含偶氮苯衍生物的脂质体及其制备方法和应用与流程
本发明涉及刺激响应载体领域,尤其涉及一种含偶氮苯衍生物的脂质体及其制备方法和应用。
背景技术:
纳米技术的突破对多个行业产生了重要的影响,尤其是对材料科学、生物技术和药物治疗。不同的纳米载体,如脂质体、聚合物、胶束和碳基纳米材料正逐渐应用到医学领域,例如治疗性药物大分子的运输(药物和基因)。但是,目前治疗性药物大分子的运输仍面临着很大的挑战,因为我们所需要的药物运输体系最好能够实现药物的定时、定点和定量释放。对用于药物运输体系,目前已经有一定的研究,例如:通过智能化微米/纳米粒子(MNPs)来实现。所述MNPs是指能够以某种具体的方式响应外部/内部刺激,进而实现内部药物的可控释放的微米/纳米粒子的总称。光照由于其强度、波长、照射时间等参数可以精确调节,并且对生物体是非侵入性的,所以,能够对光刺激进行响应的MNPs具有广阔的应用前景。
因此,需要一种能够对光刺激进行响应的含偶氮苯衍生物的脂质体。
技术实现要素:
本发明的目的在于提供了一种含偶氮苯衍生物的脂质体及其制备方法和应用,所述含偶氮苯衍生物的脂质体能够对光刺激进行响应。
为实现上述目的,本发明提供了一种含偶氮苯衍生物的脂质体,所述含偶氮苯衍生物的脂质体中的偶氮苯衍生物、磷脂和胆固醇的摩尔比为:1~2:1~2:1~2;
所述偶氮苯衍生物的结构式如下:
其中,R1为C1~6的直链烷基;R2为C12~16的直链疏水性烷基;X为:O或NH。
其中,所述含偶氮苯衍生物的脂质体还包括:包封在所述含偶氮苯衍生物的脂质体内的药物。
其中,所述药物为阿霉素。
为了进一步实现发明目的,本发明还提供了一种含偶氮苯衍生物的脂质体的制备方法,所述偶氮苯衍生物的结构式如下:
其中,R1为C1~6的直链烷基;R2为C12~16的直链疏水性烷基;X为:O或NH;
所述含偶氮苯衍生物的脂质体的制备方法包括以下步骤:
A、将摩尔比为:1~2:1~2:1~2的偶氮苯衍生物、磷脂和胆固醇溶于有机溶剂中,旋干,得脂质薄膜;
B、加入水化试剂,然后进行超声处理,得含偶氮苯衍生物的脂质体。
其中,所述有机溶剂为按2:1体积比配置的氯仿和甲醇混合液。
其中,所述水化试剂为pH=7.4的柠檬酸溶液或pH=7.4的磷酸缓冲液。
其中,所述步骤B具体为:
在瓶壁上有脂质薄膜的瓶中加入适量水化试剂,用探头式超声波细胞破碎仪超声10~40min,然后静置于25~42℃环境中,得含偶氮苯衍生物的脂质体悬液。
其中,所述含偶氮苯衍生物的脂质体的制备方法还包括以下步骤:
C、利用pH梯度法,将阿霉素包封在含偶氮苯衍生物的脂质体中,得内包阿霉素的含偶氮苯衍生物的脂质体。
其中,所述步骤C具体为:
将阿霉素加入至含偶氮苯衍生物的脂质体悬液中,充分溶解,然后用NaOH调pH至7.4,8 000~12 000g离心,所得上层液体即为内包阿霉素的含偶氮苯衍生物的脂质体。
为了进一步实现本发明的目的,本发明还提供了上述含偶氮苯衍生物的脂质体在制备药物过程中的应用。
本发明的含偶氮苯衍生物的脂质体能够对紫外光进行响应,当所述含偶氮苯衍生物的脂质体包封有药物时,可通过紫外线的照射来实现该含偶氮苯衍生物的脂质体内部药物的可控释放,从而减轻药物的副作用,提高治疗效果。本发明的含偶氮苯衍生物的脂质体的制备方法简单,效率高。
附图说明
图1是本发明第一具体制备实施例制备所得偶氮苯衍生物的结构式。
图2是本发明第二具体制备实施例制备所得偶氮苯衍生物的结构式。
图3是本发明第三具体制备实施例制备所得偶氮苯衍生物的结构式。
图4是本发明第三具体制备实施例制备所得偶氮苯衍生物在经紫外光照射后的紫外-可见吸收光谱变化图。
图5是本发明第四具体制备实施例制备所得内包阿霉素的含偶氮苯衍生物的脂质体的光控释放曲线。
具体实施方式
下面结合具体实施例对本发明做进一步的详细说明,以下实施例是对本发明的解释,本发明并不局限于以下实施例。
在本发明的第一实施例中,一种偶氮苯衍生物,结构式如下图1所示:
其中,R1为C1~6的直链烷基;R2为C12~16的直链疏水性烷基;X为:O或NH。
本发明的偶氮苯衍生物的一端的直链疏水性烷基链能够插入脂质体的磷脂双分子层中,另一端的亲水性阳离子季铵盐基团则能朝向磷脂双分子层膜外的水溶液,从而在脂质体中形成规律性的排布。本发明的偶氮苯衍生物在紫外光照射下可以发生可逆的构象改变,在未经紫外线照射时,保持反式构型,经紫外线照射后,变换成顺式构型。
优选的,所述R1为C2~4的直链烷基,即,乙烷基、丙烷基或正丁烷基。
优选的,所述R2为C12、C14或C16的直链疏水性烷基。
在本发明的第二实施例中,一种制备上述偶氮苯衍生物的方法,包括以下步骤:
A、使化合物1和化合物2反应,得化合物3;
B、将化合物3和N-溴代琥珀酰亚胺在惰性气体氛围下溶于四氯甲烷并在过氧化苯甲酰的催化下发生溴化反应,得化合物4;
C、使化合物4与第一底物反应,得所述偶氮苯衍生物;
所述化合物1的结构式如下:
所述化合物2的结构式如下:
X2-R2;
所述化合物3的结构式如下:
所述化合物4的结构式如下:
所述X1为OH或NH2;
所述X2为与X1反应形成X的COOH或COCl;
所述第一底物的结构式为:
需要说明的是,所述惰性气体包括氦气、氩气、氖气或氙气中的至少一种。
在本发明的一具体实施例中,所述X1为NH2;所述X2为COCl;所述步骤A包括以下步骤:
A1、将化合物1溶于二氯甲烷,然后加入三乙胺,得第一反应溶液;
A2、将化合物2溶于二氯甲烷,得第二反应溶液;
A3、将第二反应溶液滴入第一反应溶液中,常温搅拌过夜;
A4、除去步骤A3的产物中的二氯甲烷,得固体;
A5、将固体溶于二氯甲烷,并通过柱层析进行分离纯化,所述流动相为按1:1体积比配制的石油醚和二氯甲烷混合液,得化合物3。
需要说明的是,在所述步骤A中,二氯甲烷主要作为反应溶剂,三丁胺为催化剂;步骤A4可通过真空干燥或旋蒸实现。另外,在本方案中,化合物1中的NH2和化合物2中的COCl反应,从而生成化合物3,这种方法与通过化合物中的OH或NH2和化合物2中的COOH反应生成化合物3的方法相比,反应效率更高,制备得到化合物3的得率更高。
在上一具体实施例的基础上,本发明提出另一具体实施例,所述步骤A还包括以下步骤:
A0、将正十二烷酸加入氯化亚砜,加热至60-70℃,搅拌过夜,进而除去未反应的氯化亚砜,得化合物2。
需要说明的是,步骤A0中可通过真空干燥或旋蒸等方法除去氯化亚砜。
在本发明的另一具体实施例中,所述步骤B包括以下步骤:
B1、将化合物3、N-溴代琥珀酰亚胺和过氧化苯甲酰在氩气氛围下溶于四氯甲烷,加热回流反应12-72小时;
B2、将反应体系冷却至0℃,抽滤得粉末;
B3、用乙醚洗涤粉末,得化合物4。
需要说明的是,步骤B1中的过氧化苯甲酰为催化剂;步骤B2抽滤所得粉末为化合物4的粗产物;步骤B3中用乙醚洗涤粉末,主要是通过乙醚将未反应的杂质去除,从而起到纯化的作用。
在本发明的另一具体实施例中,所述步骤C包括以下步骤:
C1、将化合物4溶于乙醇,再加入第一底物,回流反应6-48小时;
C2、将步骤C1的产物蒸干,再用按5:1体积比配制的乙醚和二氯甲烷混合液溶解,过滤收集固体沉淀。
需要说明的是,步骤C2中,可通过旋蒸将步骤C1的产物蒸干;步骤C2所得固体沉淀为本发明所述偶氮苯衍生物的粗产物。
基于上一具体实施例,本发明提出另一具体实施例,所述步骤C还包括以下步骤:
C3、用按5:1体积比配制的乙醚和二氯甲烷混合液洗涤固体沉淀,得所述偶氮苯衍生物。
需要说明的是,所述按5:1体积比配制的乙醚和二氯甲烷混合液能够溶解未发生反应的杂质,从而起到提纯的作用。
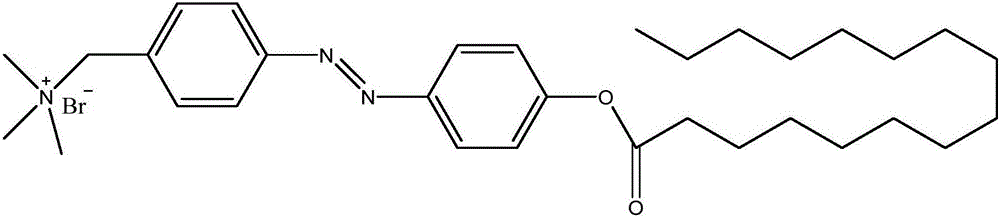
在本发明的第一具体制备实施例中,所述偶氮苯衍生物的结构式如图1所示,该偶氮苯衍生物的制备方法包括以下步骤:
S01、使化合物1(4-甲基-4’羟基偶氮苯)与化合物2(正十四烷酸)在浓硫酸的催化和加热条件下发生脂化反应,生成化合物3(4-甲基-4’正十四烷酸偶氮苯酯);
S02、将4-甲基-4’正十四烷酸偶氮苯酯、N-溴代琥珀酰亚胺和过氧化苯甲酰在氩气氛围下溶于四氯甲烷,加热回流反应24小时;
S03、将步骤S02的反应体系冷却至0℃,抽滤得粉末;
S04、用乙醚洗涤步骤S03所得粉末,除去未反应的杂质,得化合物4(4-溴甲基-4’正十四烷酸偶氮苯酯);
S05、将4-溴甲基-4’正十四烷酸偶氮苯酯溶于乙醇,然后加入三乙胺,回流反应12小时;
S06、将溶液蒸干,再用按5:1体积比配制的乙醚和二氯甲烷混合液溶解,过滤收集固体沉淀,即得图1所示偶氮苯衍生物,即4-(N,N,N-三乙胺甲基溴化物)-4’-正十四烷酸偶氮苯酯。
在本发明的第二具体制备实施例中,所述偶氮苯衍生物的结构式如图2所示,该偶氮苯衍生物的制备方法包括以下步骤:
S01、使化合物1(4-甲基-4’羟基偶氮苯)与化合物2(正十六烷酸)在浓硫酸的催化以及加热条件下发生脂化反应,生成化合物3(4-甲基-4’-正十六烷酸偶氮苯酯);
S02、将4-甲基-4’-正十六烷酸偶氮苯酯、N-溴代琥珀酰亚胺和过氧化苯甲酰在氩气氛围下溶于四氯甲烷,加热回流反应48小时;
S03、将步骤S02的反应体系冷却至0℃,抽滤得粉末;
S04、用乙醚洗涤步骤S03所得粉末,除去未反应的杂质,得化合物4(4-溴甲基-4’-正十六烷酸偶氮苯酯);
S05、将4-溴甲基-4’-正十六烷酸偶氮苯酯溶于乙醇,然后加入三甲胺,回流反应24小时;
S06、将溶液蒸干,再用按5:1体积比配制的乙醚和二氯甲烷混合液溶解,过滤收集固体沉淀,即粗产物。
S07、用按5:1体积比配制的乙醚和二氯甲烷混合液洗涤固体沉淀3次,得图2所示偶氮苯衍生物,即4-(N,N,N-三甲胺甲基溴化物)-4’-正十六烷酸偶氮苯酯。
在本发明的第三具体制备实施例中,所述偶氮苯衍生物的结构式如图3所示,该偶氮苯衍生物的制备方法包括以下步骤:
S01、将2g正十二烷酸加入10mL氯化亚砜中,65℃搅拌过夜,旋蒸出去未反应得氯化亚砜,得到2g无色油状粗产物,即化合物2(正十二酰氯);
S02、将0.62g化合物1(4-甲基-4’氨基偶氮苯)溶于20mL二氯甲烷,然后加入0.59g三乙胺,得第一反应溶液;
S03、将0.97g无色油状粗产物(正十二酰氯)溶于5mL二氯甲烷,得第二反应溶液;
S04、将第二反应溶液慢慢滴入第一反应溶液中,25℃搅拌过夜;
S05、真空干燥除去步骤S04的产物中的二氯甲烷,得黄色固体;
S06、用二氯甲烷溶解步骤S05所得黄色固体,同时加入200-300目的硅胶搅拌,旋蒸除去二氯甲烷后上样,加流动相进行柱层析,所述流动相为按1:1体积比配制的石油醚和二氯甲烷混合液;得纯化后的化合物3(4-甲基-4’正十二酰氨基偶氮苯)1.12g;
S07、将0.5g 4-甲基-4’正十二酰氨基偶氮苯、0.34g N-溴代琥珀酰亚胺和15mg过氧化苯甲酰在氩气氛围下溶于四氯甲烷,加热回流反应48小时;
S08、将步骤S07产物冷却至0℃,抽滤得黄色粉末;
S09、用乙醚洗涤黄色粉末,除去未反应的杂质,得化合物4(4-溴甲基-4’-正十二酰氨基偶氮苯);
S10、将250mg 4-溴甲基-4’-正十二酰氨基偶氮苯溶于6mL乙醇,然后加入1mL三丁胺和2mL乙醇,回流反应24小时;
S11、将步骤S10的产物蒸干,再用20mL按5:1体积比配制的乙醚和二氯甲烷混合液溶解,过滤收集橘黄色固体沉淀;
S12、用20mL按5:1体积比配制的乙醚和二氯甲烷混合液洗涤橘黄色固体沉淀三次,得120mg图3所示偶氮苯衍生物,即4-(N,N,N-三丁胺甲基溴化物)-4’-正十二酰氨基偶氮苯。
为了进一步证明本发明的偶氮苯衍生物的光响应特性,本发明的发明人设计了以下试验,以氯仿或乙醇为溶剂,将所述偶氮苯衍生物配置成约1μmol/L的溶液,并测定其紫外可见吸收光谱,然后用紫外光照射不同的时间,测各个时间点的吸收光谱,然后按不同的紫外光照射时间,分别作吸光度和波长的曲线。其中,以第三具体实施例所得偶氮苯衍生物的结果如图4所示。如图4所示,随着紫外线照射时间的延长,该偶氮苯衍生物在350nm处的吸光度值不断降低,说明该偶氮苯衍生物可以发生快速的反式-顺式异构,可作为一种光控开关。
需要说明的是,本发明的其他偶氮苯衍生物,例如上述第一具体制备实施例、第二具体制备实施例中所得的偶氮苯衍生物,在上述试验中的结果与图4类似(图未示),说明本发明的偶氮苯衍生物可以发生快速的反式-顺式异构,可作为一种光控开关。
在本发明的第三实施例中,一种制备含上述偶氮苯衍生物的脂质体的方法,包括以下步骤:
A、将摩尔比为:1~2:1~2:1~2的偶氮苯衍生物、磷脂和胆固醇溶于有机溶剂中,旋干,得脂质薄膜;
B、加入水化试剂,然后进行超声处理,得含偶氮苯衍生物的脂质体。
需要说明的是,在第三实施例中,所述磷脂为磷脂酰胆碱、磷脂酰乙醇胺、磷脂酸、磷脂酰丝氨酸、磷脂酰甘油、磷脂酰肌醇、或它们的任意组合物。所述机溶剂包括氯仿、二氯甲烷、甲醇、乙醇和乙醚中的至少一种。所述水化试剂用于给含偶氮苯衍生物的脂质体提供稳定的环境,形成脂质体悬液。
第三实施例中通过超声分散法制备含偶氮苯衍生物的脂质体,制备方法简单,效率高。
若要制备包封有药物的含上述偶氮苯衍生物的脂质体,根据药物性质的不同,可在第三实施例的方法的基础上,通过不同的处理获得。
在本发明的一个实施例中,当药物为水溶性药物时,可将适量的药物溶于活性溶液中,例如pH=7.4的磷酸盐缓冲液(PBS),然后将该溶液加入瓶壁上有脂质薄膜的瓶中,用探头式超声波细胞破碎仪超声10~40min,然后静置于25~42℃环境中静置1~6小时。将所得产物加入透析袋中,并在pH=7.4的磷酸盐缓冲液(PBS)中透析,除去未被包封的药物,最终获得内包药物的含偶氮苯衍生物的脂质体悬液。
需要说明的是,本实施例中,所述溶有药物的活性溶液的量,可根据瓶中瓶壁上的脂质薄膜的覆盖范围进行调整;所述透析袋的分子量大小根据药物的分子量进行选择。
在本发明的另一个实施例中,当药物为脂溶性药物,例如阿霉素时,可将适量的300mM的柠檬酸溶液(pH=7.4)加入瓶壁上有脂质薄膜的瓶中,用探头式超声波细胞破碎仪超声10~40min,然后静置于25~42℃环境中静置1~6小时。然后用pH梯度法包封阿霉素(DOX),获得内包阿霉素的含偶氮苯衍生物的脂质体悬液。
需要说明的是,本发明所述的内包药物可为水溶性药物,例如:盐酸吉西他滨、卡铂、盐酸阿糖胞苷、盐酸氮芥、盐酸米托蒽醌;也可为脂溶性药物,例如阿霉素、紫杉醇等。
在本发明的第四具体制备实施例中,所述偶氮苯衍生物为上述第三具体制备实施例制备所得的产物;所述药物为阿霉素。
1)将磷脂、胆固醇、偶氮苯衍生物按照1:1:1的摩尔比(各0.02mmol)一起溶于按2:1体积比配置的20mL氯仿和甲醇混合液,充分溶解后,在37℃恒温水浴下用旋转蒸发器除去有机溶剂,圆底烧瓶底形成一层均匀脂质薄膜。
2)然后加入10mL,300mM的柠檬酸溶液(pH=7.4),将脂质薄膜水化下来,接着用探头式超声波细胞破碎仪超声30min(功率100mW,on 30s,off 30s),然后置于37℃水浴中放置2小时以完成封闭过程,所得即为脂质体悬液。
3)用pH梯度法包封阿霉素(DOX):称取2mg DOX,加入到脂质体悬液中,充分溶解,用1.0M NaOH调pH至7.4。将脂质体溶液10 000g离心10min,除去超声过程中探头上掉下来的钛颗粒以及未分散的脂质,上层液体即为内包阿霉素的含偶氮苯衍生物的脂质体悬液。
为了验证所得内包阿霉素的光响应脂质体的光控释放效果,本发明的发明人设计了另外一个验证试验。
实验组:将第四具体制备实施例制备的内包阿霉素的含偶氮苯衍生物的脂质体悬液5mL分散在45mL pH=7.4的PBS缓冲液中,然后将所得的溶液放置在黑暗环境中,每小时用紫外光照射10min,每2个小时测定一次PBS缓冲液中被释放出的阿霉素含量。
对照组:将第四具体制备实施例制备的内包阿霉素的光响应脂质体5mL分散在45mL pH=7.4的PBS缓冲液中,然后将所得的溶液放置在黑暗环境中,同样每2个小时测定一次PBS缓冲液中被释放出的阿霉素含量。
实验组、对照组各做3个重复。
以每个时间点PBS缓冲液中被释放出的阿霉素含量与该PBS缓冲液中的总阿霉素量的比值为纵坐标,时间为横坐标,作曲线,结果如图5所示。
如图5所示,在紫外线光照下,内包阿霉素的光响应脂质体的磷脂双分子层中的偶氮苯化合物发生反式-顺式异构,对磷脂膜造成扰动,并在磷脂双分子层中形成通道,进而使得阿霉素被释放出,加速内包阿霉素的光响应脂质体内部的阿霉素的释放。
因此,本发明的偶氮苯化合物能够对光刺激进行响应,基于本发明的偶氮苯化合物制备的含偶氮苯衍生物的脂质体可制备成内包药物的MNPs,具有广阔的应用前景。
以上仅为本发明的优选实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书及附图内容所作的等效结构或等效流程变换,或直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!