一种新型负载大黄素用纳米粒子的制备方法与流程
本发明属于生物黏附性药物载体的技术领域,具体涉及一种新型负载大黄素用纳米粒子的制备方法。
背景技术:
壳聚糖(chitosan,cs)又名2-氨基-2-脱氧-β-d-葡糖,是甲壳素(2-乙酸氨基-2-脱氧-β-d-葡糖)脱乙酰化的产物。甲壳素又名蟹壳素、甲壳质、几丁质和壳多糖等,是一种天然的线性多糖,是甲壳纲动物外壳的重要成分,也存在于低等植物如菌、藻类的细胞壁中。由于壳聚糖无毒,且具有优良的生物相容性,在生物体内易降解等特点,被广泛应用于医用辅料领域,壳聚糖能与黏膜蛋白形成氢键和静电作用,具有良好的生物黏附性能,但这种基于非共价键的黏附作用不保证药物在指定部位的持续释放,限制了壳聚糖的应用,而壳聚糖经巯基化后,黏附性能显著提高,这是因为硫化聚合物(thiomers)能够与黏膜层形成二硫键,与黏蛋白中半胱氨酸丰富的亚区发生特异性结合。
然而巯基化聚合物到目前为止作为疏水性药物载体应用并不理想,这是由于巯基化聚合物与疏水药物分子作用很弱,常常导致药物释放、快、不持续释放、包封率较差现象。
技术实现要素:
本发明所要解决的技术问题在于针对上述现有技术的不足,提供一种新型负载大黄素用纳米粒子的制备方法,该新型巯基化纳米粒子通过分步合成的方法合成巯基化mpeg-pla-cs-mbi纳米粒子,合成过程中最后亲水性聚乙二醇单甲醚(mpeg)与聚乳酸(pla)和壳聚糖(cs)形成mpeg-pla-cs聚合物,并在此基础上通过5-氨基-2-巯基苯并咪唑(mbi)使聚合物巯基化,最终形成巯基聚合物(mpeg-pla-cs-mbi)。该巯基聚合物(mpeg-pla-cs-mbi)可负载疏水性药物如大黄素等用作缓释药物。巯基聚合物通过巯基氧化形成二硫键粘附于黏膜表面,赋予纳米粒子更强的黏附性能,达到延长药物在黏膜上的滞留时间,有利于药物分子的缓释。同时,改性后壳聚糖与药物结合形成纳米复合物后,mpeg可在复合物表面形成核-壳结构胶束,保护纳米复合物不被网状内皮系统(res系统)识别和清除,使得所制得的粒子具有表面稳定作用,能促进复合物粒子在体内的长循环目的。
为解决上述技术问题,本发明采用的技术方案是:一种新型负载大黄素用纳米粒子的制备方法,其特征在于,该方法包括以下步骤:
步骤一:将5g~20g左旋丙交酯、2g~10g聚乙二醇单甲醚和0.2g~1g异辛酸亚锡溶解于20ml二氯甲烷中,在130℃的条件反应18h后置于冰乙醚中沉淀3次,再在40℃的真空条件下干燥3天,得到第一中间产物mpeg-pla-oh;
步骤二:将10g步骤一中制备的第一中间产物mpeg-pla-oh、2g丁二酸酐和1.2g4-二甲氨基吡啶溶于100ml氯仿中,搅拌均匀后加入2ml三乙胺,在室温条件下反应3天,然后置于乙醚中沉淀3次,经过滤得到滤渣,再将所述滤渣在40℃的真空条件下干燥3天,最后得到第二中间产物mpeg-pla-cooh;
步骤三:将2.5g步骤二中制备的第二中间产物mpeg-pla-cooh溶于40ml的二氯甲烷中,再加入0.7g1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐和0.7gn-羟基琥珀酰亚胺,在室温条件下反应24h,旋蒸后溶于二甲基亚砜溶液中,再加入到添加有壳聚糖的60ml二甲基亚砜中反应24h,透析3天,冷冻干燥,得到第三中间产物mpeg-pla-cs;所述壳聚糖的添加量为0.1g~1g,脱乙酰度为85%;
步骤四:将0.5g步骤三中制备的第三中间产物mpeg-pla-cs溶于140ml水中,再加入0.3g高碘酸钠溶液,室温孵化2h后,加入300μl的乙二醇,在室温条件下反应2h,透析3天,冷冻干燥,得到第四中间产物mpeg-pla-cs-cho,并在4℃下保存;所述高碘酸钠溶液的浓度是2.14g/l;
步骤五:将0.2g~1g5-氨基-2-巯基苯并咪唑和0.2g步骤四中制备的第四中间产物mpeg-pla-cs-cho溶于在40ml二甲基亚砜溶液中,并在室温条件下孵化2h,然后加入0.2g~2g的氰基硼氢化钠,再在室温条件下反应24h~72h,透析3天,冷冻干燥,得到用于负载大黄素的巯基化mpeg-pla-cs-mbi纳米粒子,并在4℃下保存;
步骤三中所述二甲基亚砜溶液和步骤五中所述二甲基亚砜溶液均由二甲基亚砜和水按照1:1的体积比混合而成。
上述一种新型负载大黄素用纳米粒子的制备方法,其特征在于,步骤一中所述聚乙二醇单甲醚的平均分子量为1000~4000。
上述一种新型负载大黄素用纳米粒子的制备方法,其特征在于,步骤一中所述左旋丙交酯的质量为14.4g,聚乙二醇单甲醚的质量为7.6g,异辛酸亚锡的质量为0.2g。
上述一种新型负载大黄素用纳米粒子的制备方法,其特征在于,步骤三中所述壳聚糖的添加量为0.5g。
上述一种新型负载大黄素用纳米粒子的制备方法,其特征在于,步骤五中所述5-氨基-2-巯基苯并咪唑质量为0.5g。
上述一种新型负载大黄素用纳米粒子的制备方法,其特征在于,步骤五中所述氰基硼氢化钠的加入质量为0.2g。
上述一种新型负载大黄素用纳米粒子的制备方法,其特征在于,步骤五中所述反应的时间为48h。
本发明与现有技术相比具有以下优点:
1、本发明通过分步骤合成的方法合成巯基化mpeg-pla-cs-mbi纳米粒子,合成过程中最后亲水性聚乙二醇单甲醚(mpeg)与聚乳酸(pla)和壳聚糖(cs)形成mpeg-pla-cs聚合物,并在此基础上通过5-氨基-2-巯基苯并咪唑(mbi)使聚合物巯基化,最终形成巯基聚合物(mpeg-pla-cs-mbi)。巯基聚合物通过巯基氧化形成二硫键粘附于黏膜表面,赋予新型巯基化纳米粒子更强的黏附性能,达到延长药物在黏膜上的滞留时间,有利于药物分子的缓释。同时,改性后壳聚糖与药物结合形成纳米复合物后,聚乙二醇单甲醚可在复合物表面形成核-壳结构胶束,保护纳米复合物不被网状内皮系统(res系统)识别和清除,使得所制得的新型巯基化纳米粒子具有表面稳定作用,能促进复合物粒子在生物体内长循环目的。
2、采用本发明制备的巯基化纳米粒子负载大黄素后的药物包封率不小于83.6%,载药量不小于3.89%,并且具有良好的水溶性和生物降解性。
3、本发明制备的巯基化mpeg-pla-cs-mbi纳米粒子不仅限于用于负载大黄素,还可应用于负载类似于大黄素的疏水性药物,用作缓释药物的载体。
下面通过附图和实施例,对本发明的技术方案做进一步的详细描述。
附图说明
图1为本发明实施例1中制备的巯基化mpeg-pla-cs-mbi纳米粒子的hnmr谱图。
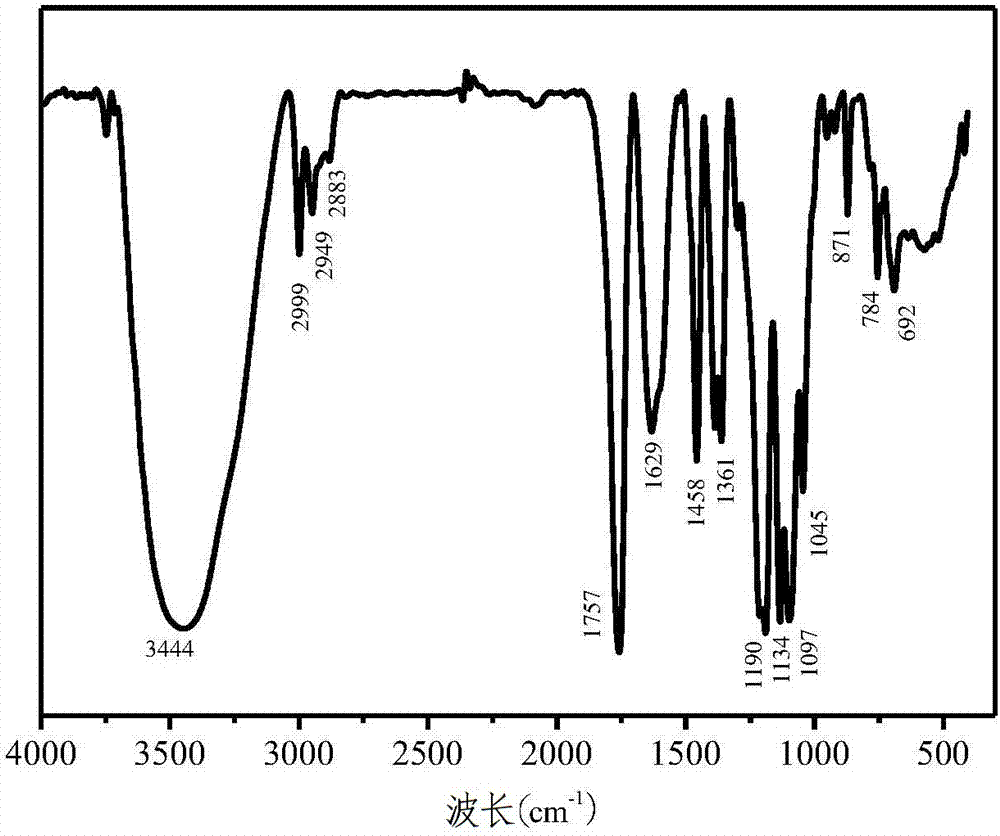
图2为本发明实施例1中制备的巯基化mpeg-pla-cs-mbi纳米粒子的共轭红外谱图。
图3为本发明实施例1中制备的巯基化mpeg-pla-cs-mbi纳米粒子在负载大黄素前后的红外谱图。
具体实施方式
实施例1
步骤一、将14.4g左旋丙交酯(l.a)、7.6g聚乙二醇单甲醚(mpeg)和0.2g异辛酸亚锡溶解于20ml二氯甲烷,在130℃反应18h后于冰乙醚中沉淀3次,40℃真空干燥3天,获得第一中间产物mpeg-pla-oh;
步骤二、将10g步骤一中制备的第一中间产物mpeg-pla-oh、2g丁二酸酐和1.2g4-二甲氨基吡啶溶于100ml氯仿中,均匀后加入2ml三乙胺,室温反应3天,乙醚沉淀3次,过滤,40℃真空干燥3天,获得第二中间产物mpeg-pla-cooh;
步骤三、将2.5g步骤二中制备的第二中间产物mpeg-pla-cooh40ml的二氯甲烷溶液与0.7g1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐和0.7gn-羟基琥珀酰亚胺,室温反应24h,旋蒸后溶于二甲基亚砜溶液,在加入到含0.5g壳聚糖60ml二甲基亚砜溶剂中,反应24h,透析3天,冷冻干燥获得第三中间产物mpeg-pla-cs;所述二甲基亚砜溶液均由二甲基亚砜和水按照1:1的体积比混合而成;
步骤四、将0.5g步骤三中制备的第三中间产物mpeg-pla-cs溶于140ml水中,加入0.3g的高碘酸钠(naio4)溶液,室温孵化2h,加入300μl的乙二醇,室温反应2h,透析3天,冷冻干燥,获得第四中间产物mpeg-pla-cs-cho,在4℃下保存;所述高碘酸钠溶液的浓度是2.14g/l;
步骤五、将0.5g5-氨基-2-巯基苯并咪唑(mbi)与0.2g步骤四中制备的第四中间产物mpeg-pla-cs-cho在40ml二甲基亚砜溶液中混合均匀,所述二甲基亚砜溶液均由二甲基亚砜和水按照1:1的体积比混合而成,2h室温孵化后,加入0.2g的nacnbh3,室温反应48h。透析3天,冷冻干燥得到巯基化mpeg-pla-cs-mbi纳米粒子,在4℃下保存。
将本实施例制备的mpeg-pla-cs-mbi纳米粒子分散于去离子水中,能够完全分散,加入溶有质量含量为40%的大黄素药物的乙醇溶液,超声处理,然后进行磁力搅拌处理,将搅拌后的混合液在8000rpm离心处理,最后对负载了药物的巯基化瓜尔胶纳米粒子进行冷冻,得到最终负载药物的纳米靶向缓控释系统。图3为本实施例制备的巯基化mpeg-pla-cs-mbi纳米粒子在负载大黄素前后的红外谱图,从图中可以看出,在图3中负载大黄素后其在436nm处有吸收,说明成功地将大黄素负载在巯基化mpeg-pla-cs-mbi纳米粒子上。最后经检测,本实施例制备的巯基化mpeg-pla-cs-mbi纳米粒子载药后的包封率为91%,载药量5.01%。
图1为本实施例制备的巯基化mpeg-pla-cs-mbi纳米粒子的hnmr谱图,图2为本实施例制备的巯基化mpeg-pla-cs-mbi纳米粒子的共轭红外谱图,从图2可以看出,分别在1757cm-1,1190cm-1,1134cm-1,1097cm-1,在1629cm-1为酰胺特征吸收峰,692cm-1,784cm-1存在证明苯环的存在,与图1的hnmr图谱中化学位移在7.08ppm,7.5ppm对应;1458cm-1,1361cm-1属于为-ch3变形震动吸收峰,2883cm-1,2949cm-1为-ch2-反伸缩振动吸收峰,在3444cm-1处存在一个较强的吸收峰,为nh的特征伸缩振动吸收峰,且只有一个为仲胺,吸收峰较强,在1045cm-1处为伯醇的特征吸收峰。化学位移在3.65ppm,5.2ppm分别对应聚乙二醇和聚乳酸上氢的位置,进一步证明改性成功。
本实施例的制备过程中亲水性聚乙二醇单甲醚(mpeg)与聚乳酸(pla)形成mpeg-pla-cs聚合物,在此基础上通过5-氨基-2-巯基苯并咪唑(mbi)使聚合物巯基化,最终形成巯基聚合物(mpeg-pla-cs-mbi)。巯基聚合物通过巯基氧化形成二硫键粘附于黏膜表面,达到延长药物在黏膜上的滞留时间,有利于药物分子的吸收,赋予制备的巯基化mpeg-pla-cs-mbi纳米粒子更强的黏附性能。同时,改性后壳聚糖与药物结合形成纳米复合物后,聚乙二醇单甲醚可在复合物表面形成核-壳结构胶束,保护纳米复合物不被网状内皮系统(res系统)识别和清除,使得所制得的制备的巯基化mpeg-pla-cs-mbi纳米粒子具有表面稳定作用,能促进复合物粒子在生物体内长循环目的。
实施例2
步骤一、将5g左旋丙交酯(l.a)、2g聚乙二醇单甲醚(mpeg)和0.6g异辛酸亚锡溶解于20ml二氯甲烷,在130℃反应18h后于冰乙醚中沉淀3次,40℃真空干燥3天,得到第一中间产物mpeg-pla-oh;
步骤二:将10g步骤一中制备的第一中间产物mpeg-pla-oh、2g丁二酸酐和1.2g4-二甲氨基吡啶溶于100ml氯仿中,均匀后加入2ml三乙胺,室温反应3天,乙醚沉淀3次,过滤,40℃真空干燥3天,获得第二中间产物mpeg-pla-cooh;
步骤三、将2.5g步骤二中制备的第二中间产物mpeg-pla-cooh40ml的二氯甲烷溶液与0.7g1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐和0.7gn-羟基琥珀酰亚胺,室温反应24h,旋蒸后溶于二甲基亚砜溶液,在加入到含0.5g壳聚糖60ml二甲基亚砜溶剂中,反应24h,透析3天,冷冻干燥获得第三中间产物mpeg-pla-cs;所述二甲基亚砜溶液均由二甲基亚砜和水按照1:1的体积比混合而成;
步骤四、将0.5g步骤三中制备的第三中间产物mpeg-pla-cs溶于140ml水中,加入0.3g的高碘酸钠(naio4)溶液,室温孵化2h,加入300μl的乙二醇,室温反应2h,透析3天,冷冻干燥,获得第四中间产物mpeg-pla-cs-cho,在4℃下保存;所述高碘酸钠溶液的浓度是2.14g/l;
步骤五、将0.2g5-氨基-2-巯基苯并咪唑(mbi)与0.2g步骤四中制备的第四中间产物mpeg-pla-cs-cho在40ml二甲基亚砜溶液中混合均匀,2h室温孵化后,加入0.5g的nacnbh3,室温反应24h。透析3天,冷冻干燥得到巯基化mpeg-pla-cs-mbi纳米粒子,在4℃下保存,所述二甲基亚砜溶液均由二甲基亚砜和水按照1:1的体积比混合而成。
将本实施例制备的mpeg-pla-cs-mbi纳米粒子分散于去离子水中,能够完全分散,加入溶有大黄素药物的40%乙醇溶液,超声处理,然后进行磁力搅拌处理,将搅拌后的混合液在8000rpm离心处理,最后对负载了药物的巯基化瓜尔胶纳米粒子进行冷冻,得到最终负载药物的纳米靶向缓控释系统。经检测,本实施例制备的巯基化mpeg-pla-cs-mbi纳米粒子载药后的包封率为86.3%,载药量4.11%。
实施例3
步骤一、将20g左旋丙交酯(l.a)、10g聚乙二醇单甲醚(mpeg)和1g异辛酸亚锡溶解于20ml二氯甲烷,在130℃反应18h后于冰乙醚中沉淀3次,40℃真空干燥3天,获得第一中间产物mpeg-pla-oh;
步骤二、将10g步骤一中制备的第一中间产物mpeg-pla-oh、2g丁二酸酐和1.2g4-二甲氨基吡啶溶于100ml氯仿中,均匀后加入2ml三乙胺,室温反应3天,乙醚沉淀3次,过滤,40℃真空干燥3天,获得第二中间产物mpeg-pla-cooh;
步骤三、将2.5g步骤二中制备的第二中间产物mpeg-pla-cooh40ml的二氯甲烷溶液与0.7g1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐和0.7gn-羟基琥珀酰亚胺,室温反应24h,旋蒸后溶于二甲基亚砜溶液,在加入到含0.5g壳聚糖60ml二甲基亚砜溶剂中,反应24h,透析3天,冷冻干燥获得第三中间产物mpeg-pla-cs;所述二甲基亚砜溶液均由二甲基亚砜和水按照1:1的体积比混合而成;
步骤四、将1g步骤三中制备的第三中间产物mpeg-pla-cs溶于140ml水中,加入0.3g的高碘酸钠(naio4)溶液,室温孵化2h,加入300μl的乙二醇,室温反应2h,透析3天,冷冻干燥,获得第四中间产物mpeg-pla-cs-cho,在4℃下保存;所述高碘酸钠溶液的浓度是2.14g/l;
步骤五、将1g5-氨基-2-巯基苯并咪唑(mbi)与0.2g步骤四中制备的第四中间产物mpeg-pla-cs-cho在40ml二甲基亚砜溶液混合均匀,所述二甲基亚砜溶液均由二甲基亚砜和水按照1:1的体积比混合而成,2h室温孵化后,加入2g的nacnbh3,室温反应72h,透析3天,冷冻干燥得到巯基化mpeg-pla-cs-mbi纳米粒子,在4℃下保存。
将本实施例制备的mpeg-pla-cs-mbi纳米粒子分散于去离子水中,能够完全分散,加入溶有大黄素药物的40%乙醇溶液,超声处理,然后进行磁力搅拌处理,将搅拌后的混合液在8000rpm离心处理,最后对负载了药物的巯基化瓜尔胶纳米粒子进行冷冻,得到最终负载药物的纳米靶向缓控释系统。经检测本实施例制备的巯基化mpeg-pla-cs-mbi纳米粒子载药后的包封率为83.6%,载药量3.89%。
以上所述,仅是本发明的较佳实施例,并非对本发明作任何限制。凡是根据发明技术实质对以上实施例所作的任何简单的修改、变更以及等效变化,均仍属于本发明技术方案的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!