器,氮气流速:0.5~2.0mL·min-1,氢气流速:5~20mL·min-1,空气流速:30~50mL·min-1,柱温为程序升温,初始温度80℃,以每分钟5℃的速度升温至200℃保持10min;分流进样:5~15:1,进样口温度:180~220℃,检测器温度:230~270℃;
(2)混合对照品溶液制备:精密称取间-乙基苏合香烯对照品、对-伞花烃对照品,分别置量瓶中;精密称取邻苯二甲酸二异丁酯对照品、对-伞花烯对照品、2,4-癸二烯醛对照品,分别置量瓶中;向各个两瓶中加入体积比为1:1的无水甲醇-乙酸乙酯的混合溶液至刻度,摇匀,作为对照品储备液;精密吸取上述对照品储备液,置同一量瓶中,加上述体积比为1:1的无水甲醇-乙酸乙酯的混合溶液稀释到刻度,作为混合对照品溶液;
(3)供试品溶液制备:精密量取药物注射液,加在C18固相萃取柱上,以每秒两滴的速度流出,弃去流出液,再用体积比为1:1的无水甲醇-乙酸乙酯的混合溶液洗脱,收集洗脱液于量瓶中,加上述体积比为1:1的无水甲醇-乙酸乙酯的混合溶液至刻度,摇匀,即得供试品溶液;
(4)测定:精密吸取混合对照品溶液、供试品溶液注入气相色谱仪测定。
所述的治疗股骨头坏死的药物组合物的检测方法,作为优选,该药物组合物由以下重量份的药味原料制成:丹参5重量份、川芎3重量份、骨碎补2重量份,该药物组合物的制备方法为:取丹参、川芎和骨碎补,混合,粉碎成粗粉,将其进行水蒸气蒸馏提取,从中提取得到水蒸气蒸馏提取物,备用;取水蒸气蒸馏提取后的药渣,加入8倍重量比的70%乙醇浸渍50h,再用70%乙醇作溶剂缓慢渗滤至滤液呈微黄色,滤液在70℃下减压浓缩得相对密度为1.25的稠膏;再加12倍水充分搅拌并静置24h使油水分离,去除沉淀物和油脂,收集水层药液,加入药用活性炭静置2h吸附药液至无色,上聚酰胺色谱柱,用水饱和正丁醇洗脱,洗脱液在70℃下减压浓缩后加入10倍重量的95%乙醇进行二次乙醇提取,乙醇提液在6℃冷藏24h后过滤,滤液经减压浓缩后,再加入10倍重量份的水进行水提,经冷藏后24h,4℃离心过滤得水提液,水提液用0.3%重量比的活性炭脱色,边加边搅拌,并加热至40℃后静止1h;经膜过滤器过滤,超滤后在40℃下减压浓缩,得提取物稠膏;将提取物稠膏、水蒸气蒸馏提取物、药用聚乙二醇和医用氯化钠混合,用注射用水稀释,滤过,灌封、灭菌、灯检、包装,即得药物注射液;
采用如下检测方法对该药物进行检测:
(1)色谱条件:色谱柱:DB-WAX毛细管色谱柱,柱长30m,内径0.53mm,膜厚1m,氢火焰离子化检测器,氮气流速:1mL·min-1,氢气流速:10mL·min-1,空气流速:40mL·min-1,柱温为程序升温,初始温度80℃,以每分钟5℃的速度升温至200℃保持10min;分流进样:10:1,进样口温度:200℃,检测器温度:250℃;
(2)混合对照品溶液制备:精密称取间-乙基苏合香烯对照品29.76mg、对-伞花烃对照品58.33mg,分别置10mL量瓶中;精密称取邻苯二甲酸二异丁酯对照品16.06mg、对-伞花烯对照品12.54mg、2,4-癸二烯醛对照品15.69mg,分别置25mL量瓶中;向各个两瓶中加入体积比为1:1的无水甲醇-乙酸乙酯的混合溶液至刻度,摇匀,作为对照品储备液;精密吸取上述对照品储备液各1mL,置同一10mL量瓶中,加上述体积比为1:1的无水甲醇-乙酸乙酯的混合溶液稀释到刻度,作为混合对照品溶液;
(3)供试品溶液制备:精密量取药物注射液10mL,加在C18固相萃取柱上,以每秒两滴的速度流出,弃去流出液,再用体积比为1:1的无水甲醇-乙酸乙酯的混合溶液9mL洗脱,收集洗脱液于10mL量瓶中,加上述体积比为1:1的无水甲醇-乙酸乙酯的混合溶液至刻度,摇匀,即得供试品溶液;
(4)测定:精密吸取混合对照品溶液、供试品溶液各1μl,注入气相色谱仪测定。
一种药物组合物在制备治疗股骨头坏死药物中的应用,该药物组合物由以下重量份的药味原料制成:丹参5重量份、川芎3重量份、骨碎补2重量份,该药物组合物的制备方法为:取丹参、川芎和骨碎补,混合,粉碎成粗粉,将其进行水蒸气蒸馏提取,从中提取得到水蒸气蒸馏提取物,备用;取水蒸气蒸馏提取后的药渣,加入8倍重量比的70%乙醇浸渍50h,再用70%乙醇作溶剂缓慢渗滤至滤液呈微黄色,滤液在70℃下减压浓缩得相对密度为1.25的稠膏;再加12倍水充分搅拌并静置24h使油水分离,去除沉淀物和油脂,收集水层药液,加入药用活性炭静置2h吸附药液至无色,上聚酰胺色谱柱,用水饱和正丁醇洗脱,洗脱液在70℃下减压浓缩后加入10倍重量的95%乙醇进行二次乙醇提取,乙醇提液在6℃冷藏24h后过滤,滤液经减压浓缩后,再加入10倍重量份的水进行水提,经冷藏后24h,4℃离心过滤得水提液,水提液用0.3%重量比的活性炭脱色,边加边搅拌,并加热至40℃后静止1h;经膜过滤器过滤,超滤后在40℃下减压浓缩,得提取物稠膏;将提取物稠膏、水蒸气蒸馏提取物、药用聚乙二醇、甘露醇和医用氯化钠混合,用注射用水稀释,滤过,灌封、灭菌、灯检、包装,即得药物注射液;
采用如下检测方法对该药物进行检测:
(1)色谱条件:色谱柱:DB-WAX毛细管色谱柱,柱长30m,内径0.53mm,膜厚1m,氢火焰离子化检测器,氮气流速:1mL·min-1,氢气流速:10mL·min-1,空气流速:40mL·min-1,柱温为程序升温,初始温度80℃,以每分钟5℃的速度升温至200℃保持10min;分流进样:10:1,进样口温度:200℃,检测器温度:250℃;
(2)混合对照品溶液制备:精密称取间-乙基苏合香烯对照品29.76mg、对-伞花烃对照品58.33mg,分别置10mL量瓶中;精密称取邻苯二甲酸二异丁酯对照品16.06mg、对-伞花烯对照品12.54mg、2,4-癸二烯醛对照品15.69mg,分别置25mL量瓶中;向各个两瓶中加入体积比为1:1的无水甲醇-乙酸乙酯的混合溶液至刻度,摇匀,作为对照品储备液;精密吸取上述对照品储备液各1mL,置同一10mL量瓶中,加上述体积比为1:1的无水甲醇-乙酸乙酯的混合溶液稀释到刻度,作为混合对照品溶液;
(3)供试品溶液制备:精密量取药物注射液10mL,加在C18固相萃取柱上,以每秒两滴的速度流出,弃去流出液,再用体积比为1:1的无水甲醇-乙酸乙酯的混合溶液9mL洗脱,收集洗脱液于10mL量瓶中,加上述体积比为1:1的无水甲醇-乙酸乙酯的混合溶液至刻度,摇匀,即得供试品溶液;
(4)测定:精密吸取混合对照品溶液、供试品溶液各1μl,注入气相色谱仪测定。
本发明提供以下实验研究资料,用以充分公开本发明的技术方案以及本发明的技术效果。
实验一:对治疗实验性股骨头坏死的处方与活性部位的药效学筛选研究
1 材料与方法
1.1 材料:
1.1.1 动物:Wistar大鼠乳鼠,60只,日龄10d,由黑龙江中医药大学动物中心提供。动物合格证号:医动字第1024-03号。实验动物均预先饲养24小时,适应实验室饲养条件。
1.1.2 试剂:醋酸泼尼松龙注射液,0.125g/ml,浙江仙琚制药厂股份有限公司。
1.1.3 仪器:双能X线骨密度仪,微循环显微观察系统,MTS生物材料试验机,美国产品。
1.1.4 药物:
(1)血塞通注射液:黑龙江珍宝岛药业股份有限公司生产,国药准字Z23020787。
(2)甲药物注射液:
处方:丹参500g、川芎300g、骨碎补200g。
制备方法:取干燥的丹参500g、川芎300g和骨碎补200g,混合,粉碎成粗粉,将其进行水蒸气蒸馏提取,从中提取得到水蒸气蒸馏提取物,备用;取水蒸气蒸馏提取后的药渣,加入8倍重量比的70%乙醇浸渍50h,再用70%乙醇作溶剂缓慢渗滤至滤液呈微黄色,滤液在70℃下减压浓缩得相对密度为1.25的稠膏;再加12倍水充分搅拌并静置24h使油水分离,去除沉淀物和油脂,收集水层药液,加入药用活性炭静置2h吸附药液至无色,上聚酰胺色谱柱,用水饱和正丁醇洗脱,洗脱液在70℃下减压浓缩后加入10倍重量的95%乙醇进行二次乙醇提取,乙醇提液在6℃冷藏24h后过滤,滤液经减压浓缩后,再加入10倍重量份的水进行水提,经冷藏后24h,4℃离心过滤得水提液,水提液用0.3%重量比的活性炭脱色,边加边搅拌,并加热至40℃后静止1h;经膜过滤器过滤,超滤后在40℃下减压浓缩,得提取物稠膏;将提取物稠膏、水蒸气蒸馏提取物、药用聚乙二醇、甘露醇和医用氯化钠混合,用注射用水稀释,滤过,灌封、灭菌、灯检、包装,即得甲药物注射液。
(3)乙药物注射液:
处方:丹参500g、川芎300g、骨碎补200g。
制备方法:取干燥的丹参500g、川芎300g和骨碎补200g,混合,粉碎成粗粉,加入8倍重量比的70%乙醇浸渍50h,再用70%乙醇作溶剂缓慢渗滤至滤液呈微黄色,滤液在70℃下减压浓缩得相对密度为1.25的稠膏;再加12倍水充分搅拌并静置24h使油水分离,去除沉淀物和油脂,收集水层药液,加入药用活性炭静置2h吸附药液至无色,上聚酰胺色谱柱,用水饱和正丁醇洗脱,洗脱液在70℃下减压浓缩后加入10倍重量的95%乙醇进行二次乙醇提取,乙醇提液在6℃冷藏24h后过滤,滤液经减压浓缩后,再加入10倍重量份的水进行水提,经冷藏后24h,4℃离心过滤得水提液,水提液用0.3%重量比的活性炭脱色,边加边搅拌,并加热至40℃后静止1h;经膜过滤器过滤,超滤后在40℃下减压浓缩,得提取物稠膏;将提取物稠膏、药用聚乙二醇、甘露醇和医用氯化钠混合,用注射用水稀释,滤过,灌封、灭菌、灯检、包装,即得乙药物注射液。
(4)丙药物注射液:
处方:丹参500g、川芎300g、骨碎补200g。
制备方法:取干燥的丹参500g、川芎300g和骨碎补200g,混合,粉碎成粗粉,将其进行水蒸气蒸馏提取,从中提取得到水蒸气蒸馏提取物,备用;取水蒸气蒸馏提取后的药渣,加入8倍重量比的70%乙醇浸渍50h,再用70%乙醇作溶剂缓慢渗滤至滤液呈微黄色,滤液在70℃下减压浓缩得相对密度为1.25的提取物稠膏;将提取物稠膏、水蒸气蒸馏提取物、药用聚乙二醇、甘露醇和医用氯化钠混合,用注射用水稀释,滤过,灌封、灭菌、灯检、包装,即得丙药物注射液。
(5)丁药物注射液:
处方:丹参500g、川芎300g、骨碎补200g。
制备方法:取干燥的丹参500g、川芎300g和骨碎补200g,混合,粉碎成粗粉,将其进行水蒸气蒸馏提取,从中提取得到水蒸气蒸馏提取物,备用;取水蒸气蒸馏提取后的药渣,加入8倍重量比的70%乙醇浸渍50h,再用70%乙醇作溶剂缓慢渗滤至滤液呈微黄色,滤液在70℃下减压浓缩得相对密度为1.25的稠膏;再加12倍水充分搅拌并静置24h使油水分离,去除沉淀物和油脂,收集水层药液,加入药用活性炭静置2h吸附药液至无色,药液经膜过滤器过滤,超滤后在40℃下减压浓缩,得提取物稠膏;将提取物稠膏、水蒸气蒸馏提取物、药用聚乙二醇、甘露醇和医用氯化钠混合,用注射用水稀释,滤过,灌封、灭菌、灯检、包装,即得丁药物注射液。
1.2 方法:
(1)分组与给药:动物分成:正常对照组、模型对照组、血塞通注射液组、甲药物注射液组、乙药物注射液组、丙药物注射液组、丁药物注射液组,共7个组,每组10只动物。正常对照组乳鼠不作任何处理。模型对照组乳鼠出生10d乳鼠肘关节以上剪除左前肢。自尾根部剪除尾部,烧灼止血后放回笼中,继续由母鼠喂养。1个月后,开始灌服醋酸泼尼松,20mg/kg体重,隔日1次,共30d。甲药物注射液组、乙药物注射液组、丙药物注射液组、丁药物注射液组、血塞注射液给药组:在大鼠灌服醋酸泼尼松同时每天静脉注射给药一次,共30d。
(2)检测指标:股骨骨密度,股骨强度、钢性,股骨湿重、干重、灰重,股骨头毛细血管分布观察,股骨头骨组织内脂肪沉积观察。
1.3 统计学处理:
各组结果以均数±标准差(±s)表示。选择Student检验进行各组间算术平均数的差异显著性检验。
结果
2.1 甲药物注射液组的股骨BMD、BMC、骨强度、骨钢性、AREA含量明显高于模型对照组,也高于乙药物注射液组、丙药物注射液组、丁药物注射液组(P<0.01,表1)。
2.2 甲药物注射液组的股骨湿重明显高于模型对照组,也高于乙药物注射液组、丙药物注射液组、丁药物注射液组,甲药物注射液还可以明显降低软骨和骨细胞脂肪着色率,明显高于模型对照,也高于乙药物注射液、丙药物注射液、丁药物注射液(P<0.05,表2)。
2.3 模型动物的股骨头内毛细血管明显稀疏,排列紊乱,单位面积内毛细血管所占比例明显下降。甲药物注射液治疗组股骨头内毛细血管接近恢复正常水平股骨头内密布毛细血管,排列也较规律。
表1 对大鼠股骨骨密度、骨矿、骨面积、骨强度和骨钢性的影响(n=10)
注:与模型对照组比较,*P<0.05,#P<0.01;与正常对照组比较,△P<0.01;与血塞通组比较,☆P<0.05,▲P<0.01
表2 对大鼠股骨重量、软骨和骨细胞脂肪着色率的影响(n=10)
注:与模型对照组比较,*P<0.05,#P<0.01;与正常对照组比较,△P<0.01;与血塞通注射液组比较,☆P<0.05,▲P<0.01
3 讨论与结论
本研究结果表明,甲药物注射液活血化瘀用于治疗股骨头坏死有较好的改善作用,能增加股骨骨密度,骨矿度,骨重量,骨强度,骨钢性,改善股骨上毛细血管紊乱,降低股骨骨细胞内脂肪着色率。同时,甲药物注射液治疗股骨头坏死作用优于乙药物注射液、丙药物注射液、丁药物注射液,提示甲药物注射液的组方及制备工艺具有明显优势,在特定的组方和特定的制备方法下,具有特别突出的有益效果。
实验二:本发明注射液安全性实验报告
为探讨本发明注射液临床用药的安全性,本研究进行了安全性实验。
1 本发明注射液药物
处方:丹参500g、川芎300g、骨碎补200g。
制备方法:取干燥的丹参500g、川芎300g和骨碎补200g,混合,粉碎成粗粉,将其进行水蒸气蒸馏提取,从中提取得到水蒸气蒸馏提取物,备用;取水蒸气蒸馏提取后的药渣,加入8倍重量比的70%乙醇浸渍50h,再用70%乙醇作溶剂缓慢渗滤至滤液呈微黄色,滤液在70℃下减压浓缩得相对密度为1.25的稠膏;再加12倍水充分搅拌并静置24h使油水分离,去除沉淀物和油脂,收集水层药液,加入药用活性炭静置2h吸附药液至无色,上聚酰胺色谱柱,用水饱和正丁醇洗脱,洗脱液在70℃下减压浓缩后加入10倍重量的95%乙醇进行二次乙醇提取,乙醇提液在6℃冷藏24h后过滤,滤液经减压浓缩后,再加入10倍重量份的水进行水提,经冷藏后24h,4℃离心过滤得水提液,水提液用0.3%重量比的活性炭脱色,边加边搅拌,并加热至40℃后静止1h;经膜过滤器过滤,超滤后在40℃下减压浓缩,得提取物稠膏;将提取物稠膏、水蒸气蒸馏提取物、药用聚乙二醇、甘露醇和医用氯化钠混合,用注射用水稀释,滤过,灌封、灭菌、灯检、包装,即得药物注射液。
2 方法
2.1 局部刺激性实验
2.1.1 实验动物
日本大耳白家兔,雌雄兼用,体重2.0~2.5Kg,由黑龙江中医药大学动物中心提供。动物合格证号:医动字第1024-03号。实验动物均预先饲养24小时,适应实验室饲养条件。
2.1.2实验方法
取健康无损伤的家兔2只,以无菌操作法分别在其左右两腿股四头肌内各注入供试品1.0mL/只,注射后48h处死动物,解剖取出股四头肌,纵向切开,观察注射部位局部刺激反应,按表3换算成相应的反应级。
表3 注射肌肉刺激反应评级标准
2.1.3 实验结果
实验结果表明:两只家兔,经肌肉注射后,家兔健康正常,腿部活动正常。家兔处死取出股四头肌后,肉眼观察给药部位,给予本发明注射液的2例股四头肌刺激反应级数各为0级。提示本品对家兔股四头肌无刺激作用。
2.2血管刺激性实验
2.2.1实验动物
日本大耳白家兔,雌雄兼用,体重2.0~2.5Kg,由黑龙江中医药大学动物中心提供。动物合格证号:医动字第1024-03号。实验动物均预先饲养24小时,适应实验室饲养条件。
2.2.2实验方法
取健康、两耳无损伤的家兔2只。用无菌操作方法于左耳耳缘静脉注射灭菌生理盐水2.0mL/Kg,于右耳耳缘静脉注射本发明注射液的浓缩液2.0mL/Kg,1次/d,连续3d。于末次注射24h后将动物放血处死,分别距注射部位进针点2cm处向心端剪下耳缘约2cm作为标本,标本用福尔马林固定,常规组织切片,观察血管周围变化、血管壁是否损伤、内皮细胞有无脱落、有无血栓形成以及其他病理变化。
2.2.3实验结果
实验结果表明:家兔左右耳肉眼观察注射部位均无异常,血管无充血,周围组织无水肿现象。病理切片观察结果表明,给予生理盐水的2例耳缘和给予本发明注射液的2例耳缘均未见血管扩张和炎细胞浸润,提示本品对家兔血管无刺激作用。
2.3过敏实验
2.3.1实验动物
健康无伤雄性豚鼠,由黑龙江中医药大学动物中心提供。动物合格证号:医动字第1024-03号。实验动物均预先饲养24小时,适应实验室饲养条件。
2.3.2实验方法
取健康无伤豚鼠6只,体重250~350g,间日腹腔注射供试品0.5mL,连续3次,然后分为两组,每组3只,分别在第1次注射后14d及21d静脉注射本品1mL进行攻击,在注射后15min内观察动物反应。如有竖毛、呼吸困难、喷嚏、干呕或咳嗽3声等现象中的两种或两种以上者,或有啰音、抽搐、虚脱或死亡现象之一者为阳性。
2.3.3实验结果
实验结果表明:本发明注射液于第1次腹腔注射后第10天和第15天进行攻击,均未发生过敏反应,表明在本次实验条件下,本发明注射液对豚鼠无全身性致敏作用结果见表4。
表4 不同时间过敏实验观察结果
“ -”表示为阴性结果 ;“ +”表示为阳性结果
2.4溶血性实验
2.4.1实验动物
日本大耳白家兔,雄性,体重2.3Kg,由黑龙江中医药大学动物中心提供。动物合格证号:医动字第1024-03号。实验动物均预先饲养24小时,适应实验室饲养条件。
2.4.2实验方法
取健康家兔1只,从颈总动脉取血约10mL,放置于200mL三角瓶中,用玻璃棒搅动血液10min,除去纤维蛋白原,使成脱纤血液,加约10倍量的生理盐水,摇匀,离心(2000r/min,5min)除去上清液,沉淀的红血球再用生理盐水如下法洗涤2~3次,至上清液不显红色为止。取沉淀的红细胞1mL,加生理盐水50mL,制得2%的兔红细胞混悬液。取试管7支,按表3配比量依次加入2%兔红细胞混悬液和生理盐水,混匀后,置于37℃恒温水浴箱30min,然后分别加入不同量的上述药液(第6管为阴性对照管,第7管为阳性对照管),摇匀,置37℃恒温水浴箱中。开始每隔15min观察1次,1h后,每隔1h观察1次,共4次。见表5。
表5 溶血实验配比量表 mL
2.4.4溶血判定标准
全溶血:溶液澄明红色,管底无细胞残留;部分溶血:溶液澄明红色或棕色,管底有少量红细胞残留;无溶血:红细胞全部下沉,上层液体无色澄明;凝集:虽无溶血,但出现红细胞凝集,振摇后不分散。
2.4.5实验结果
本发明注射液无溶血和凝集反应结果见表6。
表6 各种时段溶血实验结果
“-”表示无溶血或凝集反应,“+”表示有溶血或凝集反应
3 结论
本发明注射液经动物实验结果表明其安全性好,可供临床使用。
实验三:本发明实施例1注射剂临床观察
临床观察:男性5例,女性5例,共10例,年龄58~65岁。
临床表现:局部红肿、刺痛,疼有定处,入夜尤甚,面色暗,发质暗红,脉沉,中医诊断为气滞血瘀型,西医诊断为局部疼痛或放射性疼痛;腰向健侧弯曲:“4”字征(+);骨盆分离试验阳性;肌肉萎缩;X线片、CT片显示有骨坏死的临床表现。
临床疗效显示:患者日静脉滴注一次,每次10mL,溶于200mL 0.9%氯化钠溶液,10天为一个疗程,跟踪观察统计骨坏死总有效率为90.0%。
实验四:本发明药物注射液特征图谱及5种主要成分的含量测定
特征图谱能提供更加丰富的信息,可全面的反映药品的质量特性,因此本研究采用GC法建立了本发明药物注射液的特征图谱,结合混合对照药材为参比的模式对其进行了定性研究,同时还对本发明药物注射液中5种主要成分间-乙基苏合香烯、对-伞花烃、邻苯二甲酸二异丁酯、对-伞花烯、2,4-癸二烯醛进行了含量测定研究,为其质量控制提供了一种较为全面的手段。
1 仪器与试药
气相色谱仪:Agilent6890N(配备氢火焰离子化检测器,美国Agilent公司),电子分析天平:AE-240(瑞士Mettler公司)。C18固相萃取柱:Agilent(6mL;500mg)、DIKMA(6mL;500mg)。间-乙基苏合香烯对照品、对-伞花烃对照品、邻苯二甲酸二异丁酯对照品、对-伞花烯对照品、2,4-癸二烯醛对照品,丹参对照药材、川芎对照药材、骨碎补对照药材,均由中国食品药品检定研究院提供。无水甲醇、乙酸乙酯均为GC级。
2 方法与结果
2.1 色谱条件
色谱柱:DB-WAX毛细管色谱柱(柱长30m,内径0.53mm,膜厚1m),氢火焰离子化检测器,氮气流速:1mL·min-1,氢气流速:10mL·min-1,空气流速:40mL·min-1,柱温为程序升温,初始温度80℃,以每分钟5℃的速度升温至200℃保持10min;分流进样:10:1,进样口温度:200℃,检测器温度:250℃。见附图1、附图2、附图3。
2.2 溶液的制备
2.2.1混合对照品溶液
精密称取间-乙基苏合香烯对照品29.76mg、对-伞花烃对照品58.33mg,分别置10mL量瓶中;精密称取邻苯二甲酸二异丁酯对照品16.06mg、对-伞花烯对照品12.54mg、2,4-癸二烯醛对照品15.69mg,分别置25mL量瓶中,加无水甲醇-乙酸乙酯(1:1)的混合溶液至刻度,摇匀,作为对照品储备液。精密吸取上述对照品储备液各1mL,置同一10mL量瓶中,加上述混合溶液稀释到刻度,作为混合对照品溶液。
2.2.2 混合对照药材溶液
按处方比例称丹参对照药材50g与川芎对照药材30g、骨碎补对照药材20g,混匀,水蒸气蒸馏,收集蒸馏液,加入对-伞花烃100mg、间-乙基苏合香烯对照品75mg,超声使溶解,用5%氢氧化钠溶液调节pH至6~7,加活性炭适量,摇匀,加水至100mL,滤过,精密吸取10mL,按“2.2.3”的方法制备混合对照药材溶液。
2.2.3 供试品溶液制备
精密量取本品10mL,加在处理好的C18固相萃取柱(规格:6mL/500mg,用甲醇、水预洗)上,以每秒两滴的速度流出(必要时适当加压),弃去流出液,再用无水甲醇-乙酸乙酯(1:1)的混合溶液9mL洗脱,收集洗脱液于10mL量瓶中,加上述混合溶液至刻度,摇匀,即得。
2.3 含量测定研究
2.3.1 标准曲线制备
精密量取各对照品溶液一定量,稀释成6种浓度的系列对照品溶液,精密吸取各1μL,注入气相色谱仪,记录峰面积,由对照品浓度(X,mg·mL-1)与对照品峰面积(Y)进行回归,结果见表7。
表7 5种成分线性关系表
2.3.2方法学考察
取同一批样品(批号:151017),按“2.2.3”方法平行制备9份供试品溶液(取样量5,10,15mL各3份),分别计算5种成分含量与RSD(n=9),考察方法重复性。取同一份供试品溶液,室温下放置,分别于0,0.5,1,2,5,8,12h进样测定,计算各成分峰面积RSD(n=7),考察供试品溶液的稳定性。取同一批样品9份(取样量5mL),加入一定量的对照品溶液,考察方法的回收率。结果见表8。
表8 方法学考察结果
2.3.3 样品测定
分别测10批样品,结果见表9。
表9 10批样品测定结果(mg·mL-1,n=3)
2.4 特征图谱研究
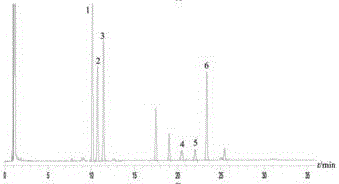
2.4.1 特征图谱的建立采用国家药典委员会相似度评价软件(2012版),以对-伞花烯为参照峰,选取15~30min之间的6个主要色谱峰为特征峰(附图4),结果10批样品特征峰相对保留时间RSD均小于1%(表10),相对峰面积见表11,以混合对照药材图谱为标准图谱,10批样品的相似度在0.914~0.995之间。
表10 样品中特征峰相对保留时间
表11 样品中特征峰相对峰面积
2.4.2方法学考察
取同一批样品,平行制备6份供试品溶液,考察方法重复性;取同一份供试品溶液,连续进样6针,考察方法精密度;取同一份样品,分别在0,1,2,4,6,8,12h进样测定,考察样品稳定性。分别计算各色谱峰相对保留时间与相对峰面积RSD,结果相对保留时间RSD均小于3.0%,相对峰面积RSD均小于5.0%,符合特征图谱要求。
3 讨论
3.1本发明药物注射液加入了甘露醇作为助溶剂,文献报道的方法中均未对其进行特别去除,为防止其对色谱柱的损害,用C18固相萃取柱对样品进行了净化处理,分别考察了洗脱溶剂与洗脱速度,经HPLC-ELSD法验证,基本能完全去除甘露醇。
3.2本发明药物注射液中正十六酸、α-侧柏烯、γ-松油烯,成分含量较高,在色谱图中其三个色谱峰所占比例较大,若采用全谱进行相似度比较,则其他成分对相似度值贡献较小,相似度均在0.99以上,因此在进行相似度评价时将这三个峰去除,选择15~30min的色谱图进行比较,以更科学地反映药品质量。
3.3 特征图谱中6个色谱峰中,1~3号峰为丹参中成分,3号峰为邻苯二甲酸二异丁酯,4号峰为对-伞花烯,5~6号峰为骨碎补中成分,5号峰为2,4-癸二烯醛;与6号峰相邻的色谱峰为溶剂峰。由于特征峰相对保留时间受色谱柱影响较大,有时在不同实验室重现性较差,所以通过混合对照药材模式与样品进行比对,方便快捷,可很好地判断各特征峰。
3.4 本研究利用GC法制定了本发明药物注射液的特征图谱及其中5个主要成分的含量测定方法,并通过固相萃取法去除了助溶剂甘露醇,操作简便,结果准确,不仅全面反映了药品内在质量,而且有利于色谱柱的保护。
实验五:药物注射剂稳定剂考察
本发明通过实验筛选了注射剂稳定剂的各项指标,以提高药品安全性。
在本发明药物注射剂中加入适量的稳定剂可以减少有关物质产生,发明人通过试验研究了D-山梨醇和甘露醇两种稳定剂对其影响,对稳定剂进行了筛选。
试验方法:
按照表12处方分别制备含D-山梨醇和甘露醇三种浓度的样品,制备步骤同实施例1,分别灌封,在121℃灭菌15分钟,并将七处方样品于光照4500LX下考察10天,考察配制溶液中有关物质的变化情况。
表12 样品的制备
由表13数据可见,当注射液中加入了D-山梨醇或甘露醇后,药液因光照的影响降低,有关物质产生减少,稳定性提高。另外单位浓度甘露醇光稳定的作用较D-山梨醇显著增高。为确保本品的质量,确定选用甘露醇浓度为10~15mg/mL作为本品中稳定剂的用量,其中作为优选为15mg/mL。
表13 研究有关物质检测结果
实验六:不同药物中活性成分的含量测定
1 待测样品
1.1 甲药物注射液:
处方:丹参500g、川芎300g、骨碎补200g。
制备方法:取干燥的丹参500g、川芎300g和骨碎补200g,混合,粉碎成粗粉,将其进行水蒸气蒸馏提取,从中提取得到水蒸气蒸馏提取物,备用;取水蒸气蒸馏提取后的药渣,加入8倍重量比的70%乙醇浸渍50h,再用70%乙醇作溶剂缓慢渗滤至滤液呈微黄色,滤液在70℃下减压浓缩得相对密度为1.25的稠膏;再加12倍水充分搅拌并静置24h使油水分离,去除沉淀物和油脂,收集水层药液,加入药用活性炭静置2h吸附药液至无色,上聚酰胺色谱柱,用水饱和正丁醇洗脱,洗脱液在70℃下减压浓缩后加入10倍重量的95%乙醇进行二次乙醇提取,乙醇提液在6℃冷藏24h后过滤,滤液经减压浓缩后,再加入10倍重量份的水进行水提,经冷藏后24h,4℃离心过滤得水提液,水提液用0.3%重量比的活性炭脱色,边加边搅拌,并加热至40℃后静止1h;经膜过滤器过滤,超滤后在40℃下减压浓缩,得提取物稠膏;将提取物稠膏、水蒸气蒸馏提取物、药用聚乙二醇、甘露醇和医用氯化钠混合,用注射用水稀释,滤过,灌封、灭菌、灯检、包装,即得甲药物注射液。
1.2 乙药物注射液:
处方:丹参500g、川芎300g、骨碎补200g。
制备方法:取干燥的丹参500g、川芎300g和骨碎补200g,混合,粉碎成粗粉,加入8倍重量比的70%乙醇浸渍50h,再用70%乙醇作溶剂缓慢渗滤至滤液呈微黄色,滤液在70℃下减压浓缩得相对密度为1.25的稠膏;再加12倍水充分搅拌并静置24h使油水分离,去除沉淀物和油脂,收集水层药液,加入药用活性炭静置2h吸附药液至无色,上聚酰胺色谱柱,用水饱和正丁醇洗脱,洗脱液在70℃下减压浓缩后加入10倍重量的95%乙醇进行二次乙醇提取,乙醇提液在6℃冷藏24h后过滤,滤液经减压浓缩后,再加入10倍重量份的水进行水提,经冷藏后24h,4℃离心过滤得水提液,水提液用0.3%重量比的活性炭脱色,边加边搅拌,并加热至40℃后静止1h;经膜过滤器过滤,超滤后在40℃下减压浓缩,得提取物稠膏;将提取物稠膏、药用聚乙二醇、甘露醇和医用氯化钠混合,用注射用水稀释,滤过,灌封、灭菌、灯检、包装,即得乙药物注射液。
1.3 丙药物注射液:
处方:丹参500g、川芎300g、骨碎补200g。
制备方法:取干燥的丹参500g、川芎300g和骨碎补200g,混合,粉碎成粗粉,将其进行水蒸气蒸馏提取,从中提取得到水蒸气蒸馏提取物,备用;取水蒸气蒸馏提取后的药渣,加入8倍重量比的70%乙醇浸渍50h,再用70%乙醇作溶剂缓慢渗滤至滤液呈微黄色,滤液在70℃下减压浓缩得相对密度为1.25的提取物稠膏;将提取物稠膏、水蒸气蒸馏提取物、药用聚乙二醇、甘露醇和医用氯化钠混合,用注射用水稀释,滤过,灌封、灭菌、灯检、包装,即得丙药物注射液。
1.4 丁药物注射液:
处方:丹参500g、川芎300g、骨碎补200g。
制备方法:取干燥的丹参500g、川芎300g和骨碎补200g,混合,粉碎成粗粉,将其进行水蒸气蒸馏提取,从中提取得到水蒸气蒸馏提取物,备用;取水蒸气蒸馏提取后的药渣,加入8倍重量比的70%乙醇浸渍50h,再用70%乙醇作溶剂缓慢渗滤至滤液呈微黄色,滤液在70℃下减压浓缩得相对密度为1.25的稠膏;再加12倍水充分搅拌并静置24h使油水分离,去除沉淀物和油脂,收集水层药液,加入药用活性炭静置2h吸附药液至无色,药液经膜过滤器过滤,超滤后在40℃下减压浓缩,得提取物稠膏;将提取物稠膏、水蒸气蒸馏提取物、药用聚乙二醇、甘露醇和医用氯化钠混合,用注射用水稀释,滤过,灌封、灭菌、灯检、包装,即得丁药物注射液。
2 测定方法
采用本发明实验四提供的检测方法对各个药物中的活性成分进行含量测定。
3 测定结果
测定结果见表14
表14 样品含量测定结果 (mg·mL-1,n=3)
4 讨论与结论
从表9中可以看出,甲药物注射液中的间-乙基苏合香烯、对-伞花烃、邻苯二甲酸二异丁酯、对-伞花烯、2,4-癸二烯醛的含量明显高于各个对比药物,这可能也是甲药物注射液在治疗股骨头坏死方面的效果优于其他药物的原因。
附图说明:
图1:对照品GC色谱图。其中,1号峰为正十六酸,2号为α-侧柏烯,3号峰为γ-松油烯,4号峰为邻苯二甲酸二异丁酯,5号峰为对-伞花烯,6号峰为2,4-癸二烯醛。
图2:对照药材GC色谱图。其中,1号峰为正十六酸,2号为α-侧柏烯,3号峰为γ-松油烯,4号峰为邻苯二甲酸二异丁酯,5号峰为对-伞花烯,6号峰为2,4-癸二烯醛。
图3:供试品GC色谱图。其中,其中,1号峰为正十六酸,2号为α-侧柏烯,3号峰为γ-松油烯,4号峰为邻苯二甲酸二异丁酯,5号峰为对-伞花烯,6号峰为2,4-癸二烯醛。
图4:10样品特征图谱叠加图。其中,1号峰为间-乙基苏合香烯,2号为对-伞花烃,3号峰为邻苯二甲酸二异丁酯,4号峰为对-伞花烯,5号峰为2,4-癸二烯醛。
具体实施方式
以下通过具体实施例对本发明进行进一步阐述:
实施例1:
处方:丹参500g、川芎300g、骨碎补200g。
制备方法:取干燥的丹参500g、川芎300g和骨碎补200g,混合,粉碎成粗粉,将其进行水蒸气蒸馏提取,从中提取得到水蒸气蒸馏提取物,备用;取水蒸气蒸馏提取后的药渣,加入8倍重量比的70%乙醇浸渍50h,再用70%乙醇作溶剂缓慢渗滤至滤液呈微黄色,滤液在70℃下减压浓缩得相对密度为1.25的稠膏;再加12倍水充分搅拌并静置24h使油水分离,去除沉淀物和油脂,收集水层药液,加入药用活性炭静置2h吸附药液至无色,上聚酰胺色谱柱,用水饱和正丁醇洗脱,洗脱液在70℃下减压浓缩后加入10倍重量的95%乙醇进行二次乙醇提取,乙醇提液在6℃冷藏24h后过滤,滤液经减压浓缩后,再加入10倍重量份的水进行水提,经冷藏后24h,4℃离心过滤得水提液,水提液用0.3%重量比的活性炭脱色,边加边搅拌,并加热至40℃后静止1h;经膜过滤器过滤,超滤后在40℃下减压浓缩,得提取物稠膏;将提取物稠膏、水蒸气蒸馏提取物、药用聚乙二醇、甘露醇和医用氯化钠混合,用注射用水稀释,滤过,灌封、灭菌、灯检、包装,即得药物注射液;
采用如下检测方法对该药物进行检测:
(1)色谱条件:色谱柱:DB-WAX毛细管色谱柱,柱长30m,内径0.53mm,膜厚1m,氢火焰离子化检测器,氮气流速:1mL·min-1,氢气流速:10mL·min-1,空气流速:40mL·min-1,柱温为程序升温,初始温度80℃,以每分钟5℃的速度升温至200℃保持10min;分流进样:10:1,进样口温度:200℃,检测器温度:250℃;
(2)混合对照品溶液制备:精密称取间-乙基苏合香烯对照品29.76mg、对-伞花烃对照品58.33mg,分别置10mL量瓶中;精密称取邻苯二甲酸二异丁酯对照品16.06mg、对-伞花烯对照品12.54mg、2,4-癸二烯醛对照品15.69mg,分别置25mL量瓶中;向各个两瓶中加入体积比为1:1的无水甲醇-乙酸乙酯的混合溶液至刻度,摇匀,作为对照品储备液;精密吸取上述对照品储备液各1mL,置同一10mL量瓶中,加上述体积比为1:1的无水甲醇-乙酸乙酯的混合溶液稀释到刻度,作为混合对照品溶液;
(3)供试品溶液制备:精密量取药物注射液10mL,加在C18固相萃取柱上,以每秒两滴的速度流出,弃去流出液,再用体积比为1:1的无水甲醇-乙酸乙酯的混合溶液9mL洗脱,收集洗脱液于10mL量瓶中,加上述体积比为1:1的无水甲醇-乙酸乙酯的混合溶液至刻度,摇匀,即得供试品溶液;
(4)测定:精密吸取混合对照品溶液、供试品溶液各1μl,注入气相色谱仪测定。
(5)测定结果:间-乙基苏合香烯含量为0.46mg·mL-1,对-伞花烃含量为0.62mg·mL-1,邻苯二甲酸二异丁酯含量为0.055mg·mL-1,为对-伞花烯含量为0.047mg·mL-1,2,4-癸二烯醛含量为0.062mg·mL-1
根据《中国药典》2010年版二部附录XIX C稳定性试验指导原则,对最优组合的实施例1制得样品进行稳定性试验考察:
加速试验:将实施例1样品,置于温度40℃、相对湿度75%的恒温恒湿箱中,放置6个月,分别于第1、2、3、6个月时取样,按照稳定性考察项目进行检测,并与0月的数据进行比较。
长期试验:将实施例1样品,置于温度25℃、相对湿度60%的条件下放置,分别于第3、6、12、18个月时取样,按照考察项目进行检测,并与0月的数据进行比较。稳定性试验结果见表15。
表15 实施例1注射液稳定性考察试验结果
由表15数据可见,本发明注射液加速试验6月与长期试验18月后,各项质量均符合规定,本发明技术方案制备的注射液质量稳定可控。
- 还没有人留言评论。精彩留言会获得点赞!